
Abstract
Autophagy is a lysosome-dependent intracellular degradative pathway, which mediates the cellular adaptation to nutrient and oxygen depletion as well as to oxidative and endoplasmic reticulum stress. The molecular mechanisms that stimulate autophagy include the activation of energy deprivation sensors, sirtuin-1 (SIRT1) and adenosine monophosphate-activated protein kinase (AMPK). These enzymes not only promote organellar integrity directly, but they also enhance autophagic flux, which leads to the removal of dysfunctional mitochondria and peroxisomes. Type 2 diabetes is characterized by suppression of SIRT1 and AMPK signaling as well as an impairment of autophagy; these derangements contribute to an increase in oxidative stress and the development of cardiomyopathy. Antihyperglycemic drugs that signal through insulin may further suppress autophagy and worsen heart failure. In contrast, metformin and SGLT2 inhibitors activate SIRT1 and/or AMPK and promote autophagic flux to varying degrees in cardiomyocytes, which may explain their benefits in experimental cardiomyopathy. However, metformin and SGLT2 inhibitors differ meaningfully in the molecular mechanisms that underlie their effects on the heart. Whereas metformin primarily acts as an agonist of AMPK, SGLT2 inhibitors induce a fasting-like state that is accompanied by ketogenesis, a biomarker of enhanced SIRT1 signaling. Preferential SIRT1 activation may also explain the ability of SGLT2 inhibitors to stimulate erythropoiesis and reduce uric acid (a biomarker of oxidative stress)—effects that are not seen with metformin. Changes in both hematocrit and serum urate are the most important predictors of the ability of SGLT2 inhibitors to reduce the risk of cardiovascular death and hospitalization for heart failure in large-scale trials. Metformin and SGLT2 inhibitors may also differ in their ability to mitigate diabetes-related increases in intracellular sodium concentration and its adverse effects on mitochondrial functional integrity. Differences in the actions of SGLT2 inhibitors and metformin may reflect the distinctive molecular pathways that explain differences in the cardioprotective effects of these drugs.
Similar content being viewed by others

Background
Autophagy is an evolutionarily-conserved intracellular degradative pathway, which mediates the cellular adaptation to stressful conditions. Autophagy involves the enclosure of unwanted cytosolic constituents by an autophagosome membrane, and the contents of these vesicles are destroyed when they fuse with lysosomes [1]. When stimulated nonselectively, autophagy recycles cellular components to generate ATP to support cells that are energy starved. Yet, autophagy can also be activated selectively in order to rid cells of accumulated debris, excessive stores of glucose and lipids, unfolded proteins, and dysfunctional or damaged organelles, which are seminal to the pathogenesis of disease [1, 2].
Triggers of and molecular pathways leading to autophagy
The primordial stimulus to autophagy is energy starvation—specifically, nutrient and oxygen deprivation. However, autophagic flux is also activated in response to a broad range of cellular stresses, including oxidative and endoplasmic reticulum stress. The most important sources of oxidative stress are dysfunctional mitochondria and peroxisomes, the two major oxygen-consuming constituents in the cell [3]. Endoplasmic reticulum stress is caused by the accumulation of misfolded proteins, glycation endproducts or fatty acid intermediates [4]. Regardless of the triggering event, autophagy is part of a wide-ranging transcriptional and metabolic shift that promotes cellular and organismal survival by prioritizing maintenance over growth [5]. Autophagy underlies the effect of starvation to prolong life in a broad range of animal species; tissue-specific overexpression of single autophagy genes is sufficient to extend lifespan [6]. Conversely, impairment of autophagy has been implicated in the pathogenesis of many human illnesses, including metabolic, cardiovascular, neurodegenerative and autoimmune diseases, and cancer [1, 2].
Nutrient and oxygen deprivation signaling promotes autophagic flux
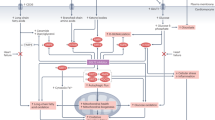
The molecular mechanisms that can activate autophagy are complex (Fig. 1). Nutrient deprivation leads to increased expression and activity of master regulator enzymes, which include sirtuin-1 (SIRT1) and adenosine monophosphate-activated protein kinase (AMPK) [7]. SIRT1 responds to levels of nicotinamide adenine dinucleotide and serves as a redox rheostat; its activation serves to support blood levels of glucose [8, 9]. AMPK is sensitive to the balance between ATP and ADP or AMP in the cytosol; its activation leads to the breakdown of energy stores, thereby promoting the generation of ATP [10]. Oxygen deprivation leads to increased expression and activity of hypoxia inducible factors (HIF-1α and HIF-2α), which promote the delivery and reduce the utilization of oxygen [11].

Effect of enhanced nutrient and oxygen deprivation signaling on autophagic flux, mitochondrial homeostasis and inflammasome activation. ATP: adenosine triphosphate
SIRT1, AMPK, HIF-1α and HIF-2α are master regulators of hundreds of genes and proteins that play a critical role in maintaining cellular homeostasis, and they can augment autophagy in cardiomyocytes and in diabetic hearts under stress [12,13,14,15]. The interplay of HIF-1α with beclin 1 promotes autophagosome formation [16], and phosphorylation of AMPK causes dissociation of the beclin 1-Bcl2 complex [12] and enhances the maturation of autophagosomes and their fusion with lysosomes [17]. In contrast, SIRT1 and HIF-2α act primarily to enhance selective autophagy, i.e., SIRT1 promotes the clearance of damaged mitochondria [18], whereas HIF-2α stimulates the degradation of dysfunctional peroxisomes [19]. Consistent with their intertwined functions, SIRT1 and HIF-2α augment and reinforce each other [20, 21].
Nutrient and oxygen deprivation signaling can mitigate oxidative stress and inflammation through mechanisms that are not autophagy-dependent
Nutrient and oxygen deprivation signaling can influence oxidative stress and inflammatory pathways in ways that may be independent of their effects to promote autophagy (Fig. 1). Both SIRT1 and AMPK act directly to maintain mitochondrial network homeostasis [22,23,24] and preserve peroxisome functionality [24, 25], and they enhance the activity of antioxidant enzymes [26]. Additionally, both SIRT1 and AMPK interact with a key subunit of NFκB to inhibit its actions, thereby attenuating activation of the NLRP3 inflammasome and muting inflammation-mediated cellular injury [27, 28]. HIF-2α shifts the cellular milieu towards an antioxidant state [29], and HIF-2α upregulation is accompanied by an anti-inflammatory macrophage polarization phenotype [30], potentially explaining why HIF-2α acts to mute the inflammatory response that underlies insulin resistance in obesity [31].
Therefore, acting through both autophagy-dependent or -independent mechanisms, the interplay of SIRT1 and HIF-2α plays a major role in ameliorating oxidative stress in the heart. Activation of SIRT1 decreases the production of reactive oxygen species [32, 33], whereas genetic or pharmacological suppression of SIRT1 markedly augments oxidative stress [34, 35]. Similarly, degradation or inhibition of HIF-2α acts to undermine antioxidant mechanisms [29, 36, 37], whereas activation of HIF-2α by cobalt chloride reduces oxidative stress in cardiac and vascular tissues [38,39,40]. If the levels of SIRT1 and HIF-2α decline, the resulting increase in oxygen free radicals acts to reactivate SIRT1 and HIF-2α signaling [41, 42], thereby limiting oxygen-mediated cellular stress.
Suppression of autophagic flux and nutrient deprivation sensor signaling in type 2 diabetes
Type 2 diabetes is characterized by hyperglycemia and hyperinsulinemia and is typically accompanied by the intracellular accumulation of glycogen and lipids. The accumulation of glycation and fatty acid intermediates undermines mitochondrial and peroxisomal stability, leading to the production of reactive oxygen species and oxidative stress [43]. The overabundance of nutrients also promotes the formation of unfolded proteins and potentially toxic lipid pools, which cause endoplasmic reticular stress [44, 45]. When these changes occur in the heart, the result is cardiomyocyte dysfunction and demise.
Although cells might be able to mitigate these metabolic, oxidative and endoplasmic reticulum stresses by stimulating autophagic flux, the stimulation of and capacity for autophagy is markedly impaired in states of energy surplus (Fig. 2) [46, 47]. Type 2 diabetes is accompanied by a decrease in the activation of SIRT1 and AMPK and by a striking suppression of autophagy [48,49,50]; these changes have been implicated in the myocardial injury and cardiomyopathy in type 2 diabetes [49,50,51]. Activation of SIRT1 alleviates oxidative stress, promotes autophagic flux, and prevents cardiomyocyte dysfunction and demise in diabetic hearts [13, 52,53,54]. Similarly, a high-fat diet acts to suppress (whereas glucose deprivation activates) HIF-2α [55,56,57], whereas upregulation of HIF-2α reduces oxidative stress and promotes autophagy in the heart [38, 39]. Thus, changes in nutrient and oxygen deprivation signaling can influence organellar stability, oxidative stress and inflammasome activation and modulate cellular dysfunction in diabetic hearts by mechanisms that are autophagy-dependent and -independent.

Derangements in energy deprivation signaling in type 2 diabetes and its implications for the development of cardiomyopathy
Interestingly, the energy surplus in type 2 diabetes may not only lead to the suppression of low-energy sensors, but changes in SIRT1, AMPK, HIF-1α and HIF-2α signaling may also contribute to glucose intolerance. Stimulation of SIRT1 and AMPK improves glycemic control, glucose transporter expression and insulin sensitivity [58,59,60,61], and intermittent hypoxia improves glycemia by causing upregulation of both AMPK and HIF-1α [61, 62]. Activation of HIF-1α enhances glycolysis, whereas HIF-2α suppresses gluconeogenesis; [63, 64] additionally, HIF-2α enhances insulin sensitivity and inhibits the actions of glucagon [64, 65]. The coordinated effects of hypoxia inducible factor signaling act to lower blood glucose, while simultaneously mediating the adaptation of cells to hypoglycemia [66]. Interestingly, the benefits of enhanced SIRT1/AMPK/HIF signaling on glucose homeostasis are likely to be mediated (at least in part) through enhanced autophagic flux, which plays a critically important role in promoting normal glucose utilization [67].
Effect of antihyperglycemic drugs on autophagic flux, nutrient deprivation signaling and cellular stress
Theoretically, any antihyperglycemic drug might increase the activity of low-energy sensors and promote autophagy simply by lowering blood glucose; however, the magnitude of the effect may be modest and be offset by other actions. Incretins and thiazolidinediones have been reported to enhance autophagy in experimental models [68,69,70], but they potentiate the release and/or response to insulin, which acts to suppress autophagy [71]. These effects may help to explain why enhanced insulin signaling adversely affects the course of heart failure [72]. In addition, dipeptidyl peptidase 4 inhibitors potentiate the actions of stromal cell-derived factor 1, which signals through its receptor CXCR4 to depress autophagic flux [73, 74].
Two glucose-lowering drugs—metformin and SGLT2 inhibitors—promote nutrient deprivation signaling and autophagic flux without enhancing insulin signaling (Fig. 3).

Mechanisms underlying the effects of glucose-lowering drugs to influence the development of cardiomyopathy. The possibility that glycosuria produced by SGLT2 inhibitors can promote renal urate excretion is not shown. AMPK: adenosine monophosphate-activated protein kinase; PGC-1α: peroxisome proliferator-activated receptor-γ coactivator-1α; SIRT1: sirtuin-1
Effects of metformin on nutrient and oxygen deprivation signaling and autophagic flux in diabetic and nondiabetic hearts under stress
Metformin promotes autophagy in hearts under stress, and this action may contribute to the effect of the drug to ameliorate cardiomyocyte dysfunction and the evolution of experimental cardiomyopathy, in the presence or absence of diabetes [75,76,77,78]. The effect of metformin to promote autophagy is primarily related to its ability to act as an agonist of AMPK [76, 79], but signaling through AMPK is capable of ameliorating oxidative stress and cardiac inflammation in ways that are independent of changes in autophagic flux [80,81,82,83]. Additionally, metformin may produce cardioprotective effects that are independent of AMPK [84], potentially by suppressing the activity of the Akt/mTOR pathway [85].
Although metformin has been postulated to interact with SIRT1 [86] several lines of evidence suggest that its capacity to promote SIRT1 signaling is modest and is not likely to mediate the cardioprotective effects of the drug. As expected from an AMPK agonist, metformin suppresses gluconeogenesis [87], but drugs that act through SIRT1 stimulate gluconeogenesis [10]. SIRT1 activation is also expected to promote erythropoiesis (since SIRT1 stimulates HIF-2α [20]); yet, metformin decreases the hematocrit [88], presumably because signaling through AMPK acts to suppress HIF-2α [89].
Effects of SGLT2 inhibitors on nutrient and oxygen deprivation signaling, autophagic flux and organellar dysfunction in cardiomyocytes
SGLT2 inhibitors cause loss of calories in the urine, and as a result of the induction of a starvation-like state [90], SGLT2 inhibitors stimulate the activity of SIRT1 [91,92,93,94] the principal sensor of glucose depletion. SGLT2 inhibitors also upregulate another nutrient deprivation sensor, peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) [95, 96], the downstream target of SIRT1 and a master regulator of mitochondrial biogenesis (Fig. 3). In addition, certain SGLT2 inhibitors (e.g., canagliflozin) directly activate AMPK; [97,98,99] empagliflozin and dapagliflozin may also promote AMPK activity, although not necessarily by a direct action or to a meaningful degree [92, 97,98,99,100,101,102]. The effects of SGLT2 inhibitors on hypoxia inducible factors in the heart have not been evaluated to date.
The action of SGLT2 inhibitors to stimulate SIRT1 (alone or in concert with other nutrient deprivation sensors) may explain the ability of these drugs to maintain mitochondrial membrane potential, preserve mitochondrial structure, restore the capacity of mitochondria to generate ATP, and mitigate mitochondrial fragmentation and DNA injury [101,102,103,104]. These benefits may be achieved by a direct salutary effect of SIRT1/AMPK/PGC-1α signaling on existing mitochondria; through autophagic clearance of injured mitochondria; and by promoting the biogenesis of healthy mitochondria (Fig. 3) [18, 22,23,24,25, 105, 106]. SGLT2 inhibitors have been shown to promote autophagic flux in diabetic hearts, thereby muting inflammation [100]. The autophagy-dependent and -independent action of SGLT2 inhibitors to maintain organellar health likely underlies their ability to ameliorate the course of experimental diabetic and nondiabetic cardiomyopathy [107,108,109].
Interestingly, intracellular sodium concentration is increased in cardiomyocytes derived from diabetic hearts [110, 111]; this perturbation may compromise the capacity of mitochondria to generate ATP and reduce the generation of reactive oxygen species [112, 113]. It is therefore noteworthy that SIRT1/AMPK signaling modulates the activity of transporters so as to promote sodium efflux out of cells [49, 114,115,116]; the resulting decrease in intracellular sodium concentrations improves mitochondrial function and antioxidant defense mechanisms, thereby preventing cell death [112]. Interestingly, SGLT2 inhibitors have been shown to decrease intracellular sodium concentration in cardiomyocytes [117]. Although this finding has been attributed to an effect on sodium-hydrogen exchange in the heart, an effect of SGLT2 inhibitors on the exchanger has yet to be demonstrated. Instead, the effect of these drugs on cytosolic sodium may possibly be the result of AMPK/SIRT1 signaling.
It is important to recognize that the effects of SGLT2 inhibitors to promote SIRT1/AMPK signaling are not cardiac specific. The loss of calorie in the urine triggers a system-wide starvation prosurvival transcriptional paradigm in a broad range of tissues [91]. Specifically, glycosuria stimulates SIRT1 in the liver and promotes hepatic gluconeogenesis, even though SGLT2 is not expressed in hepatic tissues [92]. SGLT2 inhibitors ameliorate the structural and functional derangements in the heart, liver, kidney, adipose tissue and skeletal muscle that are seen in states of energy overabundance [92, 118, 119], even though there are no measurable levels of the target protein in most of these tissues.
Distinctions between metformin and SGLT2 inhibitors with respect to energy deprivation signaling and cardioprotection
There is compelling evidence from large-scale trials that SGLT2 inhibitors reduce the risk of cardiovascular death and hospitalization for heart failure in patients with and without diabetes [120, 121]. In contrast, there is uncertainty whether metformin exerts such benefits in the clinical setting. Metformin has been associated with a reduction in heart failure events in some (but not all) epidemiological studies [122,123,124,125]; however, in these reports, metformin was compared with antihyperglycemic drugs that can increase the risk of heart failure. Furthermore, in these studies, it seems likely that metformin was preferentially prescribed to patients at low risk of heart failure [126], since physicians have worried that the drug may trigger lactic acidosis. Given the observational nature of these analyses and the lack of evidence from randomized controlled trials, the true effect of metformin on the development of heart failure in patients with type 2 diabetes remains unclear [127].
However, metformin and SGLT2 inhibitors differ with respect to their actions to promote nutrient and oxygen deprivation signaling (Fig. 3). Metformin exerts its effects primarily through the activation of AMPK; in contrast, several lines of evidence suggest that SGLT2 inhibitors exert their effects principally through SIRT1 and its downstream effectors, and not AMPK [95,96,97,98,99]. Due to the loss of calories in the urine, SGLT2 inhibitors recapitulate a starvation-like state, which signals more through SIRT than AMPK [128,129,130], since SIRT1 (and not AMPK) mediates the effects of caloric restriction to prolong survival [131]. Additionally, both fasting and SGLT2 inhibition are accompanied by hyperketonemia, and there is a close association between ketogenesis and the activation of SIRT1 [132,133,134] Ketogenesis depends on gluconeogenesis, which is stimulated by SIRT1 [1, 10] but inhibited by AMPK and metformin [135, 136]. The other major pathway leading to the formation of ketone bodies—fatty acid oxidation—also requires SIRT1 [137,138,139]. Finally, pretreatment with metformin does not attenuate the ability of empagliflozin (which does not directly activate AMPK [97, 98]) to reduce the risk of heart failure hospitalizations [140]. Therefore, in contradistinction to metformin, it appears that SGLT2 inhibitors preferentially activate SIRT1, rather than AMPK.
Differences in the pattern of nutrient deprivation signaling with metformin and SGLT2 inhibitors may also lead to different effects on intracellular sodium. As noted earlier, SGLT2 inhibitors reduce levels of cytosolic sodium in cardiomyocytes, an effect that may yield direct benefits on mitochondrial capacity and stability [112, 117]. In contrast, metformin does not ameliorate the heightened intracellular sodium concentrations seen in diabetic cardiomyocytes [110, 111, 141].
SIRT1 signaling may explain the results of statistical mediation analyses of the heart failure benefit seen in large-scale clinical trials
The likely role of SIRT1 in mediating the effects of SGLT2 inhibitors is noteworthy, since SIRT1 (but not AMPK) can stimulate HIF-2α [20, 21], the primary transactivator of the gene for erythropoietin synthesis [142]. Interestingly, SGLT2 inhibitors have been strongly linked to the enhanced production of erythropoietin and to an increase in red blood cell mass in clinical trials [121, 143,144,145]. More importantly, activation of HIF-2α can be expected to exert its own effects to promote autophagy and mute cellular stress and inflammation [19, 29,30,31]. In contrast, as a result of AMPK agonism, metformin suppresses the activity of HIF-2α [89], and thus, the drug decreases the hematocrit [88]. The potential differences in HIF-2α signaling between SGLT2 inhibitors and metformin may be clinically relevant, since (in statistical mediation analyses) the erythrocytosis produced by SGLT2 inhibitors is the most powerful predictor of the ability of these drugs to reduce the risk of serious heart failure events in large-scale clinical trials [144, 145].
Interestingly, in the mediation analyses of large-scale cardiovascular outcomes trials, the effect of SGLT2 inhibitors to decrease serum uric acid is also a major independent predictor of the drug-related reduction in serious heart failure events [144, 145]. Previous work attributed the urate-lowering effects of SGLT2 inhibitors to an effect of these drugs to simultaneously inhibit glucose and uric acid reabsorption in the proximal renal tubule [146], since glycosuria may directly enhance fractional excretion of uric acid [147]. However, urate is also a biomarker of oxidative stress in the stressed myocardium [148,149,150], i.e., the increase in reactive oxygen species in patients with diabetes leads to activation of xanthine oxidase, the enzyme that catalyzes the synthesis of uric acid [151]. Interestingly, the depletion of nicotinamide adenine dinucleotide (NAD+) in diabetes not only causes upregulation of xanthine oxidase but also downregulation of SIRT1 [152, 153]. There is an inverse relationship between the activities of SIRT1 and xanthine oxidase. Upregulation of xanthine oxidase suppresses SIRT1 [154] and inhibition of xanthine oxidase activates SIRT1 [155]; thus, serum levels of uric acid are inversely related to the activity of SIRT1 in states of energy overabundance [156]. Therefore, by enhancing SIRT1-mediated suppression of oxidative stress or by a direct consequence of SIRT1 activation [157,158,159], SGLT2 inhibitors may suppress the activity of xanthine oxidase and reduce serum uric acid [145, 160]. Thus, activation of SIRT1 may explain the observed statistical link between the urate-lowering and cardioprotective effects of SGLT2 inhibitors. In contrast, metformin (which does not enhance signaling through SIRT1) increases serum uric acid [161].
Conclusions
Heart failure is the most common and serious cardiovascular complication of type 2 diabetes, possibly because diabetes increases oxidative and endoplasmic reticulum stress in cardiomyocytes, with its attendant risks of cellular dysfunction and demise. The increase in cellular stress in the diabetic heart is related to suppression of nutrient deprivation signaling, which normally acts to maintain organellar function and promote the removal of dysfunctional mitochrondria and peroxisomes through the lysosome-dependent housekeeping process of autophagy. The downregulation of SIRT1 and AMPK has been shown to cause cardiomyopathy in experimental models of diabetes, whose features are characterized by oxidative stress and organellar dysfunction.
Both metformin and SGLT2 inhibitors activate SIRT1 and AMPK, which may explain their effect to alleviate cellular stress and ameliorate the course of experimental cardiomyopathy, benefits that are likely mediated through their actions to restore mitochondrial function, both directly and indirectly, through their actions to promote autophagy. However, the evidence supporting a heart failure benefit is substantially more compelling with SGLT2 inhibitors than with metformin. Furthermore, SGLT2 inhibitors may have important mechanistic advantages over metformin in producing cardioprotection. Specifically, they may preferentially enhance SIRT1 and HIF-2α (as reflected by ketogenesis and erythrocytosis), alleviate sources of oxidative stress (as reflected by serum uric acid levels), and reduce intracellular sodium concentration in cardiomyocytes—effects that are not seen with metformin. Therefore, differences in their action on nutrient deprivation pathways may underlie differences between metformin and SGLT2 inhibitors in their ability to reduce heart failure events in the clinical setting.
Availability of data and materials
Not applicable.
Abbreviations
- ADP:
-
Adenosine diphosphate
- Akt:
-
Protein kinase B
- AMP:
-
Adenosine monophosphate
- AMPK:
-
Adenosine monophosphate-activated protein kinase
- ATP:
-
Adenosine triphosphate
- CXCR4:
-
C-X-C chemokine receptor type 4
- HIF:
-
Hypoxia inducible factor
- HIF-1α:
-
Hypoxia inducible factor isoform 1-alpha
- HIF-2α:
-
Hypoxia inducible factor isoform 2-alpha
- mTOR:
-
Mammalian target of rapamycin
- NFκB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRP3:
-
NOD-, LRR- and pyrin domain-containing protein 3
- PGC-1α:
-
Peroxisome proliferator-activated receptor-γ coactivator-1α
- SGLT2:
-
Sodium-glucose transporter 2
- SIRT1:
-
Sirtuin-1
References
Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42.
Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125:14–24.
Marcassa E, Kallinos A, Jardine J, Rusilowicz-Jones EV, Martinez A, Kuehl S, Islinger M, Clague MJ, Urbé S. Dual role of USP30 in controlling basal pexophagy and mitophagy. EMBO Rep. 2018;19(7):e45595. https://doi.org/10.15252/embr.201745595.
Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, Mauthe M, Katona I, Qualmann B, Weis J, Reggiori F, Kurth I, Hübner CA, Dikic I. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015;522:354–8.
Leidal AM, Levine B, Debnath J. Autophagy and the cell biology of age-related disease. Nat Cell Biol. 2018;20:1338–48.
Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19:579–93.
Panieri E, Toietta G, Mele M, Labate V, Ranieri SC, Fusco S, Tesori V, Antonini A, Maulucci G, De Spirito M, Galeotti T, Pani G. Nutrient withdrawal rescues growth factor-deprived cells from mTOR-dependent damage. Aging (Albany NY). 2010;2:487–503.
Gillum MP, Erion DM, Shulman GI. Sirtuin-1 regulation of mammalian metabolism. Trends Mol Med. 2011;17:8–13. https://doi.org/10.1016/j.molmed.2010.09.005.
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8.
Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–62.
Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–74.
He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013;62:1270–81.
Wang B, Yang Q, Sun YY, Xing YF, Wang YB, Lu XT, Bai WW, Liu XQ, Zhao YX. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med. 2014;18:1599–611.
Gui L, Liu B, Lv G. Hypoxia induces autophagy in cardiomyocytes via a hypoxia-inducible factor 1-dependent mechanism. Exp Ther Med. 2016;11:2233–9.
Gyongyosi A, Terraneo L, Bianciardi P, Tosaki A, Lekli I, Samaja M. The impact of moderate chronic hypoxia and hyperoxia on the level of apoptotic and autophagic proteins in myocardial tissue. Oxid Med Cell Longev. 2018;16(2018):5786742. https://doi.org/10.1155/2018/5786742.
Lu N, Li X, Tan R, An J, Cai Z, Hu X, Wang F, Wang H, Lu C, Lu H. HIF-1α/Beclin1-mediated autophagy is involved in neuroprotection induced by hypoxic preconditioning. J Mol Neurosci. 2018;66:238–50.
Jang M, Park R, Kim H, Namkoong S, Jo D, Huh YH, Jang IS, Lee JI, Park J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci Rep. 2018;8(1):12637. https://doi.org/10.1038/s41598-018-30977-7.
Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, Mitchell JR, Croteau DL, Bohr VA. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014;157:882–96.
Walter KM, Schönenberger MJ, Trötzmüller M, Horn M, Elsässer HP, Moser AB, Lucas MS, Schwarz T, Gerber PA, Faust PL, Moch H, Köfeler HC, Krek W, Kovacs WJ. Hif-2α promotes degradation of mam-malian peroxisomes by selective autophagy. Cell Metab. 2014;20:882–97.
Chen R, Xu M, Hogg RT, Li J, Little B, Gerard RD, Garcia JA. The acetylase/deacetylase couple CREB-binding protein/Sirtuin 1 controls hypoxia-inducible factor 2 signaling. J Biol Chem. 2012;287:30800–11.
Chen R, Dioum EM, Hogg RT, Gerard RD, Garcia JA. Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J Biol Chem. 2011;286:13869–78.
Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018;19:121–35.
Tang BL. Sirt1 and the mitochondria. Mol Cells. 2016;39:87–95.
Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, Laboy R, Hirschey MD, Mair WB. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 2017;26(884–896):e5.
Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Sueyasu K, Washida N, Tokuyama H, Tzukerman M, Skorecki K, Hayashi K, Itoh H. Kidney-specific overexpression of Sirt1 protects against acute kidney injury by retaining peroxisome function. J Biol Chem. 2010;285:13045–56.
Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Washida N, Tokuyama H, Hayashi K, Itoh H. Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun. 2008;372:51–6.
Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25:1939–48.
Chen X, Li X, Zhang W, He J, Xu B, Lei B, Wang Z, Cates C, Rousselle T, Li J. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism. 2018;83:256–70.
Yuan G, Peng YJ, Reddy VD, Makarenko VV, Nanduri J, Khan SA, Garcia JA, Kumar GK, Semenza GL, Prabhakar NR. Mutual antagonism between hypoxia-inducible factors 1α and 2α regulates oxygen sensing and cardio-respiratory homeostasis. Proc Natl Acad Sci USA. 2013;110:E1788–96.
Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501.
Choe SS, Shin KC, Ka S, Lee YK, Chun JS, Kim JB. Macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes. 2014;63:3359–71.
Li T, Chen L, Yu Y, Yang B, Li P, Tan XQ. Resveratrol alleviates hypoxia/reoxygenation injury–induced mitochondrial oxidative stress in cardiomyocytes. Mol Med Rep. 2019;19:2774–80.
Bagul PK, Deepthi N, Sultana R, Banerjee SK. Resveratrol ameliorates cardiac oxidative stress in diabetes through deacetylation of NFkB-p65 and histone 3. J Nutr Biochem. 2015;26:1298–307.
Waldman M, Cohen K, Yadin D, Nudelman V, Gorfil D, Laniado-Schwartzman M, Kornwoski R, Aravot D, Abraham NG, Arad M, Hochhauser E. Regulation of diabetic cardiomyopathy by caloric restriction is mediated by intracellular signaling pathways involving ‘SIRT1 and PGC-1α’. Cardiovasc Diabetol. 2018;17(1):111. https://doi.org/10.1186/s12933-018-0754-4.
Sanz MN, Grimbert L, Moulin M, Gressette M, Rucker-Martin C, Lemaire C, Mericskay M, Veksler V, Ventura-Clapier R, Garnier A, Piquereau J. Inducible cardiac-specific deletion of Sirt1 in male mice reveals progressive cardiac dysfunction and sensitization of the heart to pressure overload. Int J Mol Sci. 2019;20(20):E5005. https://doi.org/10.3390/ijms20205005.
Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng YJ, Kumar GK, Garcia JA, Prabhakar NR. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci USA. 2009;106:1199–204.
Peng YJ, Nanduri J, Khan SA, Yuan G, Wang N, Kinsman B, Vaddi DR, Kumar GK, Garcia JA, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 2α (HIF-2α) heterozygous-null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc Natl Acad Sci USA. 2011;108:3065–70.
Yildirim O, Büyükbingöl Z. Effect of cobalt on the oxidative status in heart and aorta of streptozotocin-induced diabetic rats. Cell Biochem Funct. 2003;21:27–33.
Gallo S, Gatti S, Sala V, Albano R, Costelli P, Casanova E, Comoglio PM, Crepaldi T. Agonist antibodies activating the Met receptor protect cardiomyoblasts from cobalt chloride-induced apoptosis and autophagy. Cell Death Dis. 2014;17(5):e1185. https://doi.org/10.1038/cddis.2014.155.
Gaitanaki C, Kalpachidou T, Aggeli IK, Papazafiri P, Beis I. CoCl2 induces protective events via the p38-MAPK signalling pathway and ANP in the perfused amphibian heart. J Exp Biol. 2007;210:2267–77.
Diebold I, Flügel D, Becht S, Belaiba RS, Bonello S, Hess J, Kietzmann T, Görlach A. The hypoxia-inducible factor-2alpha is stabilized by oxidative stress involving NOX4. Antioxid Redox Signal. 2010;13:425–36.
Liu H, Colavitti R, Rovira II, Finkel T. Redox-dependent transcriptional regulation. Circ Res. 2005;97:967–74.
Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–70.
Lakshmanan AP, Harima M, Suzuki K, Soetikno V, Nagata M, Nakamura T, Takahashi T, Sone H, Kawachi H, Watanabe K. The hyperglycemia stimulated myocardial endoplasmic reticulum (ER) stress contributes to diabetic cardiomyopathy in the transgenic non-obese type 2 diabetic rats: a differential role of unfolded protein response (UPR) signaling proteins. Int J Biochem Cell Biol. 2013;45:438–47.
Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res. 2010;107:579–91.
Xu XJ, Valentine RJ, Ruderman NB. AMP-activated protein kinase (AMPK): does this master regulator of cellular energy state distinguish insulin sensitive from insulin resistant obesity? Curr Obes Rep. 2014;3:248–55.
Kurylowicz A, Owczarz M, Polosak J, Jonas MI, Lisik W, Jonas M, Chmura A, Puzianowska-Kuznicka M. SIRT1 and SIRT7 expression in adipose tissues of obese and normal-weight individuals is regulated by microRNAs but not by methylation status. Int J Obes. 2016;40:1635–42.
Wang Y, Liang B, Lau WB, Du Y, Guo R, Yan Z, Gan L, Yan W, Zhao J, Gao E, Koch W, Ma XL. Restoring diabetes-induced autophagic flux arrest in ischemic/reperfused heart by ADIPOR (adiponectin receptor) activation involves both AMPK-dependent and AMPK-independent signaling. Autophagy. 2017;13:1855–69.
Yuan Q, Zhou QY, Liu D, Yu L, Zhan L, Li XJ, Peng HY, Zhang XL, Yuan XC. Advanced glycation end-products impair Na+/K+-ATPase activity in diabetic cardiomyopathy: role of the adenosine monophosphate-activated protein kinase/sirtuin 1 pathway. Clin Exp Pharmacol Physiol. 2014;41:127–33.
Tao A, Xu X, Kvietys P, Kao R, Martin C, Rui T. Experimental diabetes mellitus exacerbates ischemia/reperfusion-induced myocardial injury by promoting mitochondrial fission: role of down-regulation of myocardial Sirt1 and subsequent Akt/Drp1 interaction. Int J Biochem Cell Biol. 2018;105:94–103.
Karbasforooshan H, Karimi G. The role of SIRT1 in diabetic cardiomyopathy. Biomed Pharmacother. 2017;90:386–92.
Ma S, Feng J, Zhang R, Chen J, Han D, Li X, Yang B, Li X, Fan M, Li C, Tian Z, Wang Y, Cao F. SIRT1 activation by resveratrol alleviates cardiac dysfunction via mitochondrial regulation in diabetic cardiomyopathy mice. Oxid Med Cell Longev. 2017;2017:4602715. https://doi.org/10.1155/2017/4602715.
Guo R, Liu W, Liu B, Zhang B, Li W, Xu Y. SIRT1 suppresses cardiomyocyte apoptosis in diabetic cardiomyopathy: an insight into endoplasmic reticulum stress response mechanism. Int J Cardiol. 2015;191:36–45.
Sulaiman M, Matta MJ, Sunderesan NR, Gupta MP, Periasamy M, Gupta M. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2010;298:H833–43.
Zeng H, Vaka VR, He X, Booz GW, Chen JX. High-fat diet induces cardiac remodelling and dysfunction: assessment of the role played by SIRT3 loss. J Cell Mol Med. 2015;19:1847–56.
Mastrocola R, Collino M, Penna C, Nigro D, Chiazza F, Fracasso V, Tullio F, Alloatti G, Pagliaro P, Aragno M. Maladaptive modulations of NLRP3 inflammasome and cardioprotective pathways are involved in diet-induced exacerbation of myocardial ischemia/reperfusion injury in mice. Oxid Med Cell Longev. 2016;2016:3480637. https://doi.org/10.1155/2016/3480637.
Heidbreder M, Qadri F, Jöhren O, Dendorfer A, Depping R, Fröhlich F, Wagner KF, Dominiak P. Non-hypoxic induction of HIF-3alpha by 2-deoxy-d-glucose and insulin. Biochem Biophys Res Commun. 2007;352:437–43.
Giri S, Rattan R, Haq E, Khan M, Yasmin R, Won JS, Key L, Singh AK, Singh I. AICAR inhibits adipocyte differentiation in 3T3L1 and restores metabolic alterations in diet-induced obesity mice model. Nutr Metab. 2006;10(3):31.
Kim MJ, An HJ, Kim DH, Lee B, Lee HJ, Ullah S, Kim SJ, Jeong HO, Moon KM, Lee EK, Yang J, Akter J, Chun P, Moon HR, Chung HY. Novel SIRT1 activator MHY2233 improves glucose tolerance and reduces hepatic lipid accumulation in db/db mice. Bioorg Med Chem Lett. 2018;28:684–8.
Yonamine CY, Pinheiro-Machado E, Michalani ML, Alves-Wagner AB, Esteves JV, Freitas HS, Machado UF. Resveratrol improves glycemic control in type 2 diabetic obese mice by regulating glucose transporter expression in skeletal muscle and liver. Molecules. 2017;22(7):E1180. https://doi.org/10.3390/molecules22071180.
Thomas A, Belaidi E, Moulin S, Horman S, van der Zon GC, Viollet B, Levy P, Bertrand L, Pepin JL, Godin-Ribuot D, Guigas B. Chronic intermittent hypoxia impairs insulin sensitivity but improves whole-body glucose tolerance by activating skeletal muscle AMPK. Diabetes. 2017;66:2942–51.
Serebrovska TV, Portnychenko AG, Drevytska TI, Portnichenko VI, Xi L, Egorov E, Gavalko AV, Naskalova S, Chizhova V, Shatylo VB. Intermittent hypoxia training in prediabetes patients: beneficial effects on glucose homeostasis, hypoxia tolerance and gene expression. Exp Biol Med. 2017;242:1542–52.
Wei K, Piecewicz SM, McGinnis LM, Taniguchi CM, Wiegand SJ, Anderson K, Chan CW, Mulligan KX, Kuo D, Yuan J, Vallon M, Morton L, Lefai E, Simon MC, Maher JJ, Mithieux G, Rajas F, Annes J, McGuinness OP, Thurston G, Giaccia AJ, Kuo CJ. A liver Hif-2α-Irs2 pathway sensitizes hepatic insulin signaling and is modulated by Vegf inhibition. Nat Med. 2013;19:1331–7.
Ramakrishnan SK, Shah YM. A central role for hypoxia-inducible factor (HIF)-2α in hepatic glucose homeostasis. Nutr Healthy Aging. 2017;4:207–16.
Taniguchi CM, Finger EC, Krieg AJ, Wu C, Diep AN, LaGory EL, Wei K, McGinnis LM, Yuan J, Kuo CJ, Giaccia AJ. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat Med. 2013;19:1325–30.
Brusselmans K, Bono F, Maxwell P, Dor Y, Dewerchin M, Collen D, Herbert JM, Carmeliet P. Hypoxia-inducible factor-2alpha (HIF-2alpha) is involved in the apoptotic response to hypoglycemia but not to hypoxia. J Biol Chem. 2001;276:39192–6.
He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, Levine B. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–5.
Zhang Y, Ling Y, Yang L, Cheng Y, Yang P, Song X, Tang H, Zhong Y, Tang L, He S, Yang S, Chen A, Wang X. Liraglutide relieves myocardial damage by promoting autophagy via AMPK-mTOR signaling pathway in Zucker diabetic fatty rat. Mol Cell Endocrinol. 2017;448:98–107.
Zhou Y, Wang H, Man F, Guo Z, Xu J, Yan W, Li J, Pan Q, Wang W. Sitagliptin protects cardiac function by reducing nitroxidative stress and promoting autophagy in Zucker diabetic fatty (ZDF) rats. Cardiovasc Drugs Ther. 2018;32:541–52.
Kato MF, Shibata R, Obata K, Miyachi M, Yazawa H, Tsuboi K, Yamada T, Nishizawa T, Noda A, Cheng XW, Murate T, Koike Y, Murohara T, Yokota M, Nagata K. Pioglitazone attenuates cardiac hypertrophy in rats with salt-sensitive hypertension: role of activation of AMP-activat-ed protein kinase and inhibition of Akt. J Hypertens. 2008;26:1669–76.
Paula-Gomes S, Gonçalves DA, Baviera AM, Zanon NM, Navegantes LC, Kettelhut IC. Insulin suppresses atrophy- and autophagy-related genes in heart tissue and cardiomyocytes through AKT/FOXO signaling. Horm Metab Res. 2013;45:849–55.
Packer M. Potentiation of insulin signaling contributes to heart failure in type 2 diabetes: a hypothesis supported by both mechanistic studies and clinical trials. JACC Basic Transl Sci. 2018;3:415–9.
Packer M. Have dipeptidyl peptidase-4 inhibitors ameliorated the vascular complications of type 2 diabetes in large-scale trials? The potential confounding effect of stem-cell chemokines. Cardiovasc Diabetol. 2018;17(1):9. https://doi.org/10.1186/s12933-017-0648-x.
Coly PM, Gandolfo P, Castel H, Morin F. The autophagy machinery: a new player in chemotactic cell migration. Front Neurosci. 2017;16(11):78. https://doi.org/10.3389/fnins.2017.00078.
Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–8.
Wang Y, Yang Z, Zheng G, Yu L, Yin Y, Mu N, Ma H. Metformin promotes autophagy in ischemia/reperfusion myocardium via cytoplasmic AMPKα1 and nuclear AMPKα2 pathways. Life Sci. 2019;225:64–71.
Sasaki H, Asanuma H, Fujita M, Takahama H, Wakeno M, Ito S, Ogai A, Asakura M, Kim J, Minamino T, Takashima S, Sanada S, Sugimachi M, Komamura K, Mochizuki N, Kitakaze M. Metformin prevents progression of heart failure in dogs: role of AMP-activated protein kinase. Circulation. 2009;119:2568–77.
Zilinyi R, Czompa A, Czegledi A, Gajtko A, Pituk D, Lekli I, Tosaki A. The cardioprotective effect of metformin in doxorubicin-induced cardiotoxicity: the role of autophagy. Molecules. 2018;23(5):E1184. https://doi.org/10.3390/molecules23051184.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74.
Wang X, Yang L, Kang L, Li J, Yang L, Zhang J, Liu J, Zhu M, Zhang Q, Shen Y, Qi Z. Metformin attenuates myocardial ischemia-reperfusion injury via up-regulation of antioxidant enzymes. PLoS ONE. 2017;12(8):e0182777. https://doi.org/10.1371/journal.pone.0182777.
Barreto-Torres G, Hernandez JS, Jang S, Rodríguez-Muñoz AR, Torres-Ramos CA, Basnakian AG, Javadov S. The beneficial effects of AMP kinase activation against oxidative stress are associated with prevention of PPARα-cyclophilin D interaction in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2015;308:H749–58.
Asensio-López MC, Lax A, Pascual-Figal DA, Valdés M, Sánchez-Más J. Metformin protects against doxorubicin-induced cardiotoxicity: involvement of the adiponectin cardiac system. Free Radic Biol Med. 2011;51:1861–71.
Daskalopoulos EP, Dufeys C, Bertrand L, Beauloye C, Horman S. AMPK in cardiac fibrosis and repair: actions beyond metabolic regulation. J Mol Cell Cardiol. 2016;91:188–200.
Xu X, Lu Z, Fassett J, Zhang P, Hu X, Liu X, Kwak D, Li J, Zhu G, Tao Y, Hou M, Wang H, Guo H, Viollet B, McFalls EO, Bache RJ, Chen Y. Metformin protects against systolic overload-induced heart failure independent of AMP-activated protein kinase α2. Hypertension. 2014;63:723–8.
Chen SC, Brooks R, Houskeeper J, Bremner SK, Dunlop J, Viollet B, Logan PJ, Salt IP, Ahmed SF, Yarwood SJ. Metformin suppresses adipogenesis through both AMP-activated protein kinase (AMPK)-dependent and AMPK-independent mechanisms. Mol Cell Endocrinol. 2017;440:57–68.
Cuyàs E, Verdura S, Llorach-Parés L, Fernández-Arroyo S, Joven J, Martin-Castillo B, Bosch-Barrera J, Brunet J, Nonell-Canals A, Sanchez-Martinez M, Menendez JA. Metformin Is a direct SIRT1-activating compound: computational modeling and experimental validation. Front Endocrinol. 2018;6(9):657. https://doi.org/10.3389/fendo.2018.00657.
Madiraju AK, Qiu Y, Perry RJ, Rahimi Y, Zhang XM, Zhang D, Camporez JG, Cline GW, Butrico GM, Kemp BE, Casals G, Steinberg GR, Vatner DF, Petersen KF, Shulman GI. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat Med. 2018;24:1384–94.
Diabetes Prevention Program Research Group. Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care. 2012;35:731–7.
Yang Q, Guo X, Yang L. Metformin enhances the effect of regorafenib and inhibits recurrence and metastasis of hepatic carcinoma after liver resection via regulating expression of hypoxia inducible factors 2α (HIF-2α) and 30 kDa HIV Tat-Interacting protein (TIP30). Med Sci Monit. 2018;24:2225–34.
Osataphan S, Macchi C, Singhal G, Chimene-Weiss J, Sales V, Kozuka C, Dreyfuss JM, Pan H, Tangcharoenpaisan Y, Morningstar J, Gerszten R, Patti ME. SGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and -independent mechanisms. JCI Insight. 2019;4(5):123130. https://doi.org/10.1172/jci.insight.123130.
Swe MT, Thongnak L, Jaikumkao K, Pongchaidecha A, Chatsudthipong V, Lungkaphin A. Dapagliflozin not only improves hepatic injury and pancreatic endoplasmic reticulum stress, but also induces hepatic gluconeogenic enzymes expression in obese rats. Clin Sci. 2019;133:2415–30.
Kim JW, Lee YJ, You YH, Moon MK, Yoon KH, Ahn YB, Ko SH. Effect of sodium-glucose cotransporter 2 inhibitor, empagliflozin, and α-glucosidase inhibitor, voglibose, on hepatic steatosis in an animal model of type 2 diabetes. J Cell Biochem. 2018. https://doi.org/10.1002/jcb.28141(Epub ahead of print).
Ying Y, Jiang C, Zhang M, Jin J, Ge S, Wang X. Phloretin protects against cardiac damage and remodeling via restoring SIRT1 and anti-inflammatory effects in the streptozotocin-induced diabetic mouse model. Aging. 2019;11:2822–35.
Mohamed HE, Asker ME, Keshawy MM, Hasan RA, Mahmoud YK. Inhibition of tumor necrosis factor-α enhanced the antifibrotic effect of empagliflozin in an animal model with renal insulin resistance. Mol Cell Biochem. 2020. https://doi.org/10.1007/s11010-020-03686-x(Epub ahead of print).
Shao Q, Meng L, Lee S, Tse G, Gong M, Zhang Z, Zhao J, Zhao Y, Li G, Liu T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc Diabetol. 2019;18(1):165. https://doi.org/10.1186/s12933-019-0964-4.
Wei D, Liao L, Wang H, Zhang W, Wang T, Xu Z. Canagliflozin ameliorates obesity by improving mitochondrial function and fatty acid oxidation via PPARα in vivo and in vitro. Life Sci. 2020;247:117414. https://doi.org/10.1016/j.lfs.2020.117414(Epub ahead of print).
Mancini SJ, Boyd D, Katwan OJ, Strembitska A, Almabrouk TA, Kennedy S, Palmer TM, Salt IP. Canagliflozin inhibits interleukin-1β-stimulated cytokine and chemokine secretion in vascular endothelial cells by AMP-activated protein kinase-dependent and -independent mechanisms. Sci Rep. 2018;8(1):5276. https://doi.org/10.1038/s41598-018-23420-4.
Hawley SA, Ford RJ, Smith BK, Gowans GJ, Mancini SJ, Pitt RD, Day EA, Salt IP, Steinberg GR, Hardie DG. The Na+/glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes. 2016;65:2784–94.
Sayour AA, Korkmaz-Icöz S, Loganathan S, Ruppert M, Sayour VN, Oláh A, Benke K, Brune M, Benkő R, Horváth EM, Karck M, Merkely B, Radovits T, Szabó G. Acute canagliflozin treatment protects against in vivo myocardial ischemia-reperfusion injury in non-diabetic male rats and enhances endothelium-dependent vasorelaxation. J Transl Med. 2019;17(1):127. https://doi.org/10.1186/s12967-019-1881-8.
Aragón-Herrera A, Feijóo-Bandín S, Otero Santiago M, Barral L, Campos-Toimil M, Gil-Longo J, Costa Pereira TM, García-Caballero T, Rodríguez-Segade S, Rodríguez J, Tarazón E, Roselló-Lletí E, Portolés M, Gualillo O, González-Juanatey JR, Lago F. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochem Pharmacol. 2019;170:113677. https://doi.org/10.1016/j.bcp.2019.113677.
Lu Q, Liu J, Li X, Sun X, Zhang J, Ren D, Tong N, Li J. Empagliflozin attenuates ischemia and reperfusion injury through LKB1/AMPK signaling pathway. Mol Cell Endocrinol. 2019. https://doi.org/10.1016/j.mce.2019.110642(Epub ahead of print).
Chang YK, Choi H, Jeong JY, Na KR, Lee KW, Lim BJ, Choi DE. Dapagliflozin, SGLT2 inhibitor, attenuates renal ischemia-reperfusion injury. PLoS ONE. 2016;11(7):e0158810. https://doi.org/10.1371/journal.pone.0158810.
Lee WC, Chau YY, Ng HY, Chen CH, Wang PW, Liou CW, Lin TK, Chen JB. Empagliflozin protects HK-2 cells from high glucose-mediated injuries via a mitochondrial mechanism. Cells. 2019. https://doi.org/10.3390/cells8091085.
Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335–46.
Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M, Yan Z. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017;8(1):548. https://doi.org/10.1038/s41467-017-00520-9.
Vainshtein A, Tryon LD, Pauly M, Hood DA. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol. 2015;308:C710–9.
Adingupu DD, Göpel SO, Grönros J, Behrendt M, Sotak M, Miliotis T, Dahlqvist U, Gan LM, Jönsson-Rylander AC. SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob-/- mice. Cardiovasc Diabetol. 2019;18(1):16. https://doi.org/10.1186/s12933-019-0820-6.
Oh CM, Cho S, Jang JY, Kim H, Chun S, Choi M, Park S, Ko YG. Cardioprotective potential of an SGLT2 inhibitor against doxorubicin-induced heart failure. Korean Circ J. 2019;49:1183–95.
Takasu T, Takakura S. Effect of ipragliflozin, an SGLT2 inhibitor, on cardiac histopathological changes in a non-diabetic rat model of cardiomyopathy. Life Sci. 2019;230:19–27.
Anzawa R, Bernard M, Tamareille S, Baetz D, Confort-Gouny S, Gascard JP, Cozzone P, Feuvray D. Intracellular sodium increase and susceptibility to ischaemia in hearts from type 2 diabetic db/db mice. Diabetologia. 2006;49:598–606.
Lagadic-Gossmann D, Feuvray D. Intracellular sodium activity in papillary muscle from diabetic rat hearts. Exp Physiol. 1991;76:147–9.
Bertero E, Prates Roma L, Ameri P, Maack C. Cardiac effects of SGLT2 inhibitors: the sodium hypothesis. Cardiovasc Res. 2018;114:12–8.
Murphy E, Eisner DA. Regulation of intracellular and mitochondrial sodium in health and disease. Circ Res. 2009;104:292–303.
Lang F, Föller M. Regulation of ion channels and transporters by AMP-activated kinase (AMPK). Channels. 2014;8:20–8.
Moopanar TR, Xiao XH, Jiang L, Chen ZP, Kemp BE, Allen DG. AICAR inhibits the Na+/H+ exchanger in rat hearts–possible contribution to cardioprotection. Pflugers Arch. 2006;453:147–56.
Vikram A, Lewarchik CM, Yoon JY, Naqvi A, Kumar S, Morgan GM, Jacobs JS, Li Q, Kim YR, Kassan M, Liu J, Gabani M, Kumar A, Mehdi H, Zhu X, Guan X, Kutschke W, Zhang X, Boudreau RL, Dai S, Matasic DS, Jung SB, Margulies KB, Kumar V, Bachschmid MM, London B, Irani K. Sirtuin 1 regulates cardiac electrical activity by deacetylating the cardiac sodium channel. Nat Med. 2017;23:361–7.
Uthman L, Baartscheer A, Bleijlevens B, Schumacher CA, Fiolet JWT, Koeman A, Jancev M, Hollmann MW, Weber NC, Coronel R, Zuurbier CJ. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na +/H + exchanger, lowering of cytosolic Na + and vasodilation. Diabetologia. 2018;61:722–6.
Xu L, Nagata N, Nagashimada M, Zhuge F, Ni Y, Chen G, Mayoux E, Kaneko S, Ota T. SGLT2 inhibition by empagliflozin promotes fat utilization and browning and attenuates inflammation and insulin resistance by polarizing M2 macrophages in diet-induced obese mice. EBioMedicine. 2017;20:137–49.
Nambu H, Takada S, Fukushima A, Matsumoto J, Kakutani N, Maekawa S, Shirakawa R, Nakano I, Furihata T, Katayama T, Yamanashi K, Obata Y, Saito A, Yokota T, Kinugawa S. Empagliflozin restores lowered exercise endurance capacity via the activation of skeletal muscle fatty acid oxidation in a murine model of heart failure. Eur J Pharmacol. 2020;5(866):172810. https://doi.org/10.1016/j.ejphar.2019.172810.
Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Furtado RHM, Bhatt DL, Leiter LA, McGuire DK, Wilding JPH, Sabatine MS. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393:31–9.
McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J, Böhm M, Chiang CE, Chopra VK, de Boer RA, Desai AS, Diez M, Drozdz J, Dukát A, Ge J, Howlett JG, Katova T, Kitakaze M, Ljungman CEA, Merkely B, Nicolau JC, O’Meara E, Petrie MC, Vinh PN, Schou M, Tereshchenko S, Verma S, Held C, DeMets DL, Docherty KF, Jhund PS, Bengtsson O, Sjöstrand M, Langkilde AM, DAPA-HF Trial Committees and Investigators. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381:1995–2008.
Tseng CH. Metformin use is associated with a lower risk of hospitalization for heart failure in patients with type 2 diabetes mellitus: a retrospective cohort analysis. J Am Heart Assoc. 2019;8(21):e011640. https://doi.org/10.1161/JAHA.118.011640.
Roumie CL, Min JY, D’Agostino McGowan L, Presley C, Grijalva CG, Hackstadt AJ, Hung AM, Greevy RA, Elasy T, Griffin MR. Comparative safety of sulfonylurea and metformin monotherapy on the risk of heart failure: a cohort study. J Am Heart Assoc. 2017;6(4):e005379. https://doi.org/10.1161/jaha.116.005379.
Romero SP, Andrey JL, Garcia-Egido A, Escobar MA, Perez V, Corzo R, Garcia-Domiguez GJ, Gomez F. Metformin therapy and prognosis of patients with heart failure and new-onset diabetes mellitus. A propensity-matched study in the community. Int J Cardiol. 2013;166:404–12.
Bromage DI, Godec TR, Pujades-Rodriguez M, Gonzalez-Izquierdo A, Denaxas S, Hemingway H, Yellon DM. Metformin use and cardiovascular outcomes after acute myocardial infarction in patients with type 2 diabetes: a cohort study. Cardiovasc Diabetol. 2019;18(1):168. https://doi.org/10.1186/s12933-019-0972-4.
Arnold SV, Echouffo-Tcheugui JB, Lam CSP, Inzucchi SE, Tang F, McGuire DK, Goyal A, Maddox TM, Sperling LS, Fonarow GC, Masoudi FA, Kosiborod M. Patterns of glucose-lowering medication use in patients with type 2 diabetes and heart failure. Insights from the Diabetes Collaborative Registry (DCR). Am Heart J. 2018;203:25–9.
Packer M. Is metformin beneficial for heart failure in patients with type 2 diabetes? Diabetes Res Clin Pract. 2018;136:168–70.
Wijngaarden MA, Bakker LE, van der Zon GC, ‘t Hoen PA, van Dijk KW, Jazet IM, Pijl H, Guigas B. Regulation of skeletal muscle energy/nutrient-sensing pathways during metabolic adaptation to fasting in healthy humans. Am J Physiol Endocrinol Metab. 2014;307:E885–95.
Vendelbo MH, Clasen BF, Treebak JT, Møller L, Krusenstjerna-Hafstrøm T, Madsen M, Nielsen TS, Stødkilde-Jørgensen H, Pedersen SB, Jørgensen JO, Goodyear LJ, Wojtaszewski JF, Møller N, Jessen N. Insulin resistance after a 72-h fast is associated with impaired AS160 phosphorylation and accumulation of lipid and glycogen in human skeletal muscle. Am J Physiol Endocrinol Metab. 2012;302:E190–200.
Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–23.
Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–2.
Elamin M, Ruskin DN, Masino SA, Sacchetti P. Ketone-based metabolic therapy: is increased NAD+ a primary mechanism? Front Mol Neurosci. 2017;14(10):377. https://doi.org/10.3389/fnmol.2017.00377.
McCarty MF, DiNicolantonio JJ, O’Keefe JH. Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med Hypotheses. 2015;85:631–9.
Vilà-Brau A, De Sousa-Coelho AL, Mayordomo C, Haro D, Marrero PF. Human HMGCS2 regulates mitochondrial fatty acid oxidation and FGF21 expression in HepG2 cell line. J Biol Chem. 2011;286:20423–30.
Lee JM, Seo WY, Song KH, Chanda D, Kim YD, Kim DK, Lee MW, Ryu D, Kim YH, Noh JR, Lee CH, Chiang JY, Koo SH, Choi HS. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J Biol Chem. 2010;285:32182–91.
Zhang X, Yang S, Chen J, Su Z. Unraveling the regulation of hepatic gluconeogenesis. Front Endocrinol (Lausanne). 2019;24(9):802. https://doi.org/10.3389/fendo.2018.00802(eCollection 2018).
Bastin J, Lopes-Costa A, Djouadi F. Exposure to resveratrol triggers pharmacological correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum Mol Genet. 2011;20:2048–57.
Liu TF, Vachharajani VT, Yoza BK, McCall CE. NAD+ -dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem. 2012;287:25758–69.
Hirschey MD, Shimazu T, Capra JA, Pollard KS, Verdin E. SIRT1 and SIRT3 deacetylate homologous substrates: AceCS1,2 and HMGCS1,2. Aging. 2011;3:635–42.
Fitchett D, Zinman B, Wanner C, Lachin JM, Hantel S, Salsali A, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE, EMPA-REG OUTCOME trial investigators. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME trial. Eur Heart J. 2016;37:1526–34.
Ségalen C, Longnus SL, Baetz D, Counillon L, Van Obberghen E. 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside reduces glucose uptake via the inhibition of Na+/H+ exchanger 1 in isolated rat ventricular cardiomyocytes. Endocrinology. 2008;149:1490–8.
Eckardt KU, Kurtz A. Regulation of erythropoietin production. Eur J Clin Invest. 2005;35(Suppl 3):13–9.
Mazer CD, Hare GMT, Connelly PW, Gilbert RE, Shehata N, Quan A, Teoh H, Leiter LA, Zinman B, Jüni P, Zuo F, Mistry N, Thorpe KE, Goldenberg RM, Yan AT, Connelly KA, Verma S. Effect of empagliflozin on erythropoietin levels, iron stores and red blood cell morphology in patients with type 2 diabetes and coronary artery disease. Circulation. 2019. https://doi.org/10.1161/circulationaha.119.044235(Epub ahead of print).
Inzucchi SE, Zinman B, Fitchett D, Wanner C, Ferrannini E, Schumacher M, Schmoor C, Ohneberg K, Johansen OE, George JT, Hantel S, Bluhmki E, Lachin JM. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA-REG OUTCOME trial. Diabetes Care. 2018;41:356–63.
Li JW, Woodward M, Perkovic V, Figtree GA, Heerspink HJL, Mahaffey KW, de Zeeuw D, Vercruysse F, Shaw W, Matthews DR, Neal B. Mediators of the effects of canagliflozin on heart failure in patients with type 2 diabetes. JACC Heart Fail. 2020;8:57–66.
Chino Y, Samukawa Y, Sakai S, Nakai Y, Yamaguchi J, Nakanishi T, Tamai I. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm Drug Dispos. 2014;35:391–404.
Novikov A, Fu Y, Huang W, Freeman B, Patel R, van Ginkel C, Koepsell H, Busslinger M, Onishi A, Nespoux J, Vallon V. SGLT2 inhibition and renal urate excretion: role of luminal glucose, GLUT9, and URAT1. Am J Physiol Renal Physiol. 2019;316:F173–85.
Romuk E, Wojciechowska C, Jacheć W, Zemła-Woszek A, Momot A, Buczkowska M, Rozentryt P. Malondialdehyde and uric acid as predictors of adverse outcome in patients with chronic heart failure. Oxid Med Cell Longev. 2019;9(2019):9246138. https://doi.org/10.1155/2019/9246138.
Wojciechowska C, Romuk E, Tomasik A, Skrzep-Poloczek B, Nowalany-Kozielska E, Birkner E, Jacheć W. Oxidative stress markers and C-reactive protein are related to severity of heart failure in patients with dilated cardio-myopathy. Mediators Inflamm. 2014;2014:147040. https://doi.org/10.1155/2014/147040.
Sakai H, Tsutamoto T, Tsutsui T, Tanaka T, Ishikawa C, Horie M. Serum level of uric acid, partly secreted from the failing heart, is a prognostic marker in patients with congestive heart failure. Circ J. 2006;70:1006–11.
Kuppusamy UR, Indran M, Rokiah P. Glycaemic control in relation to xanthine oxidase and antioxidant indices in Malaysian Type 2 diabetes patients. Diabet Med. 2005;22:1343–6.
Lee CF, Chavez JD, Garcia-Menendez L, et al. Normalization of NAD + redox balance as a therapy for heart failure. Circulation. 2016;134:883–94.
Zhang Z, Blake DR, Stevens CR, et al. A reappraisal of xanthine dehydrogenase and oxidase in hypoxic reperfusion injury: the role of NADH as an electron donor. Free Radic Res. 1998;28:151–64.
Lappas M, Andrikopoulos S, Permezel M. Hypoxanthine-xanthine oxidase down-regulates GLUT1 transcription via SIRT1 resulting in decreased glucose uptake in human placenta. J Endocrinol. 2012;213:49–57.
Kim H, Baek CH, Chang JW, Yang WS, Lee SK. Febuxostat, a novel inhibitor of xanthine oxidase, reduces ER stress through upregulation of SIRT1-AMPK-HO-1/thioredoxin expression. Clin Exp Nephrol. 2020;24:205–15.
Aşcı H, Saygın M, Yeşilot Ş, Topsakal Ş, Cankara FN, Özmen Ö, Savran M. Protective effects of aspirin and vitamin C against corn syrup consumption-induced cardiac damage through sirtuin-1 and HIF-1α pathway. Anatol J Cardiol. 2016;16:648–54.
Ryan MJ, Jackson JR, Hao Y, Williamson CL, Dabkowski ER, Hollander JM, Alway SE. Suppression of oxidative stress by resveratrol after isometric contractions in gastrocnemius muscles of aged mice. J Gerontol A Biol Sci Med Sci. 2010;65:815–31.
Huang XF, Li HQ, Shi L, Xue JY, Ruan BF, Zhu HL. Synthesis of resveratrol analogues, and evaluation of their cytotoxic and xanthine oxidase inhibitory activities. Chem Biodivers. 2008;5:636–42.
Wang J, Zhu XX, Liu L, Xue Y, Yang X, Zou HJ. SIRT1 prevents hyperuricemia via the PGC-1α/PPARγ-ABCG2 pathway. Endocrine. 2016;53:443–52.
Zhao Y, Xu L, Tian D, Xia P, Zheng H, Wang L, Chen L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: a meta-analysis of randomized controlled trials. Diabetes Obes Metab. 2018;20:458–62.
Nistala R, Raja A, Pulakat L. mTORC1 inhibitors rapamycin and metformin affect cardiovascular markers differentially in ZDF rats. Can J Physiol Pharmacol. 2017;95:281–7.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
There is only one author, who takes full responsibility for the work. The author read and approved the final manuscript.
Authors’ information
The author has written extensively about SGLT2 inhibitors and their mechanism of action, and is currently leading two of the large-scale clinical trials of SGLT2 inhibitors in heart failure (EMPEROR-Reduced and EMPEROR-Preserved). He has also co-authored leading papers on the molecular mechanisms of autophagy (Cell in 2005; Nature in 2012; and Journal of Clinical Investigation in 2015).
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable. All authors agree to publication. No permissions are needed.
Competing interests
Dr. Packer has consulted for Abbvie, Actavis, Akcea, Amgen, AstraZeneca, Boehringer Ingelheim, Cardiorentis, Daiichi Sankyo, Johnson & Johnson, NovoNordisk, Pfizer, Relypsa, Sanofi, Synthetic Biologics and Theravance.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Packer, M. Autophagy-dependent and -independent modulation of oxidative and organellar stress in the diabetic heart by glucose-lowering drugs. Cardiovasc Diabetol 19, 62 (2020). https://doi.org/10.1186/s12933-020-01041-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-020-01041-4