
Abstract
Diabetes mellitus currently affects over 350 million patients worldwide and is associated with many deaths from cardiovascular complications. Sodium–glucose co-transporter 2 (SGLT-2) inhibitors are a novel class of antidiabetic drugs with cardiovascular benefits beyond other antidiabetic drugs. In the EMPA-REG OUTCOME trial, empagliflozin significantly decreases the mortality rate from cardiovascular causes [38% relative risk reduction (RRR)], the mortality rate from all-causes (32% RRR) and the rate of heart failure hospitalization (35% RRR) in diabetic patients with established cardiovascular diseases. The possible mechanisms of SGLT-2 inhibitors are proposed to be systemic effects by hemodynamic and metabolic actions. However, the direct mechanisms are not fully understood. In this review, reports concerning the effects of SGLT-2 inhibitors in models of diabetic cardiomyopathy, heart failure and myocardial ischemia from in vitro, in vivo as well as clinical reports are comprehensively summarized and discussed. By current evidences, it may be concluded that the direct effects of SGLT-2 inhibitors are potentially mediated through their ability to reduce cardiac inflammation, oxidative stress, apoptosis, mitochondrial dysfunction and ionic dyshomeostasis.
Similar content being viewed by others

Background
Diabetes mellitus (DM) currently affects over 350 million patients globally [1]. The causes of death up to 80% in patients with type 2 DM (T2DM) are associated with cardiovascular diseases [2, 3]. Diabetic cardiomyopathy is a progressive disease which affects both cardiac structure and function in diabetic patients. These abnormalities include cardiac hypertrophy, cardiac apoptosis and necrosis, ventricular dilatation and interstitial fibrosis [4, 5] which consequently leads to both systolic and diastolic dysfunctions [6]. Metabolic disturbances, including hyperglycemia, insulin resistance and hyperlipidemia, play an important role in the diabetic cardiomyopathy process by triggering the renin–angiotensin–aldosterone system, altered lipid metabolism, inflammation, oxidative stress, mitochondrial dysfunction and endoplasmic reticulum (ER) stress [7]. Chronic exposure to these conditions make heart limited to physiological adaptation and repair capacity.
The sodium–glucose co-transporters (SGLT) are a family of active glucose transporter proteins with two major isoforms, SGLT-1 and SGLT-2 [8]. SGLT-1 expression found in the small intestine, liver, lung, kidney and heart, whereas SGLT-2 expression is predominantly found in the kidney [9]. SGLT-2 inhibitors are a novel class of antidiabetic drugs which produce glycosuric and natriuretic effects by inhibiting glucose and sodium reabsorption from the proximal convoluted tubules [10]. Some SGLT-2 inhibitors, including canagliflozin, dapagliflozin and empagliflozin, have been approved for their use in Europe and the USA [11]. Recently, SGLT-2 inhibitors have become the topic of interest due to the benefits in a cardiovascular outcome trial beyond other antidiabetic drugs. The EMPA-REG OUTCOME trial (2010–2015) showed the cardioprotective effect of empagliflozin by significantly lowering the rate of death from cardiovascular causes [38% relative risk reduction (RRR)], all-cause death (32% RRR) and heart failure hospitalization (HHF) (35% RRR) in T2DM patients with established cardiovascular diseases (CVD) [12]. These benefits of empagliflozin are expected to be class effects with SGLT-2 inhibitors. Several studies also supported this evidence [13,14,15] as summarized in Table 1. The CANVAS trial (2009–2017) showed canagliflozin significantly reduced the composite of cardiovascular-cause death, nonfatal myocardial infarction (MI) or nonfatal stroke [hazard ratio (HR) 0.86, 95% confidence interval (CI) 0.75–0.97] and HHF (HR 0.67, 95% CI 0.52–0.87) [13]. The CVD-REAL study which reported the cardiovascular effect of SGLT-2 inhibitors compared to other glucose-lowering drugs showed that SGLT-2 inhibitors could significantly decrease the rate of HHF (HR 0.49, 95% CI 0.41–0.57) and all-cause death (HR 0.61, 95% CI 0.51–0.73) [14]. Ongoing cardiovascular trials of SGLT-2 inhibitors include DECLARE-TIMI 58 (dapagliflozin, 2013–2019), VERTIS CV (ertugliflozin, 2013–2019) and RECEDE-CHF (empagliflozin, 2017–2019) may help to confirm this expectation [16,17,18].
Despite the cardiovascular benefits of SGLT-2 inhibitors, their biological mechanisms leading to cardioprotection are not fully understood. Possible mechanisms are clearly proposed to be systemic effects by hemodynamic actions via natriuresis and metabolic actions via glycosuria [19,20,21,22,23]. Natriuresis results in lowering plasma volume and blood pressure, which are subsequently decreasing cardiac preload and afterload [24,25,26,27]. This effect occurs without heart rate changes suggesting the lack of sympathetic stimulation [24, 28]. Empagliflozin also reduces arterial stiffness and vascular resistance in diabetic patients [28, 29]. In renal hemodynamics, empagliflozin attenuates albuminuria and hyperglycemia induced glomerular hyperfiltration, resulting in decreased intraglomerular hypertension [30,31,32]. The modulation of systemic and renal hemodynamics by SGLT2 inhibitors decreases cardiac workload and improves cardiac function. Glycosuria results in reducing plasma glucose concentration and subsequently decreasing glucotoxicity, a factor which leads to diabetic cardiomyopathy, as evidenced by the improvement of β cell function and insulin sensitivity [33, 34]. Interestingly, one of the benefits of SGLT-2 inhibitors beyond other hypoglycemic drugs is that they do not cause hypoglycemia, since they can enhance endogenous glucose production [33, 34]. The mechanism responsible for this benefit is the increase of plasma glucagon concentration by SGLT-2 inhibitors [33,34,35]. Dapagliflozin can directly stimulate pancreatic alpha cells for glucagon secretion [35]. Glucagon is known as the key hormone for hepatic glucose production [36, 37], enhances ketogenesis [38, 39] and improves cardiac contractility [40, 41]. Furthermore, SGLT-2 inhibitors can shift metabolism from carbohydrate to lipid [33, 42] and increase ketone body level in both animal and clinical studies [42,43,44]. The mechanisms responsible for that are potentially from compensatory mechanisms of glucose lowering drugs [45], reducing renal ketone body clearance [46] and enhancing ketone body production by glucagon action [33,34,35]. Ketone bodies are good energy source in myocardium especially in failing hearts [47, 48]. In addition, ketone bodies are associated with anti-arrhythmia and increasing mitochondrial biogenesis [49, 50]. This is known as the ketone body theory [51]. However, their benefits and mechanisms are still questionable because ketone bodies can precipitate diabetic ketoacidosis, a serious complication of diabetes [46]. Glycosuria also results in weight and fat mass reduction due to stimulation of lipid oxidation compensating for energy loss [52,53,54]. Both hemodynamic and metabolic actions of SGLT-2 inhibitors potentially work together for improving diabetic cardiomyopathy and finally resulting in cardioprotection as shown in the EMPA-REG OUTCOME trial. However, only systemic mechanisms may be not enough to explain much better cardiovascular benefits of SGLT-2 inhibitors when compared to other glucose-lowering drugs. Their direct cardiac mechanisms, even no SGLT-2 expression in the heart [9, 55, 56], may be the answers for that. Therefore, the objective of this review was to comprehensively summarize reports from in vitro, in vivo and clinical studies regarding the evidence of possible direct mechanisms responsible for cardioprotective effect of SGLT-2 inhibitors, which are independently from their systemic actions.
Effects of SGLT-2 inhibitors on cardiac structure
SGLT-2 inhibitors have been shown to improve cardiac histopathologic changes in the diabetic cardiomyopathy models of mice and rats, the heart failure model of zebrafish embryos and also the myocardial ischemic model of rats. These reports are summarized in Table 2. In 2016, Kusaka et al. studied the effect of empagliflozin in genetic prediabetes/metabolic syndrome rat model [57]. After 10 weeks of treatment, empagliflozin significantly reduced left ventricular weight, cardiomyocyte size, cardiac interstitial fibrosis and cardiac interstitial macrophage infiltration. Several reports that studied the effect of empagliflozin in genetic diabetic mouse models also suggested the improvement of cardiac morphologic changes by decreasing the cardiomyocyte cross sectional area, interstitial collagen I and III depositions, interstitial fibrosis and interstitial macrophage infiltration [58, 59]. Lin et al. also reported that empagliflozin could attenuate pericoronary arterial fibrosis and coronary arterial thickening [58]. The reduction of cardiac fibrosis was shown to be due to the attenuation of the expression of pro-fibrotic signaling pathway, serum- and glucocorticoid-regulated kinase 1 (SGK1) and epithelial sodium channel (ENaC) [59]. In another genetic diabetic mouse model, empagliflozin was given for a total of 6 weeks [60]. Although there were no significant changes in left ventricular (LV) mass and histologic myocardial fibrosis, the treatment group showed the decrease of cardiac hypertrophy and remodeling markers. Empagliflozin was found to decrease the mRNA expression of hypertrophic fetal genes including atrial natriuretic peptide/factor and beta-myosin heavy chain. It also decreased the expression of proteins associated with mitogen-activated protein kinase pathways, including extracellular signal-regulated kinases, c-Jun NH2-terminal kinases and p38, which played an important role in the development of cardiac remodeling [61]. In streptozotocin-induced diabetic cardiomyopathy rats treated with empagliflozin for 8 weeks, the attenuation of disordered cell arrays and focal necrosis was observed in a dose-dependent manner [62]. Furthermore, Ye et al. showed that the treatment of dapagliflozin for 8 weeks decreased myocardial collagen-1 and collagen-3 mRNA levels and percentage of fibrosis in genetic diabetic mice [63]. These reports suggest that the improvement of diabetic cardiomyopathy morphology is potentially to be class effects of SGLT-2 inhibitors.
In addition to the diabetic cardiomyopathy model, SGLT-2 inhibitors were also tested in models of myocardial ischemia and heart failure. We recently demonstrated that by giving dapagliflozin for 4 weeks in high fat diet induced obese-insulin resistance rats and underwent acute ischemic/reperfusion (I/R) injury by left anterior descending artery (LAD) ligation, dapagliflozin could attenuate myocardial infarct size [64]. In chronic MI rat model, Lee et al. investigated the effect of dapagliflozin treatment beginning 1 day after LAD ligation and continued for 4 weeks [65]. They found that dapagliflozin did not alter the size of an infarction, however it could attenuate myofibroblast infiltration and cardiac fibrosis. In 2017, Shi et al. tested the effect of empagliflozin in aristolochic acid induced heart failure in zebrafish embryos [66]. Zebrafish embryos treated with aristolochic acid would develop cardiac hypertrophy, bradycardia and profound cardiac failure within 3 days of age [67]. Pretreatment with empagliflozin showed the improvement of histopathologic changes including unlooping defects, cardiac edema and deformed cardiac chambers in a concentration-dependent manner [66]. Furthermore, empagliflozin could attenuate the expression of heart failure markers including atrial natriuretic peptide and brain natriuretic peptide. In a very recent clinical trial, Januzzi et al. tested the effect of 2-year canagliflozin treatment in 666 elderly T2DM patients [68]. Compared to placebo, treatment with canagliflozin delayed the rise of heart failure biomarkers including serum N-terminal pro-brain natriuretic peptide and high-sensitivity troponin I.
Effects of SGLT-2 inhibitors on cardiac function
SGLT-2 inhibitors have also been shown to improve cardiac function in diabetic cardiomyopathy models and myocardial ischemic models of mice and rats as summarized in Table 3. In genetic diabetic mice, 8-week treatment of dapagliflozin improved ejection fraction (EF) and fractional shortening [63]. It also attenuated the increase in end-systolic volume (ESV), end-diastolic volume (EDV), interventricular septal thickness in systole and diastole. Dapagliflozin also improved the E/A (early/late diastolic) ratio, EF, isovolumic relaxation time (IVRT), deceleration time (DT) and end diastolic wall thickness (EDWT) in a diabetic non-obese mouse model [69]. These reports indicated that dapagliflozin could improve both systolic and diastolic LV function in diabetic mice.
For empagliflozin, a number of reports showed its benefits preferred diastolic function to systolic function [57,58,59,60, 62]. In genetic diabetic mice, empagliflozin improved diastolic function as seen by increased septal wall motion and decreased LV filling pressure [59]. It also attenuated vascular dilating dysfunction by ameliorating the impairment of vascular endothelium-dependent relaxation in thoracic aortas [58]. Moreover, empagliflozin has been shown to improve LV diastolic function, both in relaxation and compliance, as evidenced by a decrease in E wave (mitral inflow peak velocity), E wave deceleration time, Tau (time constant for isovolumic relaxation) and end-diastolic pressure–volume relationship [60]. However, LV systolic function was not affected in this report. In a diabetic cardiomyopathy rat model, empagliflozin also improved LV function by the increase of end-systolic pressure (ESP), + dp/dt and − dp/dt (the maximal ascending rate and the maximal descending rate of left ventricular pressure, respectively) and the decrease of end-diastolic pressure (EDP) [62]. However, in the prediabetic/metabolic syndrome rat model, 10 weeks of empagliflozin treatment, which attenuated LV weight and cardiac interstitial fibrosis, did not significantly improve heart rate, blood pressure, sympathetic activity or baroreceptor function [57].
Electrophysiologically, two recent clinical studies reported the effect of SGLT-2 inhibitors on electrocardiographic parameters in patients with T2DM [68, 70]. Sato et al. retrospectively analyzed changes in indices of ventricular repolarization before and after 0.66-year treatment with SGLT-2 in 46 people with T2DM [70]. They found the heart rate and QTc interval were not changed, but QTc dispersion was significantly decreased, suggesting that SGLT-2 inhibitors could reverse ventricular repolarization heterogeneity in T2DM patients. However, inconsistent findings exist. Januzzi et al. demonstrated the negative results from a randomized control trial which tested the effect of a 2-year canagliflozin treatment compared to placebo in 666 elderly T2DM patients [68]. They found that canagliflozin did not change any electrocardiographic parameters including PR interval, QRS interval, QT/QTc or RR intervals. Therefore, more clinical trials are required to assess the effects of SGLT-2 inhibitors on the cardiovascular function.
In addition to the diabetic cardiomyopathy models, SGLT-2 inhibitors also improved cardiac function in models of myocardial ischemia. In 2018, Tanajak et al. tested the dapagliflozin effect in obese-insulin resistance rats with I/R injury [64]. Before I/R injury operation, 4-week dapagliflozin treatment already showed the improvement of E/A ratio, EF, IVRT, DT and EDWT which referred to both systolic and diastolic LV function. Furthermore, during I/R injury, rats treated with dapagliflozin gave the increase of time to the first ventricular tachycardia/fibrillation onset, the increase of gap junction protein connexin 43 expression, the decrease of arrhythmia score and the improvement of EF, stroke volume, ESP, EDP, ESV and EDV. Similar results were found in Wistar rats with MI treated with dapagliflozin for 4 weeks in which the improvement of cardiac function as evidenced by increasing the maximal rate of LV + dP/dt and − dP/dt were observed [65]. Based on the EMPA-REG OUTCOME trial, there were no significant differences in the rates of MI between the placebo and treatment groups [12]. However, as evidenced from animal studies, SGLT-2 inhibitors could potentially exert beneficial effects to decrease the severity of MI both structural and function. Future clinical studies are needed to warrant these findings from basic reports.
Potential mechanisms of SGLT-2 inhibitors responsible for cardioprotection
SGLT-2 inhibitors on cardiac inflammation
Cardiac inflammation is one of the mechanisms that leads to diabetic cardiomyopathy in diabetic patients [71,72,73]. Evidence shows that SGLT-2 inhibitors, together with systemic effects, could directly decrease cardiac inflammation. These effects of SGLT-2 inhibitors on cardiac inflammation are summarized in Table 4. In the genetic prediabetes/metabolic syndrome rat model, 10 weeks of empagliflozin treatment significantly reduced cardiac interstitial macrophage infiltration [57]. Empagliflozin could also attenuate cardiac macrophage infiltration by decreasing cell numbers in the genetic diabetes/obesity mouse model [58]. Lee et al. also reported the improvement of cardiac inflammation in Wistar rats with acute phase of MI treated with dapagliflozin for 2 days [65]. They revealed that dapagliflozin decreased inflammatory cytokines mRNA levels including IL-1β and IL-6, increased anti-inflammatory cytokine mRNA levels including IL-10, and also increased the M2/M1 phenotype macrophage ratio. Since M1 is pro-inflammatory phenotype of macrophage, whereas M2 is anti-inflammatory one [74], these findings indicated that dapagliflozin promotes macrophage polarization toward an anti-inflammatory phenotype. Furthermore, empagliflozin could attenuate the myocardial expression of pro-inflammatory genes including cyclooxygenase-2 and interleukin-1β (IL-1β) in a heart failure model of zebrafish embryos [66]. All of these findings suggest that empagliflozin can reduce cardiac inflammation in diabetic cardiomyopathy, myocardial ischemia and heart failure models. However, we cannot conclude whether these benefits are from systemic and/or direct cardiac effects. More in vitro studies are required to explore its direct role independently from systemic one.
Interestingly, a recent study revealed the direct mechanism of SGLT-2 inhibitors on cardiac inflammation reduction through the reduction of cardiac nucleotide-binding oligomerization domain-like receptor 3 (NLRP3) inflammasome [63]. The NLRP3 inflammasome is an interleukin-1β family cytokine-activating multi-protein signaling complex upregulated in the heart and associated with cardiac inflammation in T2DM, which leads to subsequent diabetic cardiomyopathy [75,76,77,78,79]. NACHT, LRR and PYD domains-containing protein 3 (NALP-3), the protein encoded by the NLRP3 gene, together with apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) form a protein complex activating caspase-1, which subsequently leads to stimulating the production of pro-inflammatory cytokines [72, 73]. Ye et al. tested the effect of 8-week dapagliflozin treatment on cardiac inflammation in genetic diabetic mice and found that dapagliflozin decreased the levels of myocardial mRNA associated with NLRP3 inflammasome and pro-inflammatory cytokines including NALP-3, ASC, caspase-1, IL-1β, IL-6 and TNFα [63]. To rule out systemic effects, they further performed in vitro experiment by incubating mouse cardiofibroblasts in media containing dapagliflozin for 16 h. Interestingly, dapagliflozin also attenuated NALP-3, ASC, caspase-1 and IL-1β mRNA levels in a dose-dependent manner [63]. Since SGLT-2 does not exist in cardiac tissue [9, 55, 56], these results suggested that these effects are unrelated from SGLT-2 and glucose reducing effects of dapagliflozin.
SGLT-2 inhibitors on cardiac oxidative stress
Oxidative stress plays an important role in the pathogenesis of cardiac hypertrophy and remodeling [80,81,82]. SGLT-2 inhibitors have been shown to act as antioxidants by decreasing cardiac oxidative stress, independently from glucose lowering effects, as evidence summarized in Table 5. In genetic prediabetes/metabolic syndrome rat model, 10 weeks of empagliflozin treatment significantly reduced superoxide levels in cardiac tissues [57]. This report showed the reduction of cardiac hypertrophy and interstitial fibrosis although no blood pressure reduction or improvement of cardiac autonomic dysfunction. Therefore, these cardioprotective effects have been attributed to the lowering of cardiac oxidative stress and inflammation. In a genetic diabetic mouse model, 10-week empagliflozin treatment could decrease cardiac and aortic superoxide levels [58]. In a diabetes/obesity mouse model, Habibi et al. showed empagliflozin (10 mg/kg/days) did not alter the levels of cardiac nitrotyrosine, advanced glycation end products (AGEs) and receptors for AGEs (RAGEs) [59]. In aortic tissues of diabetic cardiomyopathy rats, empagliflozin treatment with high dose (30 mg/kg/days) significantly decreased AGEs and RAGEs levels but was unchanged in the low dose (10 mg/kg/days) treatment group [83]. Therefore, a high dose of empagliflozin treatment is required for oxidative stress reduction.
For a model of acute myocardial ischemia, we previously demonstrated that dapagliflozin attenuated malondialdehyde in the cardiac ischemic area of obese-insulin resistant rats with I/R injury [64]. In a chronic MI model of rats, Lee et al. showed that dapagliflozin acted as an antioxidant and mediated M2 macrophage polarization through the signal transducer and activator of the transcription 3 (STAT3) mediated pathway [65]. Antioxidants, the reactive oxygen and nitrogen species (RONS) scavengers, are known to increase STAT3 activity which subsequently plays an action in M2 macrophage polarization upregulation, resulting in a decrease of cardiac inflammation [84, 85]. Moreover, myocardial ischemic rats fed with dapagliflozin for 2 days demonstrated the attenuation of superoxide and nitrotyrosine levels in cardiac tissues [65]. Dapagliflozin also increased STAT3 activity and stimulated macrophages toward an anti-inflammatory phenotype through STAT3 signaling [65]. In an ex vivo experiment using isolated hearts after 3-day infarction treated with dapagliflozin for 1 h, dapagliflozin-treated hearts still had increased STAT3 activity and IL-10 protein levels [65]. This evidence supports the role of dapagliflozin in MI as antioxidant and inflammatory modulators through direct RONS-dependent STAT3 signaling, independently from its SGLT-2 and glucose lowering effects.
SGLT-2 inhibitors on cardiac apoptosis
Cardiac apoptosis has been shown to be responsible for cardiomyocyte death during MI and heart failure [86,87,88]. Accumulating reports have demonstrated that SGLT-2 inhibitors could decrease cardiac apoptosis in diabetic models and myocardial ischemic model of mice and rats as summarized in Table 6. In genetic diabetic mouse model, dapagliflozin treatment for 8 weeks significantly attenuated apoptotic cells in the left ventricle [63]. Consistently, diabetic cardiomyopathy rats treated with empagliflozin for 8 weeks had a decreased level of apoptotic cardiomyocytes [62]. This effect appeared in a dose-dependent manner since the number of apoptotic cells in high dose treated group (30 mg/kg/days) was significant lower than that in a low dose (10 mg/kg/days) treated group. In that report [62], it has been proposed that empagliflozin protected against cardiomyocyte apoptosis by suppression of the endoplasmic reticulum stress (ERS) pathway.
ERS is one of the pathological conditions in the diabetic cardiomyopathy which activated reactive oxygen species (ROS)-mediated cell apoptosis [89]. ERS can be stimulated by situations such as hyperglycemia, hypoxia, and ROS exposure, which results in abnormal protein folding and maturation leading to apoptosis [90]. Inhibitions of ERS could attenuate myocardial apoptosis and diabetic cardiomyopathy development in streptozotocin-induced diabetic rats [91, 92]. In response to ERS, glucose-regulated protein 78 (GRP78), a major ER chaperone protein, is activated and plays a vital role in detecting the anomalous proteins [93]. Once ERS occurred, caspase-12-mediated apoptosis, which is a unique apoptosis pathway of ER, is activated [94, 95]. Then, CCAAT-enhancer-binding protein homologous protein (CHOP), a subsequent protein of the apoptotic pathway, can stimulate the caspase protein in the cytosol, leading to apoptosis [96]. CHOP can be activated by the over-transcription of activating transcription factor 4 (ATF4), tumor necrosis factor receptor-associated factor 2 (TRAF2) and X-box binding protein 1 (XBP1) [97, 98]. It has been shown that empagliflozin decreased all of mRNA and protein expressions associated with ERS including GRP78, CHOP, Caspase-12, ATF4, TRAF2 and XBP1 [62]. Interestingly, these beneficial effects are in a dose-dependent manner. Thus, another role of empagliflozin in protection against diabetic cardiomyopathy is by attenuating cardiomyocyte apoptosis through inactivating the ERS pathway. However, inconsistent findings exist. A report by Hammoudi and colleagues demonstrated that empagliflozin (10 mg/kg/days) did not alter the protein expression of the antiapoptotic molecule Bcl-2 and the pro-apoptotic protein Bax in diabetic mice [60]. These inconsistent findings could be due to different doses of drug used; empagliflozin treatment at a low dose might not show an improvement [59, 83]. Future studies using a higher dose of empagliflozin treatment should be investigated in in vivo to explore its role on an apoptotic protein expression in diabetic cardiomyopathy model.
In myocardial I/R injury model, it has been shown that treatment with dapagliflozin for 4 weeks in obese-insulin resistance rats attenuated Bax/Bcl-2 ratio and cleaved caspase 3 level when these rats underwent I/R injury [64]. By current evidences, SGLT-2 inhibitors seem to attenuate apoptotic myocardial cells in diabetic cardiomyopathy and MI, however more studies are required to explore their roles on cardiac apoptosis.
SGLT-2 inhibitors on cardiac mitochondrial function
Mitochondria are important to maintain physiological cardiac function due to their roles in energy production, calcium homeostasis and ROS production [99]. Mitochondrial dysfunction is found to be associated with the pathological progression of diabetic cardiomyopathy [100]. Impaired mitochondrial function and dynamics are observed in diabetic patients and leads to myocardial contractile dysfunction [101]. Several reports showed the attenuation of mitochondrial dysfunction by SGLT-2 inhibitor treatment [59, 64].
In a genetic diabetic mouse model, empagliflozin has been shown to attenuate ultrastructural anomalies of inter-myofibrillar mitochondria including disorganized appearance of sarcomeres, reduced matrix electron density, loss of cristae and mitochondrial fragmentation [59]. In obese-insulin resistant rats treated with dapagliflozin for 4 weeks before undergoing cardiac I/R injury, dapagliflozin attenuated the increase of mitochondrial ROS production, depolarization and mitochondrial swelling [64]. The mitochondrial morphology was also improved by attenuating mitochondrial fragmentation, loss of cristae and fusion of cristae. For mitochondrial biogenesis, dapagliflozin increased the protein expressions of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1-α) and carnitine palmitoyltransferase 1 (CPT1), which were essential proteins for the regulation of cardiac mitochondrial fatty acid oxidation [102, 103]. The expression of complex I of the electron transport chain was also increased by dapagliflozin treatment, suggesting its role in restoring the reduction of cardiac energy metabolism during cardiac I/R injury [64].
It is known that mitochondria are dynamic organelles with the balance of continual fission (division) and fusion (joining) [104]. These cycles maintain functional mitochondria by removing damaged mitochondria and facilitating apoptosis when cells are exposed to stress [105]. Dynamin-related protein 1 (DRP1) plays role in constriction of the membrane during fission whereas mitofusin 2 (MFN2) and optic atrophy 1 (OPA1) support fusion of the outer and inner membranes, respectively [100]. It has been shown that an inhibition of cardiac mitochondrial fission could protect the heart during myocardial I/R injury and cardiac arrest [106, 107]. Recently, we have demonstrated that a 4-week treatment with dapagliflozin attenuated the increase of cardiac mitochondrial fission and the decrease of mitochondrial fusion as evidenced by decreased DRP1 and increased MFN2 and OPA1 expressions in obese-insulin resistant rats undergoing cardiac I/R injury [64]. The improvement of mitochondrial dynamics observed in this study, together with the improvement of mitochondrial function, morphology, ROS production, biogenesis and protein expressions could be the mechanisms responsible for smaller infarct size with dapagliflozin treatment compared to placebo. However, these benefits are still unclear whether they are from direct cardiac and/or systemic effects. More studies are needed to explore its direct role on the heart.
SGLT-2 inhibitors on cardiac ionic homeostasis
Cardiac Ca2+ and Na+ homeostasis plays an important role in maintaining physiologic cardiac function, including rhythm and contraction [108, 109]. Intracellular Na+ and Ca2+ loading has been observed in diabetic hearts, particularly in heart failure [110,111,112]. Reduction of myocardial intracellular Na+ concentration by inhibition of Na+/Ca2+ or Na+/H+ exchangers improves heart failure and cardiac hypertrophy [113,114,115]. SGLT-2 inhibitors have been shown to improve cardiac Na+ and Ca2+, as evidence summarized in Table 7. Baartscheer and colleagues demonstrated the direct myocardial effects of empagliflozin on Na+ and Ca2+ concentration alteration, independent from SGLT-2 activity [116]. In isolated ventricular myocytes from rabbits and rats incubated with empagliflozin for 3 h, empagliflozin decreased cytoplasmic Na+ ([Na+]c) and Ca2+ ([Ca2+]c) and also increased mitochondrial Ca2+ concentration ([Ca2+]m). These effects were similar to the effect of Na+/H+ exchange (NHE) inhibitor [110]. These findings were confirmed in cells pre-treated with cariporide, a strong NHE inhibitor, in which the results showed that empagliflozin had very little effect on the [Na+]c of these cells. Therefore, despite no SGLT-2 expression in the heart [9, 55, 56], empagliflozin could have cardiac effects by decreasing myocardial [Na+]c and [Ca2+]c and increasing [Ca2+]m through the inhibition of NHE directly. Moreover, Liu and O’Rourke demonstrated that high [Na+]c could cause low [Ca2+]m through its efflux via mitochondrial Na+/Ca2+ exchanger (NCX) [117]. Increasing [Ca2+]m, could prevent sudden death in a swine heart failure model [113]. Taken together, SGLT-2 inhibitors directly inhibited myocardial NHE and consequently decreased the cytoplasmic Na+ level, leading to increased mitochondrial Ca2+ level and decreased cytoplasmic Ca2+ level through mitochondrial NCX activity. In addition, it has been shown that empagliflozin improved LV diastolic function by increasing sarcoplasmic endoplasmic reticulum Ca2+-ATPase (SERCA2a) activity in genetic diabetic mice [60]. Consistent with this report, Joubert et al. also showed that dapagliflozin increased SERCA2a function in a diabetic lipodystrophic mouse model [69]. SERCA2a is an important calcium handling protein which regulates cardiac contractility via Ca2+ reuptake into sarcoplasmic reticulum [118]. Decreased SERCA2a activity can cause abnormal Ca2+ handling and a contractile state, leading to cardiac contractile dysfunction [119]. All of these findings indicate that the improvement of cardiac calcium handling of SGLT-2 inhibitors through these mechanisms could be responsible for the protective effect in heart failure observed in the EMPA-REG OUTCOME trial.
Conclusions and perspectives
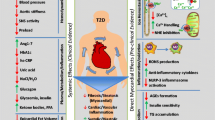
The cardioprotection of SGLT-2 inhibitors has been demonstrated in models of diabetic cardiomyopathy, heart failure and myocardial ischemia. They are seen to be effective by improving cardiac morphologic changes including cardiac hypertrophy, interstitial fibrosis, heart failure and myocardial infarct size. They also improve both systolic and diastolic LV function in diabetic cardiomyopathy and prevent cardiac arrhythmia in cardiac I/R injury. Potential mechanisms responsible for the cardioprotective effects of SGLT-2 inhibitors are through direct and systemic effects which are summarized in Fig. 1. Their systemic effects are modulated by hemodynamic actions via natriuresis and metabolic actions via glycosuria. The direct effects of SGLT-2 inhibitors could potentially mediate through their abilities to attenuate cardiac inflammation, oxidative stress, apoptosis, mitochondrial dysfunction and ionic dyshomeostasis.

Potential mechanisms responsible for cardioprotective effect of SGLT-2 Inhibitors. From current evidence from both in vitro or ex vivo experiments, SGLT-2 inhibitors have been demonstrated that they could have direct cardiac effects on inflammation [63], oxidative stress [65], and ionic dyshomeostasis [116]. Although the effects of SGLT-2 inhibitors on the attenuation of apoptosis and mitochondrial dysfunction could be direct cardiac effects [59, 62, 64], they have not been proved by either in vitro or ex vivo experiments. Italics indicate the mechanisms have not been proved by either in vitro or ex vivo experiments. SGLT-2 sodium–glucose co-transporter 2, NLRP3 nucleotide-binding oligomerization domain-like receptor 3, IL interleukin, STAT3 signal transducer and activator of transcription 3, ERS endoplasmic reticulum stress, Bcl-2 B cell chronic lymphocytic leukemia/lymphoma-2, Bax Bcl-2-associated X, PGC1-α peroxisome proliferator-activated receptor gamma coactivator 1-alpha, CPT1 carnitine palmitoyltransferase 1, ROS reactive oxygen species, NHE Na+/H+ exchange, [Na+]c cytoplasmic Na+ concentration, [Ca2+]c cytoplasmic Ca2+ concentration, [Ca2+]m mitochondrial Ca2+ concentration, SERCA2a sarcoplasmic endoplasmic reticulum Ca2+-ATPase, I/R ischemic/reperfusion
Focus on The EMPA-REG OUTCOME trial, empagliflozin decreased the rate of death from cardiovascular causes and HHF in T2DM patients with established CVD [12]. The mechanisms of SGLT-2 inhibitors responsible for these benefits could be due to their systemic as well as direct cardiac effects. SGLT-2 inhibitors can modify risk factors of major cardiovascular events including diabetes and hypertension via lowering blood glucose and blood pressure, respectively. They also directly attenuate cardiac inflammation, oxidative stress [63, 65] which lead to improving both cardiac structure and function, and finally result in decreased mortality rate from cardiovascular causes. For reducing HHF, natriuretic effect of SGLT-2 inhibitors results in lowering plasma volume and blood pressure, which are subsequently decreasing cardiac preload and afterload [24,25,26]. SGLT-2 inhibitors also directly improve cardiac calcium handling via inhibiting myocardial NHE which subsequently decrease intracellular Na+ and Ca2+ loading mostly found in heart failure [116]. Therefore, cardiac contractility and cardiac output could be improved in heart failure patients as observed in clinical trials [12,13,14,15].
Despite these growing number of reports, the cardioprotective effects in some respects such as cardiac apoptosis and mitochondrial dysfunctions are still unclear whether they are the direct effects or systemic effects of SGLT-2 inhibitors. More studies of heart failure and myocardial ischemic models are required to investigate the roles of SGLT-2 inhibitors on the heart. Although SGLT-2 does not exist in the myocardium [9, 55, 56], Park et al. reported that endothelial cells of porcine coronary artery exposed to high glucose upregulated SGLT-2 expression despite no expression in normal condition [120]. Even the role of SGLT-2 on vessels has not been understood yet. It has been shown that chronic treatment of SGLT-2 inhibitors in a diabetic mouse model attenuated vascular relaxation dysfunction and atherosclerosis in aorta and coronary artery [58, 121, 122]. More studies investigating the roles of SGLT-2 inhibitors on the heart are needed to warrant their use in the future.
Abbreviations
- [Ca2+]c :
-
cytoplasmic Ca2+ concentration
- [Ca2+]m :
-
mitochondrial Ca2+ concentration
- [Na+]c :
-
cytoplasmic Na+ concentration
- + dp/dt:
-
the maximal ascending rate of left ventricular pressure
- − dp/dt:
-
the maximal descending rate of left ventricular pressure
- AGEs:
-
advanced glycation end products
- ASC:
-
apoptosis-associated speck-like protein containing a caspase recruitment domain
- ATF4:
-
activating transcription factor 4
- CHOP:
-
CCAAT-enhancer-binding protein homologous protein
- CPT1:
-
carnitine palmitoyltransferase 1
- CVD:
-
cardiovascular diseases
- DM:
-
diabetes mellitus
- DRP1:
-
dynamin-related protein 1
- DT:
-
deceleration time
- E wave:
-
mitral inflow peak velocity
- E/A:
-
early/late diastolic
- EDP:
-
end-diastolic pressure
- EDV:
-
end-diastolic volume
- EDWT:
-
end diastolic wall thickness
- EF:
-
ejection fraction
- ENaC:
-
epithelial sodium channel
- ER:
-
endoplasmic reticulum
- ERS:
-
endoplasmic reticulum stress
- ESP:
-
end-systolic pressure
- ESV:
-
end-systolic volume
- GRP78:
-
glucose-regulated protein 78
- HHF:
-
heart failure hospitalization
- HR:
-
hazard ratio
- I/R:
-
ischemic/reperfusion
- IL-1β:
-
interleukin-1β
- IVRT:
-
isovolumic relaxation time
- LAD:
-
left anterior descending artery
- LV:
-
left ventricular
- MFN2:
-
mitofusin 2
- MI:
-
myocardial infarction
- NALP-3:
-
NACHT, LRR and PYD domains-containing protein 3
- NCX:
-
Na+/Ca2+ exchanger
- NHE:
-
Na+/H+ exchange
- NLRP3:
-
nucleotide-binding oligomerization domain-like receptor 3
- OPA1:
-
optic atrophy 1
- PGC1-α:
-
peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- RAGEs:
-
receptors for advanced glycation end products
- RONS:
-
reactive oxygen and nitrogen species
- ROS:
-
reactive oxygen species
- RRR:
-
relative risk reduction
- SERCA2a:
-
sarcoplasmic endoplasmic reticulum Ca2+-ATPase
- SGK1:
-
serum- and glucocorticoid-regulated kinase 1
- SGLT:
-
sodium–glucose co-transporters
- STAT3:
-
signal transducer and activator of the transcription 3
- T2DM:
-
type 2 diabetes mellitus
- Tau:
-
time constant for isovolumic relaxation
- TRAF2:
-
tumor necrosis factor receptor-associated factor 2
- XBP1:
-
X-box binding protein 1
References
Lotfy M, Adeghate J, Kalasz H, Singh J, Adeghate E. Chronic complications of diabetes mellitus: a mini review. Curr Diab Rev. 2017;13:3–10.
Gaede P, Lund-Andersen H, Parving H-H, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N Engl J Med. 2008;358:580–91.
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Executive summary: heart disease and stroke statistics—2016 update: a report from the american heart association. Circulation. 2016;133:447–54.
Huynh K, Bernardo BC, McMullen JR, Ritchie RH. Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol Ther. 2014;142:375–415.
Schilling JD, Mann DL. Diabetic cardiomyopathy: bench to bedside. Heart Fail Clin. 2012;8:619–31.
González-Vílchez F, Ayuela J, Ares M, Pi J, Castillo L, Martín-Durán R. Oxidative stress and fibrosis in incipient myocardial dysfunction in type 2 diabetic patients. Int J Cardiol. 2005;101:53–8.
Tate M, Grieve DJ, Ritchie RH. Are targeted therapies for diabetic cardiomyopathy on the horizon? Clin Sci Lond Engl. 1979;2017(131):897–915.
Ferrannini E. Sodium–glucose co-transporters and their inhibition: clinical physiology. Cell Metab. 2017;26:27–38.
Vrhovac I, Balen Eror D, Klessen D, Burger C, Breljak D, Kraus O, et al. Localizations of Na(+)-d-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflugers Arch. 2015;467:1881–98.
Verbrugge FH. Role of SGLT2 inhibitors in patients with diabetes mellitus and heart failure. Curr Heart Fail Rep. 2017;14:275–83.
Tomlinson B, Hu M, Zhang Y, Chan P, Liu Z-M. Evaluation of the pharmacokinetics, pharmacodynamics and clinical efficacy of empagliflozin for the treatment of type 2 diabetes. Expert Opin Drug Metab Toxicol. 2017;13:211–23.
Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–28.
Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644–57.
Kosiborod M, Cavender MA, Fu AZ, Wilding JP, Khunti K, Holl RW, et al. Lower risk of heart failure and death in patients initiated on sodium–glucose cotransporter-2 inhibitors versus other glucose-lowering drugs: the CVD-REAL Study (comparative effectiveness of cardiovascular outcomes in new users of sodium–glucose cotransporter-2 inhibitors). Circulation. 2017;136:249–59.
Gautam S, Agiro A, Barron J, Power T, Weisman H, White J. Heart failure hospitalization risk associated with use of two classes of oral antidiabetic medications: an observational, real-world analysis. Cardiovasc Diabetol. 2017;16:93.
von Lewinski D, Kolesnik E, Wallner M, Resl M, Sourij H. New antihyperglycemic drugs and heart failure: synopsis of basic and clinical data. Biomed Res Int. 2017;2017:1253425.
Raz I, Mosenzon O, Bonaca MP, Cahn A, Kato ET, Silverman MG, et al. DECLARE-TIMI 58: participants’ baseline characteristics. Diab Obes Metab. 2018;20:1102–10.
Mordi NA, Mordi IR, Singh JS, Baig F, Choy A-M, McCrimmon RJ, et al. Renal and Cardiovascular Effects of Sodium–Glucose cotransporter 2 (SGLT2) inhibition in combination with loop Diuretics in diabetic patients with Chronic Heart Failure (RECEDE-CHF): protocol for a randomised controlled double-blind cross-over trial. BMJ Open. 2017;7:e018097.
Tanaka A, Node K. Emerging roles of sodium–glucose cotransporter 2 inhibitors in cardiology. J Cardiol. 2017;69:501–7.
Rajasekeran H, Lytvyn Y, Cherney DZI. Sodium–glucose cotransporter 2 inhibition and cardiovascular risk reduction in patients with type 2 diabetes: the emerging role of natriuresis. Kidney Int. 2016;89:524–6.
Abdul-Ghani M, Del Prato S, Chilton R, DeFronzo RA. SGLT2 inhibitors and cardiovascular risk: lessons learned from the EMPA-REG OUTCOME study. Diab Care. 2016;39:717–25.
Heerspink HJL, Perkins BA, Fitchett DH, Husain M, Cherney DZI. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation. 2016;134:752–72.
Marx N, McGuire DK. Sodium–glucose cotransporter-2 inhibition for the reduction of cardiovascular events in high-risk patients with diabetes mellitus. Eur Heart J. 2016;37:3192–200.
Baker WL, Smyth LR, Riche DM, Bourret EM, Chamberlin KW, White WB. Effects of sodium–glucose co-transporter 2 inhibitors on blood pressure: a systematic review and meta-analysis. J Am Soc Hypertens JASH. 2014;8(262–275):e9.
Mazidi M, Rezaie P, Gao H-K, Kengne AP. Effect of sodium–glucose cotransport-2 inhibitors on blood pressure in people with type 2 diabetes mellitus: a systematic review and meta-analysis of 43 randomized control trials with 22 528 patients. J Am Heart Assoc. 2017;6:e004007.
Baker WL, Buckley LF, Kelly MS, Bucheit JD, Parod ED, Brown R, et al. Effects of sodium–glucose cotransporter 2 inhibitors on 24-hour ambulatory blood pressure: a systematic review and meta-analysis. J Am Heart Assoc. 2017;6:e005686.
Lambers Heerspink HJ, de Zeeuw D, Wie L, Leslie B, List J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diab Obes Metab. 2013;15:853–62.
Chilton R, Tikkanen I, Cannon CP, Crowe S, Woerle HJ, Broedl UC, et al. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diab Obes Metab. 2015;17:1180–93.
Cherney DZ, Perkins BA, Soleymanlou N, Har R, Fagan N, Johansen OE, et al. The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc Diabetol. 2014;13:28.
Vallon V, Gerasimova M, Rose MA, Masuda T, Satriano J, Mayoux E, et al. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol. 2014;306:F194–204.
Cherney D, Lund SS, Perkins BA, Groop P-H, Cooper ME, Kaspers S, et al. The effect of sodium glucose cotransporter 2 inhibition with empagliflozin on microalbuminuria and macroalbuminuria in patients with type 2 diabetes. Diabetologia. 2016;59:1860–70.
Cherney DZI, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, et al. Renal hemodynamic effect of sodium–glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. 2014;129:587–97.
Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, Heise T, et al. Metabolic response to sodium–glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest. 2014;124:499–508.
Merovci A, Solis-Herrera C, Daniele G, Eldor R, Fiorentino TV, Tripathy D, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124:509–14.
Bonner C, Kerr-Conte J, Gmyr V, Queniat G, Moerman E, Thévenet J, et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat Med. 2015;21:512–7.
DeFronzo RA, Ferrannini E. Regulation of hepatic glucose metabolism in humans. Diab Metab Rev. 1987;3:415–59.
Matsuda M, Defronzo RA, Glass L, Consoli A, Giordano M, Bressler P, et al. Glucagon dose-response curve for hepatic glucose production and glucose disposal in type 2 diabetic patients and normal individuals. Metabolism. 2002;51:1111–9.
Schade DS, Eaton RP. Glucagon regulation of plasma ketone body concentration in human diabetes. J Clin Invest. 1975;56:1340–4.
Keller U, Schnell H, Sonnenberg GE, Gerber PP, Stauffacher W. Role of glucagon in enhancing ketone body production in ketotic diabetic man. Diabetes. 1983;32:387–91.
Méry PF, Brechler V, Pavoine C, Pecker F, Fischmeister R. Glucagon stimulates the cardiac Ca2+ current by activation of adenylyl cyclase and inhibition of phosphodiesterase. Nature. 1990;345:158–61.
Ali S, Ussher JR, Baggio LL, Kabir MG, Charron MJ, Ilkayeva O, et al. Cardiomyocyte glucagon receptor signaling modulates outcomes in mice with experimental myocardial infarction. Mol Metab. 2015;4:132–43.
Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, et al. Shift to fatty substrate utilization in response to sodium–glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes. 2016;65:1190–5.
Devenny JJ, Godonis HE, Harvey SJ, Rooney S, Cullen MJ, Pelleymounter MA. Weight loss induced by chronic dapagliflozin treatment is attenuated by compensatory hyperphagia in diet-induced obese (DIO) rats. Obes Silver Spring Md. 2012;20:1645–52.
Suzuki M, Takeda M, Kito A, Fukazawa M, Yata T, Yamamoto M, et al. Tofogliflozin, a sodium/glucose cotransporter 2 inhibitor, attenuates body weight gain and fat accumulation in diabetic and obese animal models. Nutr Diab. 2014;4:e125.
Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids. 2004;70:243–51.
Taylor SI, Blau JE, Rother KI. SGLT2 inhibitors may predispose to ketoacidosis. J Clin Endocrinol Metab. 2015;100:2849–52.
Janardhan A, Chen J, Crawford PA. Altered systemic ketone body metabolism in advanced heart failure. Tex Heart Inst J. 2011;38:533–8.
Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, et al. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133:698–705.
Snorek M, Hodyc D, Sedivý V, Durišová J, Skoumalová A, Wilhelm J, et al. Short-term fasting reduces the extent of myocardial infarction and incidence of reperfusion arrhythmias in rats. Physiol Res. 2012;61:567–74.
Srivastava S, Kashiwaya Y, King MT, Baxa U, Tam J, Niu G, et al. Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J Off Publ Fed Am Soc Exp Biol. 2012;26:2351–62.
Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: a “Thrifty Substrate” hypothesis. Diab Care. 2016;39:1108–14.
Neeland IJ, McGuire DK, Chilton R, Crowe S, Lund SS, Woerle HJ, et al. Empagliflozin reduces body weight and indices of adipose distribution in patients with type 2 diabetes mellitus. Diab Vasc Dis Res. 2016;13:119–26.
Bolinder J, Ljunggren Ö, Kullberg J, Johansson L, Wilding J, Langkilde AM, et al. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab. 2012;97:1020–31.
Yokono M, Takasu T, Hayashizaki Y, Mitsuoka K, Kihara R, Muramatsu Y, et al. SGLT2 selective inhibitor ipragliflozin reduces body fat mass by increasing fatty acid oxidation in high-fat diet-induced obese rats. Eur J Pharmacol. 2014;727:66–74.
Zhou L, Cryan EV, D’Andrea MR, Belkowski S, Conway BR, Demarest KT. Human cardiomyocytes express high level of Na+/glucose cotransporter 1 (SGLT1). J Cell Biochem. 2003;90:339–46.
Chen J, Williams S, Ho S, Loraine H, Hagan D, Whaley JM, et al. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diab Ther. 2010;1:57–92.
Kusaka H, Koibuchi N, Hasegawa Y, Ogawa H, Kim-Mitsuyama S. Empagliflozin lessened cardiac injury and reduced visceral adipocyte hypertrophy in prediabetic rats with metabolic syndrome. Cardiovasc Diabetol. 2016;15:157.
Lin B, Koibuchi N, Hasegawa Y, Sueta D, Toyama K, Uekawa K, et al. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc Diabetol. 2014;13:148.
Habibi J, Aroor AR, Sowers JR, Jia G, Hayden MR, Garro M, et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc Diabetol. 2017;16:9.
Hammoudi N, Jeong D, Singh R, Farhat A, Komajda M, Mayoux E, et al. Empagliflozin improves left ventricular diastolic dysfunction in a genetic model of type 2 diabetes. Cardiovasc Drugs Ther. 2017;31:233–46.
Liang L, Jiang J, Frank SJ. Insulin receptor substrate-1-mediated enhancement of growth hormone-induced mitogen-activated protein kinase activation. Endocrinology. 2000;141:3328–36.
Zhou Y, Wu W. The sodium–glucose co-transporter 2 inhibitor, empagliflozin, protects against diabetic cardiomyopathy by inhibition of the endoplasmic reticulum stress pathway. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2017;41:2503–12.
Ye Y, Bajaj M, Yang H-C, Perez-Polo JR, Birnbaum Y. SGLT-2 inhibition with dapagliflozin reduces the activation of the Nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc Drugs Ther. 2017;31:119–32.
Tanajak P, Sa-Nguanmoo P, Sivasinprasasn S, Thummasorn S, Siri-Angkul N, Chattipakorn SC, et al. Cardioprotection of dapagliflozin and vildagliptin in rats with cardiac ischemia-reperfusion injury. J Endocrinol. 2018;236:69–84.
Lee T-M, Chang N-C, Lin S-Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic Biol Med. 2017;104:298–310.
Shi X, Verma S, Yun J, Brand-Arzamendi K, Singh KK, Liu X, et al. Effect of empagliflozin on cardiac biomarkers in a zebrafish model of heart failure: clues to the EMPA-REG OUTCOME trial? Mol Cell Biochem. 2017;433:97–102.
Huang C-C, Monte A, Cook JM, Kabir MS, Peterson KP. Zebrafish heart failure models for the evaluation of chemical probes and drugs. Assay Drug Dev Technol. 2013;11:561–72.
Januzzi JL, Butler J, Jarolim P, Sattar N, Vijapurkar U, Desai M, et al. Effects of canagliflozin on cardiovascular biomarkers in older adults with type 2 diabetes. J Am Coll Cardiol. 2017;70:704–12.
Joubert M, Jagu B, Montaigne D, Marechal X, Tesse A, Ayer A, et al. The sodium–glucose cotransporter 2 inhibitor dapagliflozin prevents cardiomyopathy in a diabetic lipodystrophic mouse model. Diabetes. 2017;66:1030–40.
Sato T, Miki T, Ohnishi H, Yamashita T, Takada A, Yano T, et al. Effect of sodium–glucose co-transporter-2 inhibitors on impaired ventricular repolarization in people with Type 2 diabetes. Diab Med J Br Diab Assoc. 2017;34:1367–71.
Nilsson J, Bengtsson E, Fredrikson GN, Björkbacka H. Inflammation and immunity in diabetic vascular complications. Curr Opin Lipidol. 2008;19:519–24.
De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–9.
Dixit VD. Nlrp3 inflammasome activation in type 2 diabetes: is it clinically relevant? Diabetes. 2013;62:22–4.
Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35.
Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010;11:897–904.
Lee H-M, Kim J-J, Kim HJ, Shong M, Ku BJ, Jo E-K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204.
Fuentes-Antrás J, Ioan AM, Tuñón J, Egido J, Lorenzo O. Activation of toll-like receptors and inflammasome complexes in the diabetic cardiomyopathy-associated inflammation. Int J Endocrinol. 2014;2014:847827.
Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE. 2014;9:e104771.
Singh LP. The NLRP3 inflammasome and diabetic cardiomyopathy: editorial to: “Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model” by Beibei Luo et al. Cardiovasc Drugs Ther. 2014;28:5–6.
Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501.
Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertens Dallas Tex. 1979;2007(49):241–8.
Yamamoto E, Lai Z-F, Yamashita T, Tanaka T, Kataoka K, Tokutomi Y, et al. Enhancement of cardiac oxidative stress by tachycardia and its critical role in cardiac hypertrophy and fibrosis. J Hypertens. 2006;24:2057–69.
Oelze M, Kröller-Schön S, Welschof P, Jansen T, Hausding M, Mikhed Y, et al. The sodium–glucose co-transporter 2 inhibitor empagliflozin improves diabetes-induced vascular dysfunction in the streptozotocin diabetes rat model by interfering with oxidative stress and glucotoxicity. PLoS ONE. 2014;9:e112394.
Wang T, Mao X, Li H, Qiao S, Xu A, Wang J, et al. N-Acetylcysteine and allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via adiponectin and attenuated myocardial postischemic injury in diabetes. Free Radic Biol Med. 2013;63:291–303.
Jadhav A, Tiwari S, Lee P, Ndisang JF. The heme oxygenase system selectively enhances the anti-inflammatory macrophage-M2 phenotype, reduces pericardial adiposity, and ameliorated cardiac injury in diabetic cardiomyopathy in Zucker diabetic fatty rats. J Pharmacol Exp Ther. 2013;345:239–49.
Xia P, Liu Y, Cheng Z. Signaling pathways in cardiac myocyte apoptosis. Biomed Res Int. 2016;2016:9583268.
Takemura G, Kanoh M, Minatoguchi S, Fujiwara H. Cardiomyocyte apoptosis in the failing heart—a critical review from definition and classification of cell death. Int J Cardiol. 2013;167:2373–86.
Takemura G, Fujiwara H. Morphological aspects of apoptosis in heart diseases. J Cell Mol Med. 2006;10:56–75.
Liu Z-W, Zhu H-T, Chen K-L, Dong X, Wei J, Qiu C, et al. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol. 2013;12:158.
Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5.
Ji Y, Zhao Z, Cai T, Yang P, Cheng M. Liraglutide alleviates diabetic cardiomyopathy by blocking CHOP-triggered apoptosis via the inhibition of the IRE-α pathway. Mol Med Rep. 2014;9:1254–8.
Wu T, Dong Z, Geng J, Sun Y, Liu G, Kang W, et al. Valsartan protects against ER stress-induced myocardial apoptosis via CHOP/Puma signaling pathway in streptozotocin-induced diabetic rats. Eur J Pharm Sci Off J Eur Fed Pharm Sci. 2011;42:496–502.
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91.
Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18:444–52.
Tong Q, Wu L, Jiang T, Ou Z, Zhang Y, Zhu D. Inhibition of endoplasmic reticulum stress-activated IRE1α-TRAF2-caspase-12 apoptotic pathway is involved in the neuroprotective effects of telmisartan in the rotenone rat model of Parkinson’s disease. Eur J Pharmacol. 2016;776:106–15.
Liu H, Baliga R. Endoplasmic reticulum stress-associated caspase 12 mediates cisplatin-induced LLC-PK1 cell apoptosis. J Am Soc Nephrol JASN. 2005;16:1985–92.
Sari FR, Widyantoro B, Thandavarayan RA, Harima M, Lakshmanan AP, Zhang S, et al. Attenuation of CHOP-mediated myocardial apoptosis in pressure-overloaded dominant negative p38α mitogen-activated protein kinase mice. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2011;27:487–96.
Cong X-Q, Piao M-H, Li Y, Xie L, Liu Y. Bis(maltolato)oxovanadium(IV) (BMOV) attenuates apoptosis in high glucose-treated cardiac cells and diabetic rat hearts by regulating the unfolded protein responses (UPRs). Biol Trace Elem Res. 2016;173:390–8.
Hernández-Reséndiz S, Buelna-Chontal M, Correa F, Zazueta C. Targeting mitochondria for cardiac protection. Curr Drug Targets. 2013;14:586–600.
Galloway CA, Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid Redox Signal. 2015;22:1545–62.
Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. 2014;130:554–64.
Lucas E, Vila-Bedmar R, Arcones AC, Cruces-Sande M, Cachofeiro V, Mayor F, et al. Obesity-induced cardiac lipid accumulation in adult mice is modulated by G protein-coupled receptor kinase 2 levels. Cardiovasc Diabetol. 2016;15:155.
Duncan JG. Peroxisome proliferator activated receptor-alpha (PPARα) and PPAR gamma coactivator-1alpha (PGC-1α) regulation of cardiac metabolism in diabetes. Pediatr Cardiol. 2011;32:323–8.
Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–84.
Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5.
Ong S-B, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–22.
Sharp WW, Beiser DG, Fang YH, Han M, Piao L, Varughese J, et al. Inhibition of the mitochondrial fission protein dynamin-related protein 1 improves survival in a murine cardiac arrest model. Crit Care Med. 2015;43:e38–47.
Bartos DC, Grandi E, Ripplinger CM. Ion channels in the heart. Compr Physiol. 2015;5:1423–64.
Priest BT, McDermott JS. Cardiac ion channels. Channels. 2015;9:352–9.
Baartscheer A, Schumacher CA, van Borren MMGJ, Belterman CNW, Coronel R, Fiolet JWT. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57:1015–24.
Pogwizd SM, Sipido KR, Verdonck F, Bers DM. Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc Res. 2003;57:887–96.
Lambert R, Srodulski S, Peng X, Margulies KB, Despa F, Despa S. Intracellular Na+ concentration ([Na+]i) is elevated in diabetic hearts due to enhanced Na+-glucose cotransport. J Am Heart Assoc. 2015;4:e002183.
Liu T, Takimoto E, Dimaano VL, DeMazumder D, Kettlewell S, Smith G, et al. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ Res. 2014;115:44–54.
Baartscheer A, Hardziyenka M, Schumacher CA, Belterman CNW, van Borren MMGJ, Verkerk AO, et al. Chronic inhibition of the Na+/H+—exchanger causes regression of hypertrophy, heart failure, and ionic and electrophysiological remodelling. Br J Pharmacol. 2008;154:1266–75.
Baartscheer A, Schumacher CA, van Borren MMGJ, Belterman CNW, Coronel R, Opthof T, et al. Chronic inhibition of Na+/H+—exchanger attenuates cardiac hypertrophy and prevents cellular remodeling in heart failure. Cardiovasc Res. 2005;65:83–92.
Baartscheer A, Schumacher CA, Wüst RCI, Fiolet JWT, Stienen GJM, Coronel R, et al. Empagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits. Diabetologia. 2017;60:568–73.
Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–88.
Kho C, Lee A, Hajjar RJ. Altered sarcoplasmic reticulum calcium cycling–targets for heart failure therapy. Nat Rev Cardiol. 2012;9:717–33.
Kawase Y, Hajjar RJ. The cardiac sarcoplasmic/endoplasmic reticulum calcium ATPase: a potent target for cardiovascular diseases. Nat Clin Pract Cardiovasc Med. 2008;5:554–65.
Park SH, Khemais-Benkhiat S, Idris-Khodja N, Amoura L, Abbas M, Auger C, et al. Upregulation of sodium–glucose cotransporter 2 (SGLT2) expression in cultured senescent endothelial cells and in arterial sites at risk in vivo in rats. Arch Cardiovasc Dis Suppl. 2018;10:224.
Han Y, Cho Y-E, Ayon R, Guo R, Youssef KD, Pan M, et al. SGLT inhibitors attenuate NO-dependent vascular relaxation in the pulmonary artery but not in the coronary artery. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1027–36.
Leng W, Ouyang X, Lei X, Wu M, Chen L, Wu Q, et al. The SGLT-2 inhibitor dapagliflozin has a therapeutic effect on atherosclerosis in diabetic ApoE−/− mice. Mediators Inflamm. 2016;2016:6305735.
Authors’ contributions
All authors wrote, revised and approved the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was supported by the Thailand Research Fund Grant RTA6080003 (SCC), the NSTDA Research Chair Grant from the National Science and Technology Development Agency Thailand (NC), and the Chiang Mai University Center of Excellence Award (NC).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lahnwong, C., Chattipakorn, S.C. & Chattipakorn, N. Potential mechanisms responsible for cardioprotective effects of sodium–glucose co-transporter 2 inhibitors. Cardiovasc Diabetol 17, 101 (2018). https://doi.org/10.1186/s12933-018-0745-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-018-0745-5