
Abstract
Background
Endothelial dysfunction plays a critical role in the development of type 2 diabetes (T2DM). T-cadherin (T-cad) has gained recognition as a regulator of endothelial cell (EC) function. The present study examined whether T-cad deficiency increases vascular vulnerability in T2DM.
Methods
Vascular segments were isolated from WT or T-cad knockout mice. Endothelial function, total NO accumulation, and the expression of T-cad related proteins were determined.
Results
Ach and acidified NaNO2 induced similar vasorelaxation in WT groups. T-cad KO mice exhibited normal response to acidified NaNO2, but manifested markedly reduced response to Ach. NO accumulation was also decreased in T-cad KO group. T-cad expression was reduced in WT mice fed 8 weeks of high fat diet (HFD). Furthermore, exacerbated reduction of vasorelaxation was observed in T-cad KO mice fed 8 weeks of HFD.
Conclusions
In the current study, we provide the first in vivo evidence that T-cadherin deficiency causes endothelial dysfunction in T2DM vascular segments, suggesting the involvement of T-cad deficiency in T2DM pathogenesis.
Similar content being viewed by others

Background
Type 2 diabetes mellitus (T2DM) affects approximately 100 million people worldwide [1]. Diabetic vascular complications are responsible for the majority of morbidity and mortality in the diabetic population. These complications can be arbitrarily divided into micro- and macrovascular complications. Macrovascular complications are associated with accelerated atherosclerosis, resulting in premature ischemic heart disease, increased risk of cerebrovascular disease, and severe peripheral vascular disease [2].
Endothelial dysfunction is characterized by deficiency of nitric oxide (NO) production in response to normal secretion signals. A critical component of atherosclerosis development, endothelial dysfunction is a characteristic abnormality observed in diabetes [3–5]. The bioavailability of NO, produced by endothelial NO synthase (eNOS), represents a key marker of vascular health. Down-regulated NO may contribute to atherogenic predisposition. Many metabolic derangement factors cause endothelial dysfunction by affecting the balance of NO production during diabetes development [6–8]. Therefore, clarification of the mechanisms responsible for endothelial dysfunction in diabetes, and identification of the therapeutic interventions that may improve endothelial function hold great potential in reducing cardiovascular complications and overall death in diabetic patients.
T-cadherin (T-cad) is a unique member of the cadherin family. Found on the cellular surface, it possesses no intracellular domain. T-cadherin content is maximal in the aorta, carotid, iliac, and renal arteries, and in the heart. Known to regulate neuronal growth during embryogenesis, T-cad has additionally gained recognition as a regulator of endothelial cell (EC) function [9–11].
In vivo expression of T-cad is increased in human atherosclerotic lesions and experimental restenosis. In vitro expression of T-cad is upregulated in proliferating endothelial and smooth muscle cells, as well as endothelial cells during oxidative and endoplasmic reticulum stress [11, 12]. Clinical evidence underlines the association between T-cad and hypoadiponectinemia, with increased risk of various metabolic diseases [13, 14]. Together, these observations strongly suggest the involvement of T-cad in diabetic vasculopathic state development. However, direct evidence that supported T-cadherin deficiency exists in diabetic endothelial dysfunction pathogenesis is currently lacking.
Therefore, the aims of the present study were: (1) to determine whether T-cad deficiency is involved in the development of T2DM, (2) to determine whether T-cad deficiency may cause endothelial dysfunction in descending aortic vascular segments (the most frequent location of atherosclerosis development), (3) to determine whether endothelial dysfunction is exacerbated by T-cad deficiency in descending aortic vascular segments in T2DM, and (4) to elucidate the responsible underlying mechanisms.
Methods
Determination of vasorelaxation in aortic tissue
Adult male T-cad knockout mice (T-cad KO) or their wild type littermates (WT) were used in all study experiments. All experiments were performed in adherence to the NIH Guidelines on the Use of Laboratory Animals, and approved by the Thomas Jefferson University Committee on Animal Care. After animals were anesthetized with 3% isoflurane, descending aortas were removed and placed into cold Krebs buffer solution [NaCl 118, KCl 4.75, CaCl2·2H2O 2.54, KH2PO4 1.19, MgSO4·7H2O 1.19, NaHCO3 25, and glucose 10.0 (mM)] Aorta were carefully cut into 2–3 segments (each 2–3 mm length); surrounding fat and tissue were debrided. Segments were suspended upon stainless steel hooks, and aerated (95% O2 and 5% CO2) in 37 °C 5 ml K–H tissue baths. Aortic rings were connected to FORT-10 force transducers (WPI, Sarasota, FL) for MacLab data acquisition. Segments were first stretched to generate 2.5 mN force, followed by 0.5 mN increments every 15 min until achieving 4 mN total force. As force was increased to 8mN, the Krebs buffer solution was replaced with a buffer with increased potassium content (HK solution) for vessel ring sustainability. Once 8 mN force was achieved, the tissue bath was replaced with normal Krebs buffer. During the period of force decrease from 8 to 4 mN, epinephrine (a potent vasoconstrictor) was added to the tissue bath in 2 concentrations (10−5 and 10−4 M) in sequence within 2 min of each other, to induce vasoconstriction. After ~5 min, the vasorelaxant acetylcholine (concentrations of 10−6, 10−5, 10−4, 10−3 and 10−2 M) were added to the tissue bath in immediate succession. The percent relaxation from the peak of the epinephrine-induced contraction to the nadir point of acetylcholine-induced relaxation was recorded on MacLab.
Determination of NO accumulation from aortic segments
To determine total NO accumulation, isolated aortic segments were placed in culture medium and incubated in a cell culture incubator (5% CO2, 37 °C). After 8 h of incubation, segments were subjected to homogenization. Protein concentrations were determined via BCA method (Pierce Chemical). Medium was transferred into the wells of 96-well plate, and incubated with nitrate reductase and cofactors for 20 min at 37 °C to reduce NO3 to NO2. Samples (50 μl) were then injected into a water-jacketed, oxygen-free purge vessel containing 5 ml of 20 mM potassium iodide in glacial acetic acid to reduce NO2 to NO. Resultant chemiluminescence from the reaction of NO and ozone was detected by a nitric oxide analyzer (SIEVERS NOA 280I; Sievers, Boulder, CO). Detector signals were collected and analyzed by a PC-based data recording and processing system. To determine the NO content of the culture medium, calculations of the slope of the regression analysis were performed. The amount of NOx released was expressed in nmol/mg protein.
Determination of NOx production from aortic segments
To detect endothelial NOx production, isolated aortic segments were placed in 6-well plates with 500 μl culture medium. Epinephrine (10−4 M) was added to each well. 3 min later, acetylcholine concentrations (10−6, 10−5, 10−4, 10−3 and 10−2 M) were added in immediate succession. Resultant medium nitrite (NO2 −) and nitrate (NO3 −) levels were determined by chemiluminescence NO detector (Siever 280i NO Analyzer).
Quantitation of tissue nitrotyrosine content
Nitrotyrosine content, the accepted footprint of in vivo peroxynitrite (ONOO−) formation, was determined by ELISA described in our previous publication [15]. In brief, aortic segments were homogenized in lysis buffer and centrifuged for 10 min at 12,000g at 4 °C. Supernatants were collected and protein concentrations were determined. Tissue samples from aortic segments, were applied to disposable sterile ELISA plates, and incubated overnight with primary antibody against nitrotyrosine (05–233, Millipore, USA). After extensive wash and incubation with the peroxidase-conjugated secondary antibody, the peroxidase reaction product was generated using TMB solution. The optical density was measured at 450 nm with a SpectraMax-Plus microplate spectrophotometer. The amount of nitrotyrosine content in tissue samples was calculated using standard curves generated from nitrated BSA containing known amounts of nitrotyrosine, and expressed as pmol/mg protein.
Immunoblotting
Aortic tissue homogenate proteins were separated on SDS-PAGE gels, transferred to PVDF membranes, and Western blotted with monoclonal antibody against eNOS, Ser1177 phosphorylated eNOS (Becton–Dickinson, USA), and polyclonal antibody against Akt and Ser473 phosphorylated Akt (Cell Signaling Technology, USA). PVDF membranes were incubated with horseradish peroxide-conjugated anti-rabbit or anti-mouse IgG antibodies (Cell Signaling Technology, USA) for 2 h. The blot was developed using Super-Signal Reagent (Pierce) and visualized with a Kodak Image Station 4000R. Blot densities were analyzed (Gelpro32 software).
Real-time PCR
Total RNA was extracted from aortic segments via RNeasy Mini Kit (QIAGEN, USA). Expression analysis of the reported genes was performed by real-time PCR via commercial kit (4367659, AB) and the ABI 7500 Sequence Detection System, using SYBR GREEN as a double-stranded DNA-specific dye. GAPDH served as endogenous control. Table 1 lists the primers for mRNA expression analysis by real-time PCR.
Statistical analysis
All values in the text, table, and figures are presented as mean ± SEM of n independent experiments. Data (except Western blot density) were subjected to t test (two groups) or ANOVA (three or more groups) followed by Bonferoni correction for post hoc t test. Western blot densities were analyzed by the Kruskal–Wallis test followed by Dunn’s post test. Probabilities of 0.05 or less were considered to be statistically significant.
Results
T-cad mRNA and protein expression decreased in WT mice fed 8 weeks HFD
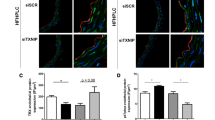
We first determined T-cad expression during T2DM condition. In an established diet-induced T2DM model, we fed WT mice high fat diet for 8 weeks, and determined both the mRNA and protein levels of T-cadherin [16]. Both mRNA and protein levels of T-cadherin were reduced compared to control, suggesting T-cadherin deficiency occurs during type 2 diabetes (Fig. 1).

T-cad mRNA and protein expression were reduced in WT mice fed 8 weeks HFD. T-cad expression as measured by Western blot and real time PCR a representative Western blots; b Western blot density analysis (n = 4); c Real time PCR analysis (n = 3).**P < 0.01 vs. WT
T-cad deficiency induced endothelial dysfunction
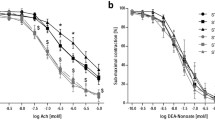
We next performed a vascular ring experiment to directly address this relationship. In WT aortic rings, administration of Ach (an endothelial-dependent NO donor) resulted in concentration-dependent vasorelaxation, with no significant difference compared to the effects elicited by acidified NaNO2 (an endothelial-independent NO donor). At maximal concentration (100 μM), Ach and acidified NaNO2 induced 79.7 and 85.9% vasorelaxation respectively, demonstrating preserved vascular ring endothelial function post-preparation protocols. However, aortic rings from T-cad KO mice exhibited markedly reduced vasorelaxation by Ach (endothelial-dependent stimulation) treatment compared to that induced by acidified NaNO2 (endothelial-independent NO donor) treatment. Furthermore, addition of 100 μM Ach resulted in only 54.5% relaxation (P < 0.01 vs. WT) in T-cad KO aortic ring; 100 μM acidified NaNO2 induced 82.3% relaxation (P > 0.05 vs. WT) (Fig. 2). Together, these results demonstrate T-cad deficiency resulted in severe endothelial dysfunction in the tested aortic segments.

T-cad deficiency induced endothelial dysfunction. Concentration-dependent vasorelaxation of aortic vascular rings in response to a Ach, an endothelium-dependent vasodilator, and b acidified NaNO2, an endothelium-independent vasodilator. Top representative figures; Bottom analysis result. Each mouse aorta generated 2–3 rings, n = 8–12 rings/group. WT wild type litternates, T-cad KO T-cadherin knockout mice. *P < 0.05 vs. WT
T-cad deficiency-induced endothelial dysfunction exacerbated in T2DM
The above experiments verified T-cad deficiency occurs in T2DM, and T-cad deficiency was associated with vascular segment endothelial dysfunction. Next, we determined the state of endothelial function in T-cad KO animals under in-viro or in vivo diabetic environment. We first utilized an in vitro model of diabetes, employing high glucose (HG, 25.5 mmol/l), high fat (HF, palmitate 300 μmol/l) treatment. After incubating aortic rings from WT and T-cad KO mice in HGHF (2.5 h), vasorelaxation was impaired in both animal types. However, the reduction in concentration-dependent vasorelaxation was markedly increased in T-cad KO mice compared to WT. To better mimic in vivo T2DM, we next fed WT mice and T-cad KO mice 8 weeks of HFD diet as previously described [17]. Again, ring vasorelaxation was reduced in both WT and T-cad KO mice. Vasorelaxation reduction was more pronounced in T-cad KO mice compared to WT mice (Fig. 3). Collectively, these results suggest that endothelial dysfunction caused by T-cad deficiency was exacerbated in the T2DM condition.

T-cad deficiency induced-endothelial dysfunction was exacerbated in T2DM. Concentration-dependent vasorelaxation of aortic vascular rings subjected to a high glucose/high fat treatment (high glucose: 25.5 m mol/l; high fat/palmitate: 300 μ mol/l) and b high fat diet alone for 8 weeks. Top representative figures; Bottom analysis result. Each mouse aorta generated 2–3 rings, n = 6–7 rings/group. WT Wild type litternates, T-cad KO T-cadherin knockout mice. **P < 0.01 vs. WT
Total NO accumulation and NOX production significantly decreased in vascular segments from T-cad KO animals
Next, total NO accumulation and NOx production were determined. Aortic segments were isolated from WT and T-cad KO animals, and incubated with cell culture medium. Culture media was collected after 8 h. Total NO accumulation in aortic segments from T-cad KO mice was significantly decreased compared to WT (Fig. 4). To measure direct NO release after Ach stimulation, total NOx production was determined. As expected, total NOx production was markedly decreased in T-cad KO mice compared to WT. Together, these results demonstrate that T-cad deficiency was associated with reduced NO production, which may be responsible in part for the observed state of endothelial dysfunction.

Basal NO production was significantly decreased in vascular segments from T-cad deficient animals. a NO concentration in culture medium after 8 h incubation of aortic segments from WT or T-cadherin knockout mice. b Concentration of nitric oxide in medium after Ach stimulation. Each mouse aorta generated 2 aortic segments. n = 8 segments/group. *P < 0.05 vs. WT
Phosphorylation of Akt, but not eNOS, was significantly reduced in T-cad KO aortic tissue
To study the underlying mechanisms, we performed Western blot. Akt phosphorylation was significantly increased in T-cad KO mice compared to WT (Fig. 5), consistent with known current research. However, eNOS phosphorylation in T-cad KO mice was slightly increased (not to significant degree) compared to WT. These results suggest that reduction of NO bioavailability caused by T-cad deficiency may induce phosphorylation of the Akt-eNOS pathways in compensatory manner, without significant effect.

Phosphorylation of Akt, but not eNOS, was significantly reduced in aortic tissue from T-cad KO animals. Expression of Akt, p-Akt, eNOS and p-eNOS in aortic tissues obtained from WT or T-cadherin knockout mice. Left representative Western blots. Right summary of density analysis of p-Akt/Akt (a) and p-eNOS/eNOS (b), n = 3 mice/group, *P < 0.05 vs. WT
Akt inactivation increases caspase-3 activity in HUVECs
To further investigate the contributive mechanisms of reduced NO bioactivity, we cultured and subjected human umbilical vein endothelial cells (HUVECs) to Akt inhibitor (A6730, Sigma, (10 μM) treatment for 24 h. Akt phosphorylation and caspase-3 activity were determined. Akt phosphorylation was significantly decreased, whereas caspase-3 activity was elevated in HUVECs subjected to Akt inhibitor treatment compared to control (Fig. 6). These results strongly suggest Akt inhibition-induced endothelial cell apoptosis contributes to reduced NO bioactivity in T-cad KO aortic tissue.

Akt inactivation increased caspase-3 activity in HUVECs. a Representative Western blots of p-Akt and Akt expression in HUVECs with or without Akt inhibitor; b Caspase-3 activity, using Ac-DEVD-pNA as substrate, n = 3, *P < 0.05 vs. WT
Increased nitric oxide inactivation exacerbated endothelial dysfunction in T-cad KO mice
We next determined whether the endothelial dysfunction observed in T-cad deficient animals is related to increased superoxide production and nitric oxide inactivation. Tissue nitrotyrosine level was determined, the accepted in vivo footprint of peroxynitrite (ONOO−), the product of NO and superoxide. Superoxide production was markedly increased in T-cad KO animals, and nitrotyrosine was significantly increased in T-cad KO mice compared to WT (Fig. 7). These results support augmented NO inactivation as a primary cause of endothelial dysfunction in T-cad deficient animals.

Increased nitric oxide inactivation exacerbated endothelial dysfunction in T-cad KO mice. a Aortic nitrotyrosine content determined by ELISA; b quantitative assay of superoxide production determined by lucigenin-enhanced chemiluminescence, n = 3–4 mice/group, *P < 0.05 vs. WT, **P < 0.01 vs. WT
Discussion
Adiponectin (APN) is a multifunctional adipocytokine of adipose tissue origin. APN exists in three isoforms (trimer, hexamer, and multimer). Plasma APN concentration (which consists of mostly the latter two high molecular weight isoforms) highly correlates with the development of T2DM. Numerous studies have shown that plasma adiponectin level is decreased in individuals with T2DM, indicating APN plays an important role in pathogenesis of T2DM [18–20]. T-cad has been identified as an important receptor of APN, particularly for the high molecular weight isoform [21–24]. Both plasma APN concentration and tissue APN expression are closely related with T-cad [14]. Additionally, T-cad itself is implicated with the pathogenesis of T2DM, but with uncertain role [25, 26]. In the present study, we have provided the first in vivo evidence that T-cad expression is decreased in the aorta from T2DM animals (Fig. 1). This data is consistent with Matsuda’s study demonstrating T-cad expression is positively correlated with plasma APN concentration, which is markedly decreased in T2DM patients [14].
Atherosclerosis is the basis for the vascular pathologies of T2DM. Endothelial dysfunction plays a key role in this process, and the ongoing underlying mechanisms [27–29]. Many studies have demonstrated T-cad plays a central multifunctional role in the vascular system [30–34]. Heretofore, no direct in vivo functional evidence exists demonstrating the role T-cad plays in endothelial dysfunction. We next performed a vascular ring experiment to study the relationship between T-cad and endothelial dysfunction. Our results demonstrated significantly reduced vasorelaxation in T-cad KO mice compared to WT, consistent with numerous in vitro studies (Fig. 2) [35, 36], suggesting T-cad deficiency may cause severe endothelial dysfunction. To investigate whether T2DM exacerbates endothelial dysfunction already caused by T-cad deficiency, we fed mice high-fat diet for 8 weeks to mimic T2DM. Our resultant data provide the first in vivo functional evidence T-cad deficiency exacerbates severe endothelial dysfunction in the T2DM condition (Fig. 3).
NO, produced by eNOS, is a critical vasodilator in the vascular system. NO bioavailability is a surrogate marker of endothelial cell function. To obtain further evidence supporting endothelial dysfunction occurs in T-cad KO vascular segments, we next performed total NO accumulation and NOx production. Our results confirmed that NO production and NOx production were both reduced in T-cad deficient animals (Fig. 4), suggesting T-cad deficiency was associated with reduced NO production, which may be responsible in part for the observed state of endothelial dysfunction.
Interestingly, phosphorylation of Akt was significantly decreased in T-cad KO animals, while phosphorylation of eNOS was grossly unchanged compared to WT (Fig. 5). At the beginning, we thought this reduction in Akt phosphorylation may induce the reduction in eNOS phosphorylation, which is a classical factor to mediate NO production [37]. But our eNOS results, which are consistent with the data of others, did not support this hypothesis [9]. The PI3 k/Akt pathway is highly associated with apoptosis [38]. The reduction in Akt phosphorylation may be contributive to apoptosis induction in EC, which partially explains why NO and NOx production were reduced in EC in vascular segments in T-cad KO mice. Therefore, we utilized Akt inhibitor to attenuate Akt phosphorylation and found caspase-3 activity was significantly increased in HUVECs subjected to Akt inhibitor (Fig. 6). Collectively, our results, which is consistent with the data of others [9, 35], suggested Akt inhibition induced apoptosis, but not reduced eNOS phosphorylation, contributes to decreased NOx production in ECs. Further studies directly addressing this phenomenon are expected.
Experimental and clinical data have demonstrated augmented superoxide production and the resultant superoxide/nitric oxide bi-radical reaction is the primary cause for NO inactivation under pathological conditions. Newly produced NO is oxidized to NO2 − and NO3 − rapidly under physiological conditions, reducing NO bio-availability and its vasorelaxative potential, termed NO inactivation. In the presence of superoxide, NO forms peroxynitrite (ONOO−), a toxic molecule that modifies tyrosine residues in proteins to create nitrotyrosine, leaving a footprint detectable in vivo [39]. In our present study, we have demonstrated both increased superoxide and nitrotyrosine content in T-cad KO animals (Fig. 7).
Conclusions
We have provided the first in vivo evidence that T-cad deficiency causes endothelial dysfunction in T2DM model vascular segments, supporting T-cad deficiency involvement in the pathogenesis of T2DM complications. Supplanting T-cad deficiency may be a potential therapeutic avenue in the prevention and amelioration of vascular injury in the diabetic population.
Abbreviations
- Ac-DEVD-pNA:
-
N-Acetyl-Asp-Glu-Val-Asp-p-Nitroanilide
- Ach:
-
acetylcholine
- APN:
-
adiponectin
- BCA:
-
bicinchoninic acid
- EC:
-
endothelial cell
- eNOS:
-
endothelial NO synthase
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- HF:
-
high fat
- HG:
-
high glucose
- HFD:
-
high fat diet
- NO:
-
nitric oxide
- NO2 − :
-
nitrite
- NO3 − :
-
nitrate
- ONOO− :
-
peroxynitrite
- T2DM:
-
type 2 diabetes mellitus
- T-cad:
-
T-cadherin
- T-cad KO:
-
T-cadherin knockout
- WT:
-
wild type
Reference
Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus—present and future perspectives. Nat Rev Endocrinol. 2011;8(4):228–36.
Creager MA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Circulation. 2003;108(12):1527–32.
Pieper GM, Meier DA, Hager SR. Endothelial dysfunction in a model of hyperglycemia and hyperinsulinemia. Am J Physiol. 1995;269(3 Pt 2):H845–50.
Bohlen HG, Lash JM. Topical hyperglycemia rapidly suppresses EDRF-mediated vasodilation of normal rat arterioles. Am J Physiol. 1993;265(1 Pt 2):H219–25.
Tesfamariam B, Brown ML, Deykin D, Cohen RA. Elevated glucose promotes generation of endothelium-derived vasoconstrictor prostanoids in rabbit aorta. J Clin Invest. 1990;85(3):929–32.
Arnal JF, Dinh-Xuan AT, Pueyo M, Darblade B, Rami J. Endothelium-derived nitric oxide and vascular physiology and pathology. Cell Mol Life Sci. 1999;55(8–9):1078–87.
Cosentino F, Hishikawa K, Katusic ZS, Luscher TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation. 1997;96(1):25–8.
McVeigh GE, Brennan GM, Johnston GD, McDermott BJ, McGrath LT, Henry WR, Andrews JW, Hayes JR. Impaired endothelium-dependent and independent vasodilation in patients with type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1992;35(8):771–6.
Philippova M, Joshi MB, Pfaff D, Kyriakakis E, Maslova K, Erne P, Resink TJ. T-cadherin attenuates insulin-dependent signalling, eNOS activation, and angiogenesis in vascular endothelial cells. Cardiovasc Res. 2012;93(3):498–507.
Philippova M, Ivanov D, Joshi MB, Kyriakakis E, Rupp K, Afonyushkin T, Bochkov V, Erne P, Resink TJ. Identification of proteins associating with glycosylphosphatidylinositol-anchored T-cadherin on the surface of vascular endothelial cells: role for Grp78/BiP in T-Cadherin-dependent cell survival. Mol Cell Biol. 2008;28(12):4004–17.
Ivanov D, Philippova M, Antropova J, Gubaeva F, Iljinskaya O, Tararak E, Bochkov V, Erne P, Resink T, Tkachuk V. Expression of cell adhesion molecule T-cadherin in the human vasculature. Histochem Cell Biol. 2001;115(3):231–42.
Kuzmenko YS, Kern F, Bochkov VN, Tkachuk VA, Resink TJ. Density- and proliferation status-dependent expression of T-cadherin, a novel lipoprotein-binding glycoprotein: a function in negative regulation of smooth muscle cell growth? FEBS Lett. 1998;434(1–2):183–7.
Pfaff D, Schoenenberger AW, Dasen B, Erne P, Resink TJ, Philippova M. Plasma T-cadherin negatively associates with coronary lesion severity and acute coronary syndrome. Eur Heart J Acute Cardiovasc Care. 2015;4(5):410–8.
Matsuda K, Fujishima Y, Maeda N, Mori T, Hirata A, Sekimoto R, Tsushima Y, Masuda S, Yamaoka M, Inoue K, et al. Positive feedback regulation between adiponectin and T-cadherin impacts adiponectin levels in tissue and plasma of male mice. Endocrinology. 2015;156(3):934–46.
Cao Y, Tao L, Yuan Y, Jiao X, Lau WB, Wang Y, Christopher T, Lopez B, Chan L, Goldstein B, et al. Endothelial dysfunction in adiponectin deficiency and its mechanisms involved. J Mol Cell Cardiol. 2009;46(3):413–9.
Zheng Q, Yuan Y, Yi W, Lau WB, Wang Y, Wang X, Sun Y, Lopez BL, Christopher TA, Peterson JM, et al. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler Thromb Vasc Biol. 2011;31(11):2616–23.
Yan W, Zhang H, Liu P, Wang H, Liu J, Gao C, Liu Y, Lian K, Yang L, Sun L, et al. Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1alpha signaling contributing to increased vulnerability in diabetic heart. Basic Res Cardiol. 2013;108(3):329.
Sheng T, Yang K. Adiponectin and its association with insulin resistance and type 2 diabetes. J Genet Genom. 2008;35(6):321–6.
Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116(7):1784–92.
Fukuda S, Hirata A, Nishizawa H, Nagao H, Kashine S, Kimura T, Inoue K, Fujishima Y, Yamaoka M, Kozawa J, et al. Systemic arteriosclerosis and eating behavior in Japanese type 2 diabetic patients with visceral fat accumulation. Cardiovasc Diabetol. 2015;14(1):8.
Hartge MM, Kintscher U, Unger T. Endothelial dysfunction and its role in diabetic vascular disease. Endocrinol Metab Clin North Am. 2006;35(3):551–60.
Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci USA. 2004;101(28):10308–13.
Kostopoulos CG, Spiroglou SG, Varakis JN, Apostolakis E, Papadaki HH. Adiponectin/T-cadherin and apelin/APJ expression in human arteries and periadventitial fat: implication of local adipokine signaling in atherosclerosis? Cardiovasc Pathol. 2014;23(3):131–8.
Parker-Duffen JL, Nakamura K, Silver M, Kikuchi R, Tigges U, Yoshida S, Denzel MS, Ranscht B, Walsh K. T-cadherin is essential for adiponectin-mediated revascularization. J Biol Chem. 2013;288(34):24886–97.
Nicolas A, Aubert R, Bellili-Munoz N, Balkau B, Bonnet F, Tichet J, Velho G, Marre M, Roussel R, Fumeron F. T-cadherin gene variants are associated with type 2 diabetes and the fatty liver index in the French population. Diabetes Metab. 2016;1262–3636(16):30408–16.
Tyrberg B, Miles P, Azizian KT, Denzel MS, Nieves ML, Monosov EZ, Levine F, Ranscht B. T-cadherin (Cdh13) in association with pancreatic beta-cell granules contributes to second phase insulin secretion. Islets. 2011;3(6):327–37.
Rask-Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab. 2013;17(1):20–33.
Capellini VK, Celotto AC, Baldo CF, Olivon VC, Viaro F, Rodrigues AJ, Evora PR. Diabetes and vascular disease: basic concepts of nitric oxide physiology, endothelial dysfunction, oxidative stress and therapeutic possibilities. Curr Vasc Pharmacol. 2010;8(4):526–44.
Shimabukuro M, Higa N, Masuzaki H, Sata M, Ueda S. Impact of individual metabolic risk components or its clustering on endothelial and smooth muscle cell function in men. Cardiovasc Diabetol. 2016;15(1):77.
Semina EV, Rubina KA, Sysoeva VY, Rutkevich PN, Kashirina NM, Tkachuk VA. Novel mechanism regulating endothelial permeability via T-cadherin-dependent VE-cadherin phosphorylation and clathrin-mediated endocytosis. Mol Cell Biochem. 2014;387(1–2):39–53.
Andreeva AV, Han J, Kutuzov MA, Profirovic J, Tkachuk VA, Voyno-Yasenetskaya TA. T-cadherin modulates endothelial barrier function. J Cell Physiol. 2009;223(1):94–102.
Semina EV, Rubina KA, Rutkevich PN, Voyno-Yasenetskaya TA, Parfyonova YV, Tkachuk VA. T-cadherin activates Rac1 and Cdc42 and changes endothelial permeability. Biochemistry (Mosc). 2009;74(4):362–70.
Ivanov D, Philippova M, Allenspach R, Erne P, Resink T. T-cadherin upregulation correlates with cell-cycle progression and promotes proliferation of vascular cells. Cardiovasc Res. 2004;64(1):132–43.
Andreeva AV, Kutuzov MA, Tkachuk VA, Voyno-Yasenetskaya TA. T-cadherin is located in the nucleus and centrosomes in endothelial cells. Am J Physiol Cell Physiol. 2009;297(5):C1168–77.
Joshi MB, Ivanov D, Philippova M, Erne P, Resink TJ. Integrin-linked kinase is an essential mediator for T-cadherin-dependent signaling via Akt and GSK3 in endothelial cells. FASEB J. 2007;21(12):3083–95.
Joshi MB. T-cadherin protects endothelial cells from oxidative stress-induced apoptosis. FASEB J. 2005;19(12):1737–9.
Vanhoutte PM, Shimokawa H, Tang EHC, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol. 2009;196(2):193–222.
Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813(11):1978–86.
Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271(5 Pt 1):C1424–37.
Authors’ contributions
HW and AA contributed equally to this work. WJY performed the real-time PCR. RS provided the gene knockout mouse. WBL and YJW were major contributors in writing the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The analyzed datasets of the current study are available from the corresponding author upon request. All results from experiments, regardless of n, have been statistically verified by appropriate statistical measures as indicated in methods.
Ethics approval and consent to participate
Adult male T-cad knockout mice (T-cad KO) or their wild type littermates (WT) were used in all study experiments. All experiments were performed in adherence to the NIH Guidelines on the Use of Laboratory Animals, and approved by the Thomas Jefferson University Committee on Animal Care.
Funding
This work was supported by the Program for National Science Fund for Distinguished Young Scholars of China (Grant No. 81225001), the National Key Basic Research Program of China (973 Program, Grant No. 2013CB531204), the Key Science and Technology Innovation Team in Shaanxi Province (Grant No. 2014KCT-19), the Program for Changjiang Scholars and Innovative Research Team in University (Grant No.PCSIRT-14R08), the National Science Funds of China (Grants Nos. 81170186, 81470478, and 81400201), the Major Science and Technology Project of China “Significant New Drug Development” (Grant No.2012ZX09J12108-06B), and the Fourth Military Medical University’s Young Talent Project (First Level).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Han Wang and Anastasia Ambrosio contributed equally to this work
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, H., Tao, L., Ambrosio, A. et al. T-cadherin deficiency increases vascular vulnerability in T2DM through impaired NO bioactivity. Cardiovasc Diabetol 16, 12 (2017). https://doi.org/10.1186/s12933-016-0488-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-016-0488-0