
Abstract
Cardiovascular disease (CVD) is one of the most common diabetes-associated complications, as well as a leading cause for death in type 2 diabetes patients (T2D). Despite the well-known correlation between the two, up until the 2008 FDA industry guidance for licensing of new anti-hyperglycemic drugs, which required an investigation of cardiovascular outcomes (CVO) of glucose-lowering agents, only a few studies had looked into the relationship between glucose lowering drugs and cardiovascular (CV) risk. Thereafter, CVOT design has focused on non-inferiority short-term studies on high-risk patient populations aiming at capturing CV safety issues. Despite the wealth of information and useful data provided by CVOTs, this approach still suffers from certain limitations. The present review will condense the main results of the most recently completed CVOTs, reflect on the lessons learned, discuss on the issues presented by current CVOT design and offer some suggestions for improvement.
Similar content being viewed by others

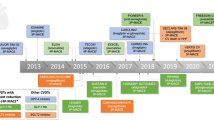
Background
Among diabetes-related complications, cardiovascular disease (CVD) stands as the leading cause for mortality and adverse outcomes in patients with type 2 diabetes (T2D). More than 60 % die from CVD while an even greater proportion suffer serious CV-associated complications [1]. T2D implies a two to fourfold increase in the risk of coronary heart disease and a decreased life expectancy (6–7 years less) in comparison with people without diabetes [2]. Despite this clear correlation between diabetes and negative CV outcomes, it is still not clear whether glycemic control per se would have any effect on reducing CVD risk in T2D [3–6]. Moreover, CV safety of glucose-lowering drugs was not thoroughly investigated until the 2008 US Food and Drug Administration (FDA) [7] and subsequent European Medicines Agency (EMA) requirement [8] that all new therapies for diabetes undergo a rigorous assessment of CV safety through large-scale cardiovascular outcome trials (CVOT).
Before the publication of the FDA and EMA regulations, several trials assessing CV risks of glucose-lowering interventions had already been performed, if only with concerns in respect to design since they were aimed towards an improvement of glycemic control and outcome analysis [6]. For instance, in 1970 the first multicenter, head to head trial (University Group Diabetes Program) of T2D glucose-lowering treatments assessing CV outcomes was interrupted, as all oral drugs (tolbutamide, phenformin) seemed to increase CV risk in comparison to placebo or insulin [9–11]. However, this trial was grossly underpowered and therefore results often contested. Later, the 1977 UKPDS trial randomized patients to either standard or intensive diabetes care with either insulin, sulphonylurea or metformin. After 10 years, there was a significant reduction of MI risk and all-cause mortality in the intensive therapy group with any of the three drugs. However, the reduction of CV-associated risk was greater with metformin (39 % MI, 36 % all-cause) than with insulin or sulphonylurea (15 % MI, 13 % all-cause) [12]. A later meta-analysis of randomized trials using metformin found highly diverse results in terms of mortality risk increase/reduction as well as possible CV deleterious effects of a metformin/sulphonylurea combination [13], which were found to be greatly diminished 10 years after the end of the study and no longer statistically significant [14].
Other trials have found no differences in CV risk between glucose-lowering treatment interventions, as was the case for the HEART2D [15] trial, which compared basal and prandial insulin treatment strategies or the BARI 2D [16] trial, that compared insulin-sensitizing and insulin-providing treatment strategies in patients with T2D and CVD. However, the HEART2D trial was clearly underpowered, and a post hoc analysis seems to suggest a positive effect of controlling postprandial hyperglycemia in some subgroups of subjects, like older patients [17, 18]. The more recent ORIGIN trial, [19] which randomized patients with prediabetes and T2D patients with CVD risk factors to either insulin glargine or standard glucose control did also not find any differences to the primary CV outcome between treatment groups.
Several compounds have been suggested to increase CV risk in diabetes. For instance, several inter-related meta-analyses infer that rosiglitazone might raise MI and heart failure (HF) risk [20, 21]. Despite the RECORD trial [22, 23] only showed an excess HF risk without any conclusive results on MI, a meta-analysis including RECORD data still concludes that the high risk/benefit ratio of rosiglitazone does not support its use for diabetes treatment [21, 24]. The PROactive trial [25] on the CV safety of the addition to usual care of pioglitazone versus placebo found a slight trend toward a combined primary CV end-point—CVD and interventions in all vascular beds reduction—(10 % reduction, p = 0.095) and a significant 16 % reduction in the secondary end-point (MI, stroke, all-cause mortality). However, increased HF rates and a number of severe associated adverse events have hindered its use in daily practice [26, 27].
The requirements for CVOTs described in the aforementioned 2008 FDA guideline include, among others [28]:
-
For outcome clinical trials, in order to exclude unacceptable CV risk, a two-sided 95 % CI upper boundary of 1.8 risk ratio (pre-approval) and/or 1.3 risk ratio (post-approval) for major adverse events (MACE) versus control group is required.
-
To satisfy the new statistical requirements, CV event analysis might include a meta-analysis of all placebo-controlled, add-on (drug vs. placebo, plus standard therapy) and active-controlled trials, and/or an additional single, large, safety CVOT can be conducted. This, alone or in addition to other trials, needs to satisfy the upper bound mentioned above before approval.
-
Patient selection should focus on high-risk populations, including those with advanced disease, elderly and those with renal impairment.
-
Trials must include at least 2 years of CV safety data.
-
A prospective independent adjudication of CV events in phase 2 and 3 studies must also be performed. These CV events include CV mortality, myocardial infarction (MI) and stroke, and possibly hospitalization for ACS, urgent revascularization and other end-points.
Figure 1 includes a representation of possible scenarios for approval of new glucose lowering drugs depending on the hazard ratios (HR) for CV risk. An upper bound of the two-sided 95 percent confidence interval for the estimated increased risk above the non-inferiority boundary of 1.3 as well as underpowered studies prevents FDA approval. Surely, the need for full compliance with FDA/EMA requirements on CV safety for approval of new glucose lowering drugs has implied a significant increase of CVOTs in the last decade [28].

Confidence interval (CI) bars indicated by FDA guideline. Shown are five examples of hazard ratios (HR) and the upper limit of the 95 % CI of a development plan and regulatory consequences of each outcome. S superiority, NI non-inferiority, I inferiority, UP underpowered
Results from early trials evaluating CV outcomes under glucose-lowering therapies could not ascertain a clear relationship between HbA1c target levels, hypoglycemia incidence and CV risk, despite a tendency for intense glucose control being beneficial in the long-term [6, 29–39]. Therefore, to avoid confounding results derived from glycemic values and the drugs themselves, CVOTs started after the 2008 FDA/EMA regulation have focused on maintaining glycemic equipoise, generally in the context of standard diabetes care [40].
In the present review, we will summarize the latest results of CVOTs on glucose-lowering agents started after the 2008 FDA Guideline as well as present an outline of ongoing CVOTs. Furthermore, we will review their influence on present glucose lowering therapy decision-making as well as comment on CVOT design limitations and potential venues for improvement.
Summary of results of recently completed CVOTs
Since the FDA and EMA guidance request for CV safety for new antihyperglycemic drugs, over 15 medium/long-term CVOT have been initiated (see Table 1). From those, results for seven are already available while the remaining will be due by 2020 latest. In comparison to clinical trials on anti-hyperglycemic drugs performed prior to 2008, patient numbers have considerably increased (more than five times on average). So has the average number of countries per trial (1.6 times average), helping produce wider range data on other ethnic groups as well as in practice variation [6, 41, 42], while follow-up time remains on an average of 2.5 years [43–50].
Hereon we summarize the findings of all CVOTs started after the 2008 FDA guideline published to date, namely the SAVOR-TIMI, TECOS, ELIXA, EXAMINE, EMPA-REG OUTCOME, LEADER and SUSTAIN-6 trials [44–52]. Despite the focus on high-risk patients (a requirement for CVOT design), which poses a problem for extrapolation of results to the general patient population, the criteria for patient selection varied from trial to trial. For instance, age requirements of EXAMINE and EMPA-REG OUTCOME included all patients over 18 years old, while in other trials minimum age ranged between 30 and 50 years old. Cardiovascular risk also differed in each trial. While for most a preexisting CVD or CVD risk factors were necessary for enrolment, in the EXAMINE and ELIXA trials only patients already recovering from an acute coronary syndrome (ACS) were included in the study. For a detailed view on patient selection criteria, see Table 2. Moreover, an important aspect of CVOTs is that the evaluation of CV safety of the new glucose lowering drugs takes place in the background of diabetes and CVD standard care. This poses an important difference with respect to early trials like the UKPDS, performed before modern blood pressure reducing drugs; statins and an active attitude to coronary revascularization were part of routine care. Therefore, in Table 3 we have summarized the baseline concomitant medication of patients enrolled in trials started after 2008.
For clarity, results for each outcome will be split into distinct sections, starting by the primary composite end-point and then proceeding to each of the possible CV outcomes evaluated by these trials: MI, unstable angina (UA), CV death and HF. Finally, we will review a few other relevant safety end-points, namely: pancreatitis, hypoglycemia occurrence, and renal events/microvascular effects
-
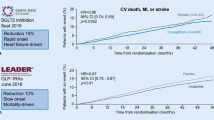
Primary MACE composite end-point Diverse individual elements are included in the primary composite end-point for each CVOT, as shown in Table 1. However, CV death, myocardial infarction and stroke are all common elements to primary composite CVOT end-points. In addition, the TECOS and ELIXA trials included hospitalization for UA in the primary MACE. Corresponding data in Table 4 shows that for saxagliptin (SAVOR-TIMI), sitagliptin (TECOS), lixisenatide (ELIXA) and alogliptin (EXAMINE) treatment, occurrence of the primary composite end-point did not differ from placebo groups, thus confirming non-inferiority of the new treatments in CV safety under the particular conditions of each of the trials. In the EMPA-REG OUTCOME trial, however, the primary outcome occurred in 10.5 % in the pooled empagliflozin group and in 12.1 % of the placebo group (empagliflozin group (HR 0.86; 95 % CI 0.74–0.99; p = 0.04 for superiority), demonstrating therefore not only non-inferiority versus placebo but superiority [49]. A similar result was observed in LEADER, where the primary outcome occurred in significantly fewer patients in the liraglutide group than in the control group (13 vs. 14.9 %; HR 0.87; 95 % CI 0.78–0.97; p = 0.01 for superiority), but only for patients with established CVD (subgroup analysis) [51]. It is important to note, however, that both in LEADER and EMPA-REG OUTCOME, the lesser occurrence in the primary composite end-point was largely driven by a reduction in cardiovascular mortality. Results from the recently published SUSTAIN-6 trial have also shown superiority for semaglutide versus placebo in the primary composite outcome (6.6 vs. 8.9 % of patients, respectively; HR: 0.74, 95 % CI 0.58–0.95; p < 0.001), however, in contrast to EMPA-REG OUTCOME and LEADER, results were not driven by a decrease of risk of cardiovascular death, but of non-fatal stroke occurrence (in 1.6 and 2.7 %, respectively (HR 0.61; 95 % CI 0.38–0.99; p = 0.04) [52].
Table 4 Comparison of outcome results from terminated CVOTs in comparison to placebo -
Cardiovascular death In all terminated trials, treatment with the new agent did not increase CV death compared to placebo treatment. In addition, in the EMPA-REG OUTCOME and LEADER trials, the treatment group showed a reduced incidence of CV death in comparison to placebo [49, 51, 53, 54].
-
Fatal/non-fatal myocardial infarction An important CV outcome to measure given the increased MI risk implied by diabetes [55], therefore its inclusion in all primary composite MACE end-points. Data from the six trials published to date has shown that all glucose-lowering treatments tested are non-inferior to placebo when it comes to MI. For a more detailed comparison of hazard rates, see Table 4.
-
Stroke In general, the third basic element of primary composite MACE end-points. So far, considering the published results of the aforementioned six trials, none of the new glucose-lowering drugs tested increases stroke occurrence in comparison to placebo. However, in EMPA-REG OUTCOME a trend towards an increased stroke incidence was reported [49]. Conversely to EMPA-REG OUTCOME, in SUSTAIN-6, a significant reduction of stroke rates was reported for patients under semaglutide in comparison to the placebo group [52]. For more data on hazard rates, see Table 4.
-
Hospitalization for UA The importance of this end-point varied among trials. While TECOS and ELIXA included UA in their primary end-points; and SAVOR-TIMI, EMPA-REG OUTCOME and EXAMINE included it as part of the secondary composite end-point, LEADER and SUSTAIN-6 included it as part of an extended primary composite end-point. As it happened with MI or stroke risk, UA rates did not increase under any of the treatments investigated when compared to placebo. Extended information is available on Table 4.
-
Hospitalization for HF As shown in Table 4, rates of hospitalization for HF did not differ between placebo and treatment groups in the EXAMINE, TECOS, ELIXA or SUSTAIN-6 trials, and LEADER showed a non-significant decrease of hospitalization for HF in patients treated with liraglutide [51]. Yet, treatment with saxagliptin (SAVOR-TIMI) was found to increase hospitalization rates for HF (3.5 vs. 2.8 %; HR 1.27; 95 % CI 1.07–1.51; p = 0.007). This effect was independent of age, as confirmed by a later analysis on efficacy and safety in older patients [43]. Conversely, in EMPA-REG OUTCOME treatment with empagliflozin reduced the number of patients hospitalized for HF (2.8 vs. 4.5 %; HR 0.61; 95 % CI 0.47–0.79; p < 0.001) and improved other HF outcomes like the composite endpoint of CV death or hospitalization for HF (5.7 vs. 8.5 %; HR 0.66; 95 % CI 0.55–0.79; p < 0.001) [45, 53].
-
Serious hypoglycemic events As part of the serious adverse event report, the rate of serious hypoglycemic events suffered by patients under treatment with the new glucose lowering drugs was investigated. Even though rates were similar to placebo in all CVOTs, and major hyperglycemia events did not differ between saxagliptin (SAVOR-TIMI) treatment and placebo, hypoglycemia occurrence generally increased with saxagliptin in combination with sulphonylureas or insulin. This effect was consistent across all age ranges analyzed [43]. On the contrary, treatment with liraglutide reduced severe hypoglycemic events in comparison to placebo (rate ratio: 0.69; 95 % CI 0.51–0.93; p = 0.02), which might be due to a reduced need for insulin co-therapy [51]. In SUSTAIN-6, the rates of severe hypoglycemia did not significantly differ between the two semaglutide-dose treatment groups and placebo [semaglutide 0.5 mg and 1.0 mg 191 (23.1 %) and 178 (21.7 %), respectively], placebo 0.5 and 1.0 mg [177 (21.5 %) and 173 (21.0 %)] [52].
-
Pancreatic effects Regarding the possible association between incretin-based therapies and adverse pancreatic effects, CVOTs evaluated whether these new antihyperglycemic agents increased the risk for pancreatitis. Acute pancreatitis occurred slightly more often in the treatment groups than with placebo when employing saxagliptin (SAVOR-TIMI), sitagliptin (TECOS), alogliptin (EXAMINE) or lixisenatide (ELIXA), and even when no significant differences between groups could be found, a meta-analysis on trials on DPP-4 inhibitors showed a marginally higher risk of pancreatitis associated with DPP-4 treatment [56]. In LEADER and SUSTAIN-6, incidence of pancreatitis was lower, even if not statistically significant, in the intervention group than in the placebo group [51, 52].
-
Renal events and/or microvascular effects Definitions for renal events were different for each trial. While in the ELIXA and TECOS trials there is no specification of the type of renal events [44], in EXAMINE only initiation of dialysis is reported [47]. A broader renal end-point including doubling of creatinine level, initiation of dialysis, renal transplantation or creatinine >6.0 mg/dl was used in the SAVOR-TIMI trial [43]. Regardless of end-point definition, none of these trials found differences between treatment and placebo with respect to renal function. Moreover, a further examination of the EMPA-REG OUTCOME trial regarding renal outcomes, found that addition of empagliflozin to standard treatment was associated with a slower progression of kidney disease (empagliflozin HR 0.61; CI 95 % 0.53–0.70; p < 0.001) and lower rates of clinically relevant renal events than placebo [57]. In the LEADER trial, a composite renal and retinal microvascular outcome was investigated. The renal outcome involved the new onset of macroalbuminuria or the doubling of the serum creatinine level and an eGFR ≤45 ml/min/1.73 m2, the need for continuous renal-replacement therapy or death from renal disease. The incidence of the composite microvascular outcome was lower with liraglutide, mainly due to a significantly lower rate of nephropathy events (HR 0.78; 95 % CI 0.67–0.92; p = 0.003) [51]. In SUSTAIN-6, the investigated renal outcome was defined as new or worsening of nephropathy and consisted on persistent macroalbuminuria, persistent doubling of the serum creatinine level and eGFR ≤45 ml/min/1.73 m2, or the need for continuous renal-replacement therapy. Based on that definition, semaglutide treated patients had a significantly lower risk than placebo treated patients (3.8 vs. 6.1 %, respectively; HR 0.64; 95 % CI 0.46–0.88; p = 0.005). Conversely, and somehow unexpectedly, retinopathy-derived complications (blindness, vitreous hemorrhage, or conditions requiring treatment with an intravitreal agent or photocoagulation) were significantly more often reported in the treatment group as in the placebo (3.0 vs. 1.8 %, respectively; HR 1.76; 95 % CI 1.11–2.78; p = 0.02) [52].
In general, the previous analysis shows that new glucose lowering drugs comply with FDA/EMA requirements for CV safety regardless of class. Moreover, some of them like empagliflozin, liraglutide or semaglutide even demonstrated beneficial effects over CV death risk, stroke and/or HF risk [49, 51–53].
Discussion
CVOT trials completed after 2008 showed that new glucose lowering agents like the DPP-4 inhibitors saxagliptin, alogliptin, and sitagliptin and the GLP-1 receptor agonist lixisenatide are safe with respect to CV outcomes in high CV risk patient populations with long T2D duration (for more details on patient selection, see Table 2) under standard care for both CVD and diabetes. In addition, the LEADER study has shown that liraglutide, a GLP-1 receptor agonist, is not only safe but that is also capable of reducing CV risk and the incidence of cardiovascular-related death [51]. Furthermore, recently published results from SUSTAIN-6 have proven another GLP-1 receptor agonist, semaglutide, superior to placebo in reducing the risk of a cardiovascular composite primary end-point, driven by a significant reduction of stroke risk [52]. Moreover, treatment with the SGLT-2 inhibitor empagliflozin was not only non-inferior to placebo but also significantly reduced CV risk -as shown by the composite primary and secondary outcomes- and a composite outcome of HF hospitalization and CV death [53, 54].
Regardless of the CV safety of all anti-hyperglycemic agents tested, one trial on DPP-4 inhibitors, SAVOR-TIMI, found a significantly higher risk for HF in the treatment group and another, EXAMINE a trend towards such outcome. In contrast, there were no such concerns in the TECOS trial. Differences to baseline patient characteristics, as well as to trial design make it difficult to compare results from these trials. Moreover, the molecular structure differs among DPP-4 inhibitors and so does their safety profile. As a result, the FDA recently issued a safety warning on saxagliptin and alogliptin increasing the risk of heart failure, particularly in patients who already have heart or kidney disease [58]. Despite recent meta-analyses of randomized clinical trials including results of SAVOR-TIMI and EXAMINE suggested an increased risk of hospitalization due to HF in T2D patients [59–62], others have found no difference in hospitalization rates for HF between treatment with saxagliptin compared with sitagliptin or with DPP-4 inhibitors compared with other classes of anti-diabetes agents [63, 64].
The analyses of results of the aforementioned CVOTs have been very useful for treatment decision-making and patient safety in diabetes [65]. Not only were these trials capable of proving CV safety, but three of them, EMPA-REG OUTCOME, LEADER and SUSTAIN-6 showed cardiovascular benefits even when they were primarily designed for non-inferiority. However, it is important to note that these results are so far only valid for the particular patient groups enrolled in the studies, and that it is not clear how translatable they are to the general patient population. Furthermore, a comparison among results from CVOT is overall difficult, among other reasons because the definition of CVD risk and/or CVD is different for each trial, and with it the degree of severity of prior disease of enrolled patients highly variable. Other reasons limiting comparability among CVOTs, especially in terms of event rates, apart from the aforementioned differences in baseline patient characteristics, are the variable trial duration and the diverse definitions of the primary end-point. In addition, another obstacle for compared evaluation of trials evaluating cardiovascular outcomes before and after FDA 2008 regulation is that the routine care background from those trials is somehow dissimilar. In general, despite the great advance for the clinical practice meant by new CVOTs, there is still room for improvement [66, 67]. Trial design could still benefit from the introduction of new strategies to improve the applicability of trial results to daily clinical practice, as was agreed by the members of the first CVOT Summit of the Diabetes and CVD (D&CVD) EASD Study Group [68].
Among the recommendations stand the necessary consensus on primary end-point definition, which should be a 3-point MACE comprising cardiovascular death, non-fatal MI and non-fatal stroke. Another important point is that these cardiovascular outcomes differ greatly in their pathophysiology: while MI has a thrombotic origin [69], CV death results mostly from arrhythmia [70] and stroke can either be a product of thrombotic origin or hemorrhagic [71, 72]. These differences should be taken into account when designing and analyzing composite MACE end-points, because a positive/neutral effect in one of the components does not necessarily mean an improvement in the others, especially when considering their particular pathophysiology, as exemplified by the results of the various components of the primary composite end-point in EMPA-REG OUTCOME [49, 53]. Moreover, and especially regarding the disparate results on HF risk in DPP-4 inhibitor trials, HF risk should be investigated more closely by CVOTs [68, 73].
A major issue of CVOT design to date is patient selection criteria. Disease duration is a potential confounding factor that is not sufficiently controlled [74]. On the other hand, extrapolating CV results from this patient population to a broader one can be challenging, especially in case of superiority to placebo. To solve this matter, potential solutions could be: increasing patient retention/adherence to treatment over longer follow-up periods, promote large-scale patient enrolment by involving patient advocacy groups and modifying trial design to new approaches that minimize patient numbers and provide closer to real-world data in the standard health care system [6, 68].
Another limitation of the present CVOTs is that trial duration is too short to evaluate real-life, long-term outcomes [74], plus incurring in an unjustified high cost per patient given the limited results they provide. The extreme cost of CVOTs make them only accessible to industry and hinders an independent CV risk review [6]. To increase the amount of available data by enabling extensive follow-up and reduce trial related costs, an alternative would be to make use of comprehensive electronic health record databases with extended functionality [75, 76].
Maintaining glycemic equipoise, by addition of the test agent to standard care, has resulted in general in modest HbA1c reductions, which combined with the short follow-up time of most studies, makes it hard to positively ascertain CV benefit of these glucose-lowering drugs [40]. On the other hand, maintaining glycemic equipoise but aiming to longer follow-up times might still result in CV improvement by incidental effects from these drugs other than glycemic control, as was the case in the EMPA-REG OUTCOME trial.
Most CVOTs started after the 2008 FDA/EMA guideline analyze drugs of the SGLT-2 inhibitor, DPP-4-inhibitor, or GLP-1 receptor agonist class. Even when the ORIGIN trial already focused on the evaluation of insulin gargline versus standard care [19], since the FDA mandate only one CVOT study is investigating CV risks of insulin treatment, the ongoing DEVOTE trial on insulin glargine versus insulin degludec. To date there is not a single CVO trial on metformin or sulphonylurea alone. Considering that metformin is a first line treatment for T2D [77] and that sulphonylurea and insulin are also very common therapeutic tools in diabetes [78], more CVOTs on these drugs are essential.
Furthermore, present CVOTs are usually simple, placebo controlled, non-inferiority trials and generally lacking of head to head comparisons. Exceptionally, the ongoing CAROLINA trial includes a head to head comparison of safety issues of linagliptin, a DPP-4 inhibitor, versus a sulphonylurea (glimepiride) [79]. In the future, trial design should be aimed at matching results of several different treatment versus a reduced placebo group and ideally, under usual care. This strategy will not only allow for a direct treatment comparison but also enable a better assessment of treatment heterogeneity and possible drug interactions in the real population under standard of care [6, 68]. This strategy was followed by a recently terminated cohort study comparing head to head CV safety of GLP-1 receptor agonists to DPP-4 inhibitors, sulfonylureas, or insulin in addition to metformin, in a similar fashion to real-world conditions [80].
Despite including analysis of adverse outcomes other than CV risks, in the future a more thorough examination of microvascular complications, renal, kidney and pancreatic effects as well as cancer occurrence should be an integral component of a CVOT design [68]. Already a number of trials have been designed with this concept in mind. For instance, an ongoing clinical trial (NCT02380521) examines the effect of exenatide once weekly, a GLP-1 receptor agonist, on several CV risk markers like subclinical atherosclerosis, endothelial dysfunction, oxidative stress and atherogenic lipoproteins, which are also indicative of potential microvascular complications [81]. The ongoing CARMELINA trial (NCT01897532) also aims to characterize renal microvascular outcomes of linagliptin (DPP-4 inhibitor) on T2D patients at high CV risk. Moreover, another ongoing trial (CANVAS-R), focuses on the renal outcomes of canagliflozin (a SGLT-2 inhibitor) treatment on T2D patients at risk for CVD [82].
Conclusion
Since the 2008 FDA/EMA regulations demanded an investigation of CV outcomes for newly developed glucose-lowering agents, a number of CVOTs have been completed and their results published. These trials, in general, have shown that glucose-lowering drugs do not increase CV risks over placebo levels, and even that some drugs, as empagliflozin, semaglutide or liraglutide, can actually lead to cardiovascular protection. However, despite satisfying the requirements of regulatory agencies when it comes to demonstrating not incrementing CV risk beyond a certain safety level, current CVOTs suffer still from certain design flaws that hinder their potential. Head to head comparisons, broader patient population groups, long-term analysis and an expansion of safety end-points, etc. would serve to improve CVOT design and expand its applicability spectrum.
Abbreviations
- CVOT:
-
cardiovascular outcome trial
- CV:
-
cardiovascular
- CVD:
-
cardiovascular disease
- T2D:
-
type 2 diabetes
- MI:
-
myocardial infarction
- CI:
-
confidence interval
- MACE:
-
major adverse cardiovascular event
- UA:
-
unstable angina
- HR:
-
hazard ratio
- EMA:
-
European Medicine Agency
- FDA:
-
Federal Drug Agency
- GLP-1:
-
glucagon-like-peptide 1
- SGLT-2:
-
sodium–glucose cotransporter-2
- DPP-4:
-
dipeptidyl-peptidase-4
References
Gregg EW, et al. Mortality trends in men and women with diabetes, 1971 to 2000. Ann Intern Med. 2007;147(3):149–55.
Seshasai SR, et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med. 2011;364(9):829–41.
Low Wang CC, et al. Clinical update: cardiovascular disease in diabetes mellitus: atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus—mechanisms, management, and clinical considerations. Circulation. 2016;133(24):2459–502.
Ferrannini E, DeFronzo RA. Impact of glucose-lowering drugs on cardiovascular disease in type 2 diabetes. Eur Heart J. 2015;36(34):2288–96.
Scheen AJ, Charbonnel B. Effects of glucose-lowering agents on vascular outcomes in type 2 diabetes: a critical reappraisal. Diabetes Metab. 2014;40(3):176–85.
Holman RR, Sourij H, Califf RM. Cardiovascular outcome trials of glucose-lowering drugs or strategies in type 2 diabetes. Lancet. 2014;383(9933):2008–17.
FDA. Guidance for industry diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. Washington, DC: US Department of Health and Human Services; 2008.
EMA. Guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500129256.pdf. Accessed 10 Jan 2016.
Meinert CL, et al. A study of the effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. II. Mortality results. Diabetes. 1970;19:789–830.
Goldner MG, Knatterud GL, Prout TE. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. 3. Clinical implications of UGDP results. JAMA. 1971;218(9):1400–10.
Knatterud GL, et al. Effects of hypoglycemic agents on vascular complications in patients with adult-onset diabetes. IV. A preliminary report on phenoformin results. JAMA. 1971;217(6):777–84.
UPDSU Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet. 1998;352(9131):837–53.
Boussageon R, et al. Reappraisal of metformin efficacy in the treatment of type 2 diabetes: a meta-analysis of randomised controlled trials. PLoS Med. 2012;9(4):e1001204.
Holman RR. Post trial monitoring results of the UKPDS sulfonylurea plus metformin substudy. Munich: EASD; 2013.
Raz I, et al. Effects of prandial versus fasting glycemia on cardiovascular outcomes in type 2 diabetes: the HEART2D trial. Diabetes Care. 2009;32(3):381–6.
Frye RL, et al. A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med. 2009;360(24):2503–15.
Ceriello A. Postprandial hyperglycemia and cardiovascular disease: is the HEART2D study the answer? Diabetes Care. 2009;32(3):521–2.
Raz I, et al. Post hoc subgroup analysis of the HEART2D trial demonstrates lower cardiovascular risk in older patients targeting postprandial versus fasting/premeal glycemia. Diabetes Care. 2011;34(7):1511–3.
Gerstein HC, et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med. 2012;367(4):319–28.
Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356(24):2457–71.
Nissen SE, Wolski K. Rosiglitazone revisited: an updated meta-analysis of risk for myocardial infarction and cardiovascular mortality. Arch Intern Med. 2010;170(14):1191–201.
Home PD, et al. Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet. 2009;373(9681):2125–35.
Mahaffey KW, et al. Results of a reevaluation of cardiovascular outcomes in the RECORD trial. Am Heart J. 2013;166(2):240–9.
Bourg CA, Phillips BB. Rosiglitazone, myocardial ischemic risk, and recent regulatory actions. Ann Pharmacother. 2012;46(2):282–9.
Dormandy JA, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (prospective pioglitazone clinical trial in macrovascular events): a randomised controlled trial. Lancet. 2005;366(9493):1279–89.
Marks DH. Drug utilization, safety and clinical use of Actos and Avandia. Int J Risk Saf Med. 2013;25(1):39–51.
Scheen AJ. Outcomes and lessons from the PROactive study. Diabetes Res Clin Pract. 2012;98(2):175–86.
Hirshberg B, Raz I. Impact of the US Food and Drug Administration cardiovascular assessment requirements on the development of novel antidiabetes drugs. Diabetes Care. 2011;34(Suppl 2):S101–6.
Dluhy RG, McMahon GT. Intensive glycemic control in the ACCORD and ADVANCE trials. N Engl J Med. 2008;358(24):2630–3.
Gaede P. Intensive glucose control and cardiovascular disease in type 2 diabetes—should we change the recommended target for glycated hemoglobin? Commentary to ACCORD and ADVANCE trials. Pol Arch Med Wewn. 2008;118(11):619–21.
Patel A, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358(24):2560–72.
Heller SR. A summary of the ADVANCE trial. Diabetes Care. 2009;32(Suppl 2):S357–61.
Turnbull FM, et al. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia. 2009;52(11):2288–98.
Brown A, Reynolds LR, Bruemmer D. Intensive glycemic control and cardiovascular disease: an update. Nat Rev Cardiol. 2010;7(7):369–75.
Riddle MC. Effects of intensive glucose lowering in the management of patients with type 2 diabetes mellitus in the action to control cardiovascular risk in diabetes (ACCORD) trial. Circulation. 2010;122(8):844–6.
Macisaac RJ, Jerums G. Intensive glucose control and cardiovascular outcomes in type 2 diabetes. Heart Lung Circ. 2011;20(10):647–54.
Koska J, et al. The effect of intensive glucose lowering on lipoprotein particle profiles and inflammatory markers in the veterans affairs diabetes trial (VADT). Diabetes Care. 2013;36:2408–14.
Pistrosch F, Hanefeld M. Hypoglycemia and cardiovascular disease: lessons from outcome studies. Curr Diab Rep. 2015;15(12):117.
Wang P, et al. HbA1c below 7 % as the goal of glucose control fails to maximize the cardiovascular benefits: a meta-analysis. Cardiovasc Diabetol. 2015;14:124.
Hirshberg B, Katz A. Insights from cardiovascular outcome trials with novel antidiabetes agents: what have we learned? An industry perspective. Curr Diab Rep. 2015;15(11):87.
Bethel MA, Sourij H. Impact of FDA guidance for developing diabetes drugs on trial design: from policy to practice. Curr Cardiol Rep. 2012;14(1):59–69.
Bethel MA, Sourij H. Positive impact of revised FDA guidance on clinical trial design in diabetes. Diabetes. 2012;61(Suppl1):A264.
Leiter LA, et al. Efficacy and safety of saxagliptin in older participants in the SAVOR-TIMI 53 trial. Diabetes Care. 2015;38(6):1145–53.
Pfeffer MA, et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373(23):2247–57.
Scirica BM, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369(14):1317–26.
Udell JA, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes and moderate or severe renal impairment: observations from the SAVOR-TIMI 53 trial. Diabetes Care. 2015;38(4):696–705.
White WB, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369(14):1327–35.
Zannad F, et al. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet. 2015;385(9982):2067–76.
Zinman B, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373(22):2117–28.
Green JB, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373(3):232–42.
Marso SP, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311–22.
Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016. doi:10.1056/NEJMoa1607141.
Fitchett D, et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: results of the EMPA-REG OUTCOME(R) trial. Eur Heart J. 2016;37(19):1526–34.
Zinman B, et al. Rationale, design, and baseline characteristics of a randomized, placebo-controlled cardiovascular outcome trial of empagliflozin (EMPA-REG OUTCOME). Cardiovasc Diabetol. 2014;13:102.
Stevens RJ, et al. Risk factors for myocardial infarction case fatality and stroke case fatality in type 2 diabetes: UKPDS 66. Diabetes Care. 2004;27(1):201–7.
Alves C, Batel-Marques F, Macedo AF. A meta-analysis of serious adverse events reported with exenatide and liraglutide: acute pancreatitis and cancer. Diabetes Res Clin Pract. 2012;98(2):271–84.
Wanner C, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med. 2016;375:323–34.
FDA. Diabetes medications containing saxagliptin and alogliptin: drug safety communication—risk of heart failure. 2016. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm494252.htm. Accessed 13 Apr 2016.
Monami M, Dicembrini I, Mannucci E. Dipeptidyl peptidase-4 inhibitors and heart failure: a meta-analysis of randomized clinical trials. Nutr Metab Cardiovasc Dis. 2014;24(7):689–97.
Udell JA, et al. Glucose-lowering drugs or strategies and cardiovascular outcomes in patients with or at risk for type 2 diabetes: a meta-analysis of randomised controlled trials. Lancet Diabetes Endocrinol. 2015;3(5):356–66.
Wu S, et al. Dipeptidyl peptidase-4 inhibitors and cardiovascular outcomes: meta-analysis of randomized clinical trials with 55,141 participants. Cardiovasc Ther. 2014;32(4):147–58.
Li L, et al. Dipeptidyl peptidase-4 inhibitors and risk of heart failure in type 2 diabetes: systematic review and meta-analysis of randomised and observational studies. BMJ. 2016;352:i610.
Fu AZ, Johnston S, Sheehan J, Ghannam A, Tsai K, Cappell K. Risk of hospitalization for heart failure with dipeptidyl peptidase-4 inhibitors vs sulfonylureas and with saxagliptin vs sitagliptin in a US claims database. Diabetes. 2015;64(Suppl 1A):42.
Giorda CB, et al. Hospitalisation for heart failure and mortality associated with dipeptidyl peptidase 4 (DPP-4) inhibitor use in an unselected population of subjects with type 2 diabetes: a nested case–control study. BMJ Open. 2015;5(6):e007959.
Ryden L, Shahim B, Mellbin L. Clinical implications of cardiovascular outcome trials in type 2 diabetes: from DCCT to EMPA-REG. Clin Ther. 2016;38(6):1279–87.
Smith RJ, Goldfine AB, Hiatt WR. Evaluating the cardiovascular safety of new medications for type 2 diabetes: time to reassess? Diabetes Care. 2016;39(5):738–42.
Zannad F, et al. Assessment of cardiovascular risk of new drugs for the treatment of diabetes mellitus: risk assessment vs. risk aversion. Eur Heart J Cardiovasc Pharmacother. 2016;2(3):200–5.
Schnell O, et al. Report from the 1st cardiovascular outcome trial (CVOT) summit of the diabetes & cardiovascular disease (D&CVD) EASD study group. Cardiovasc Diabetol. 2016;15(1):33.
Montecucco F, Carbone F, Schindler TH. Pathophysiology of ST-segment elevation myocardial infarction: novel mechanisms and treatments. Eur Heart J. 2016;37(16):1268–83.
El-Sherif N, et al. Pathophysiology, risk stratification, and management of sudden cardiac death in coronary artery disease. Cardiol J. 2010;17(1):4–10.
Koo J. The Latest information on intracranial atherosclerosis: diagnosis and treatment. Interv Neurol. 2015;4(1–2):48–50.
Manea MM, et al. Brain-heart axis—review article. J Med Life. 2015;8(3):266–71.
McMurray JJ, et al. Heart failure: a cardiovascular outcome in diabetes that can no longer be ignored. Lancet Diabetes Endocrinol. 2014;2(10):843–51.
Johansen OE. Interpretation of cardiovascular outcome trials in type 2 diabetes needs a multiaxial approach. World J Diabetes. 2015;6(9):1092–6.
Hess CN, et al. Embedding a randomized clinical trial into an ongoing registry infrastructure: unique opportunities for efficiency in design of the study of access site for enhancement of percutaneous coronary intervention for women (SAFE-PCI for Women). Am Heart J. 2013;166(3):421–8.
Lauer MS, D’Agostino RB Sr. The randomized registry trial—the next disruptive technology in clinical research? N Engl J Med. 2013;369(17):1579–81.
An H, He L. Current understanding of metformin effect on the control of hyperglycemia in diabetes. J Endocrinol. 2016;228(3):R97–106.
Inzucchi SE, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38(1):140–9.
Marx N, et al. Design and baseline characteristics of the cardiovascular outcome trial of linagliptin versus glimepiride in type 2 diabetes (CAROLINA(R)). Diab Vasc Dis Res. 2015;12(3):164–74.
Patorno E, et al. Comparative cardiovascular safety of glucagon-like peptide-1 receptor agonists versus other antidiabetic drugs in routine care: a cohort study. Diabetes Obes Metab. 2016;18(8):755–65.
Rizzo M. Exenatide once weekly, cardiovascular risk and type-2 diabetes, in NCT02380521, clinicaltrials.gov, editor. 2015.
Janssen Research & Development L. A study of the effects of canagliflozin (JNJ-28431754) on renal endpoints in adult participants with type 2 diabetes mellitus (CANVAS-R), in NCT01989754, Clinicaltrials.gov, editor. 2014.
Authors’ contributions
OS was responsible for conception and design, drafting of the manuscript, and the final approval of the version to be published. ES, LR and AC contributed specialized input especially regarding their fields of expertise, and critically revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank Dr. R. Garcia-Verdugo for her involvement in our scientific discussions, and support on manuscript edition and formatting.
Competing interests
L.R has been a member of advisory boards and/or speaker for AstraZeneca, Bayer, Boehringer-Ingelheim, Sanofi; and received research support from the Swedish Heart–Lung Foundation, Swedish Diabetes Foundation, Karolinska Institutet,Private Foundations, Stockholm County Council, Swedish Medical Assembly,Bayer and Boehringer-Ingelheim. E. S. has received lecturing honoraria and consultation fees from AstraZeneca, Bayer, Boehringer Ingelheim, Merck-Serono, MSD/Merck, Novartis, Sanofi. A.C. has been a member of advisory boards for Astra Zeneca, Bayer Healthcare, Boehringer Ingelheim, Bristol Myers Squibb, Danone, DOC Generici, Eli Lilly, Janssen, Medtronic, Merck Sharp & Dome, Novartis, Novo Nordisk, OM Pharma, Roche Diagnostics, Sanofi, Takeda and Unilever; has been a consultant for Bayer Pharma, Lifescan, Mendor, Novartis and Roche Diagnostics; given lectures for Astra Zeneca, Bayer Healthcare, Bayer Pharma, Boehringer Ingelheim, Bristol Myers Squibb, Eli Lilly, Merck Sharp & Dome, Mitsubishi, Novartis, Novo Nordisk, Nutricia, Sanofi, Servier and Takeda; and received research grants from Mitsubishi, Novartis and Novo Nordisk. O. S. has acted as member of advisory boards and/or given lectures under support from Abbott, Astra Zeneca, Bayer Healthcare, Boehringer-Ingelheim, Eli Lilly, Medtronic, Novartis, Roche Diagnostics, Sanofi; and is CEO and founder of Sciarc GmbH.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated during the current study.
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Schnell, O., Rydén, L., Standl, E. et al. Current perspectives on cardiovascular outcome trials in diabetes. Cardiovasc Diabetol 15, 139 (2016). https://doi.org/10.1186/s12933-016-0456-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-016-0456-8