
Abstract
Background
Cardiovascular disease (CVD) is the leading cause of morbidity and mortality in patients with diabetes mellitus (DM). To identify the most effective treatment for CVD, it is paramount to understand the mechanism behind cardioprotective therapies. Although metformin has been shown to reduce CVD in Type-2 DM clinical trials, the underlying mechanism remains unexplored. CD34+ cell-based therapies offer a new treatment approach to CVD. The aim of this study was to investigate the effect of metformin on the angiogenic properties of CD34+ cells under conditions mimicking acute myocardial infarction in diabetes.
Methods
CD34+ cells were cultured in 5.5 or 16.5 mmol/L glucose ± 0.01 mmol/L metformin and then additionally ± 4 % hypoxia. The paracrine function of CD34+ cell-derived conditioned medium was assessed by measuring pro-inflammatory cytokines, vascular endothelial growth factor A (VEGFA), and using an in vitro tube formation assay for angiogenesis. Also, mRNA of CD34+ cells was assayed by microarray and genes of interest were validated by qRT-PCR.
Results
Metformin increased in vitro angiogenesis under hyperglycemia–hypoxia and augmented the expression of VEGFA. It also reduced the angiogenic-inhibitors, chemokine (C–X–C motif) ligand 10 (CXCL10) and tissue inhibitor of metalloproteinase 1 (TIMP1) mRNAs, which were upregulated under hyperglycemia–hypoxia. In addition metformin, increased expression of STEAP family member 4 (STEAP4) under euglycemia, indicating an anti-inflammatory effect.
Conclusions
Metformin has a dual effect by simultaneously increasing VEGFA and reducing CXCL10 and TIMP1 in CD34+ cells in a model of the diabetic state combined with hypoxia. Therefore, these angiogenic inhibitors are promising therapeutic targets for CVD in diabetic patients. Moreover, our data are commensurate with a vascular protective effect of metformin and add to the understanding of underlying mechanisms.
Similar content being viewed by others


Background
Cardiovascular disease (CVD) remains the major cause of morbidity and mortality worldwide [1]. The increased risk of CVD in patients with diabetes is well established, and their disease progression is greater than for non-diabetic patients despite similar interventions [2]. CVD is the major cause of mortality in diabetic patients accounting for more than 50 % of all fatalities [2]. Furthermore, the outcome of CVD interventions in patients with diabetes is much worse in comparison with non-diabetic individuals [3, 4]. Impaired angiogenesis is an important factor leading to the development of diabetes associated CVD [5, 6]. Thus, there is an increasing demand for an investigation of new therapeutic targets and treatment approaches directed at restoring angiogenesis in diabetic CVD cases.
CD34+ cells alone or as part of bone marrow aspirate have been used as novel stem cell therapies in the treatment of acute myocardial infarction (MI) or heart failure with beneficial effect [7]. A study in SCID mice after experimental MI showed that the primary mode of action of CD34+ cells is paracrine [8]. Paracrine secretion and tissue repair were reported to be impaired in CD34+ cells from diabetic patients in comparison to non-diabetic or healthy volunteers [9–11]. Patients with diabetes have increased circulating levels of inflammatory markers including tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and chemokine (C–C motif) ligand 2 (CCL2) [12]. In addition, increased levels of inflammatory markers predict cardiovascular risk in diabetic patients [13].
Metformin is the anti-diabetic drug that reduced CVD in Type-2 diabetes mellitus (DM) clinical trial (UKPDS) [14]. Although clinical studies provide plausible evidence that metformin improves endothelial function [15–17], the underlying mechanism is not clearly understood. In the current proof of concept study, we used human CD34+ cells to understand the beneficial effect of metformin on angiogenesis at 3 h of hypoxia, as this is a therapeutic window in the management of acute myocardial infarction. As umbilical cord blood (UCB) is a non-invasive source of CD34+ cells with a higher number of primitive CD34+ cells than bone marrow we used UCB derived CD34+ cells in our experiments [18, 19].
We reasoned that understanding the cardioprotective mechanism behind metformin therapy will underpin the identification of potential new therapeutic targets and treatment approaches to CVD, particularly in diabetic patients.
Methods
Tissue supply
Human umbilical cord blood was collected in 250 mL blood collection bags (Macropharma, Tourcoing, France) for the isolation of CD34+ cells. Human umbilical cords were collected and preserved in 50 mL conservation buffer containing Dulbecco’s phosphate buffer saline without Ca+2 and Mg+2, 200 U/mL penicillin, 200 µg/mL streptomycin, and 2.5 µg/mL fungizone (Gibco, Life Technologies, Paisley, UK).
The study was approved by NHS Health Research Authority, NRES Committee North East-Sunderland, UK (12/NE/0044) and subjects gave informed written consent.
CD34+ cell culture
CD34+ cells were isolated from three different UCB collections per condition (10 conditions studied) using Lymph prep protocol and CD34 Microbeads separation according to the manufacturer’s instructions (Miltenyi Biotic Inc., Bergisch Gladbach, Germany). The purity of isolated CD34+ cells was assessed by flow cytometry (FACSCanto II, BD, Bioscience, San Jose, CA). All samples showed purity >95 %.
Approximately 1 × 106 CD34+ cells per condition were cultured in M199 medium supplemented with 2.0 mmol/L l-glutamate, 100 Units/mL penicillin, 100 μg/mL streptomycin, 2.5 µg/mL fungizone (Gibco, Life Technologies, Paisley, UK), 0.25 % (v/v) human serum albumin (Octapharma, Manchester, UK), 15 mM HEPES, 1.35 g/L NaHCO3 (GE Healthcare, Little Chalfont, UK), 100 ng/mL Flt3-L, and 100 ng/mL stem cell factor (PerproTech, Rocky Hill, NJ, USA).
CD34+ cells were incubated in a culture medium with 5.5 mmol/L (euglycemia) or 16.5 mmol/L (hyperglycemia) glucose concentrations (Sigma-Aldrich, Dorset, UK). Glucose at 16.5 mmol/L simulates a diabetic state but did not impair cellular growth, whereas increased the expression of endothelial adhesion molecules, consistent with hyperglycemia-mediated vascular inflammation [20–22]. The cells were simultaneously incubated in the presence or absence of 0.01 mmol/L metformin (Sigma-Aldrich, Dorset, UK) for 24 h. The metformin concentration used was selected according to the peak plasma concentration and metformin pharmacokinetics reported earlier in diabetic patients [23]. Subsequently, cells were subjected additionally to 4 % hypoxia (Heracell™ 150i, ThermoScientific) for 3 h to simulate cellular and molecular responses following ischemia [24].
Meso scale discovery (MSD) assay
The culture medium was collected from 5 × 105 CD34+ cells exposed to the above mentioned conditions and assayed using K15025C human Pro-inflammatory II 4-Plex, K151A0H Custom V-PLEX Human Biomarkers (Meso Scale Discovery, Rockville, MD) in accordance with the manufacturer protocol. Plates were read with MSD Sector Imager 2400 and data were analyzed by MSD Discovery Workbench version 2.0 software. The data were normalized against the complete culture medium, and three samples were processed in duplicates.
HUVEC cultures
HUVEC were harvested from three independent umbilical cords as previously described by us [21]. All experiments were performed at passage two and 80 % confluency.
In vitro Matrigel tube formation assay
HUVEC (2.0 × 104 cells) were serum starved overnight in M199 complete medium with 0.25 % FBS. An in vitro tube formation assay was performed as previously described with minor modifications [25]. EBM-2 medium containing 14 µmol/L sunitinib malate (VEGFA inhibitor, Sigma-Aldrich, Dorset, UK) was used as a negative control, and this concentration showed no toxicity in HUVEC cell cultures as illustrated in Additional file 1: Figure S1. Subsequently, the cells were incubated at 37 °C and 5 % CO2 (OkoLab, NA, Italy) for 24 h in a chamber that was connected to a camera (Hamamatsu Orca ER, Hamamatsu City, Japan). Tube formation was examined by phase-contrast microscopy (Nikon Eclipse Tie, Tokyo, Japan) using software based autofocus (NIS Elements V4.0, Nikon, Tokyo, Japan). Images were acquired every hour. All conditions in each experiment were assessed in duplicate, and tube length was measured as the mean summed length of capillary-like structures in 2 wells. Three independent experiments were performed for each condition. The tube length was measured using Adobe Acrobat Professional version 8 software by measuring long tubes first and then the small branches to cover the whole image. The analysis was conducted blinded.
Total RNA extraction
Total RNA from CD34+ cells was extracted using the RNeasy Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The cell lysates from three different CD34+ cultures per condition were pooled before proceeding to an RNeasy Mini spin column. On-column DNase digestion with RNase-Free DNase set (QIAGEN) was performed. The quality and quantity of RNA was assessed using a NanoDrop 2000c Spectrophotometer (Thermo Scientific, Wilmington, DE, USA). The integrity of RNA samples was assessed by using Agilent 2100 Bioanalyzer (Santa Clara, CA, USA) yielding high RNA Integrity Numbers (RIN) between 7.0 and 9.7.
Microarray experiments and gene expression analysis
Microarray experiments were performed using Affymetrix (Santa Clara, CA, USA) Human Gene 1.0 ST arrays according to manufacturer’s instructions with minor modifications [21, 26]. Two technical replicates were hybridized for each experimental condition resulting in a total of 20 microarray experiments.
Affymetrix CEL files were imported to Partek Genomic Suite version 6.6 (Partek Inc., MO, USA). The data were normalized using Robust Multichip Average (RMA) normalization. Principal component analysis (PCA) was performed on all probes to visualize high-dimensional data. By default, expression values were filtered for statistical significance using Benjamini and Hochberg’s False Discovery Rate (FDR), with an FDR-unadjusted p value <0.05. ANOVA was performed using the commonly employed p values <0.05 and cut off fold change (FC) ≥1.5 as described by Peart et al. [27] and Raouf et al. [28].
Two-dimensional average linkage hierarchical clustering was performed for the differentially expressed genes using Spearman’s correlation as a similarity matrix. The microarray data generated in this study are in compliance with MIAME (http://www.mged.org/Workgroups/MIAME/miame.html) guidelines. The complete dataset and associated experimental information were submitted to NCBI’s Gene Expression Omnibus (GEO) and were accessible through accession number GSE46262. Ingenuity pathway analysis (IPA) software version 9 (Ingenuity, Redwood City, CA, USA) was employed to enable exploring the Canonical Pathways that may be increased or decreased based on activation or inhibition of molecules within that pathway. Additionally, IPA assisted in detecting the interactive molecular and cellular functions affected in each condition.
Quantitative RT-PCR
Approximately 100 ng of total RNA from each sample was converted to cDNA using SuperScript VILO™ cDNA Synthesis Kit (Life Technologies, Paisley, UK) in a final volume of 20 μL. The cDNA product was quantified by hydrolysis probe real-time PCR performed with TaqMan® Universal Master Mix II and assayed on a 7900HT Fast Real-time PCR system (Life Technologies) according to the manufacturer’s recommended conditions. Expression of the following genes were determined using the TaqMan gene expression assays (Life Technologies): CCL2: Hs00234140_m1, CCL5: Hs00174575_m1, C–X–C motif chemokine 10 (CXCL10): Hs01124251_g1, hepatocyte growth factor (HGF): Hs00900070_m1, IL-1α: Hs00174092_m1, IL-6: Hs00985639_m1, IL-8: Hs00174103_m1, tissue metallopeptidase inhibitor 1 (TIMP1): Hs00171558_m1, selectin P (SELP): Hs00927900_m1. All samples were run in triplicate, and average values were calculated. Three independent reverse transcriptions were tested for each gene. The Comparative Ct method (ΔΔCt) was used to quantify expression of the target genes, which were normalized to the endogenous control gene for the large ribosomal protein P0 (RPLP0) [21] (Catalog number 432631, Life Technologies).
Statistical analysis
Results are presented as mean ± SEM, and statistical analysis was performed using one-way ANOVA followed by post hoc analysis using Fisher’s least significant difference (LSD) test for the in vitro Matrigel tube formation assay, qRT-PCR, and MSD assay. Calculations were performed using IBM SPSS software version 21.0 (SPSS Inc, NY). A p value <0.05 was considered statistically significant.
Results
Secretion of pro-inflammatory and pro-angiogenic cytokines by CD34+ cells
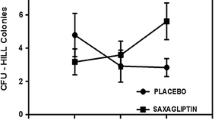
Measured concentrations of the pro-inflammatory cytokines IL-1β, IL-6, IL-8, and TNF-α; were very low in conditioned medium (CM) of CD34+ cells under all conditions studied. However, the pro-angiogenic factor VEGFA was found to be significantly increased in CM collected from CD34+ cells treated with hyperglycemia (2.0-fold, p < 0.001) versus euglycemia, whereas no change was observed under hyperglycemia combined with hypoxia for 3 h versus hyperglycemia. The CM from CD34+ cells treated with metformin displayed augmented levels of VEGFA either under euglycemia (1.6-fold, p = 0.008), euglycemia combined with hypoxia (1.4-fold, p = 0.037), and hyperglycemia combined with hypoxia (1.2-fold, p = 0.037) compared with the metformin-untreated condition (Fig. 1).

Expression of pro-angiogenic factor VEGFA in CD34+ cell-derived conditioned media. The levels of VEGFA were assayed in three independent biological replicates using the MSD technique. Results are presented as ±SEM and were statistically analyzed using one-way ANOVA followed by Fisher’s LSD test. Data for effects of hypoxia and hyperglycemia were compared with control (5.5 mmol/L), and data for cells treated with metformin were compared with the corresponding metformin-untreated condition. *P < 0.05, **P < 0.01, ***P < 0.001
The effect of metformin on in vitro angiogenic function of CD34+ cell-derived CM
Metformin significantly increased tube formation by 30.7 % (p = 0.04) in HUVEC incubated with CD34+ CM derived from cells treated with hyperglycemia–hypoxia compared with the condition without metformin (Fig. 2a highlighted with red and b). However, under other conditions metformin did not cause any significant increase in tube formation.

Effect of CM from CD34+ cells exposed to different conditions on in vitro angiogenic assays. a HUVEC were plated on Matrigel with CM from 2.0 × 105 CD34+ cells treated with euglycemia, euglycemia and hypoxia, hyperglycemia or hyperglycemia and hypoxia in the presence and absence of metformin. EBM-2 medium containing the VEGF inhibitor sunitinib (14 µmol/L) was used as a negative control for the assay. The highlighted image shows the greatest tube length, which was achieved in HUVEC incubated with CM-derived from CD34+ cells treated with hyperglycemia and hypoxia in the presence of metformin. b Tube length at 6 h was expressed as a percentage of the tube length of HUVEC treated with 5.5 mmol/L glucose CM (n = 3); *P < 0.05 compared pairwise, i.e., condition with metformin versus metformin-untreated condition
Metformin drives a gene expression profile consistent with an anti-inflammatory action under euglycemia and euglycemia-hypoxia
The transcriptomic analysis of CD34+ cells showed that 313 genes passed the cutoff FC of 1.5 and p value <0.05 under euglycemia and metformin compared to euglycemia alone. The top associated molecular and cellular functions predicted using IPA were inflammatory response (p = 1.52E−06) and binding of EC (p = 1.56E−06), which were inhibited by metformin. The corresponding downregulated genes are highlighted in Table 1.
One hundred genes were differentially expressed in CD34+ cells (cutoff FC of 1.5 and p value <0.05) under euglycemia and hypoxia compared to euglycemia alone. The top molecular and cellular functions affected as predicted by IPA are reported in Additional file 1: Table S1. Metallothionein 2A (MT2A) was upregulated (1.7-fold, p = 4.70E−03) under hypoxia which was previously indicated as one of hypoxia inducible genes [29].
Metformin treatment of CD34+ cells exposed to euglycemia and hypoxia caused differential expression of 21 genes compared with the metformin-untreated condition. The top related molecular functions predicted by IPA were: cell death/survival (p = 7.23E−04), and cellular growth/proliferation (p = 7.23E−04). Genes associated with these functions were IL-5 (1.52-fold, p = 7.05E−03), serine peptidase inhibitor, Kazal type 7 (putative) (SPINK7, 1.60-fold, p = 1.48E−02), and transducer of ERBB2, 2 (TOB2, −1.53-fold, p = 4.62E−02).
Metformin had no effect on inflammatory response, cell movement, or adhesion under euglycemia-hypoxia as demonstrated in Additional file 1: Figure S2.
Metformin drives a gene expression profile consistent with improved cell survival in CD34+ cells under hyperglycemia
Analysis of microarray data revealed that 370 genes passed the cutoff FC of 1.5 and p value <0.05 under hyperglycemia versus euglycemia. Approximately 35 % of the genes were upregulated, and 65 % were downregulated. The uppermost 30 differentially expressed genes are listed in Additional file 1: Table S2. The top molecular functions predicated by IPA were cell to cell signaling and interaction (p = 1.80E−17), cellular growth/proliferation (p = 1.21E−12), inflammatory response (p = 1.59E−12), cellular movement (p = 2.60E−11) which were all predicted to be inhibited (Additional file 1: Table S3). The main canonical pathways affected were atherosclerosis signaling (p = 3.2E−07), T helper cell differentiation (p = 6.6E−04), IL-6 signaling (p = 2.1E−03) (Additional file 1: Table S4).
Metformin treatment of CD34+ cells under hyperglycemia led to differential expression of 65 genes compared with hyperglycemia alone. Approximately 40 % of the genes were upregulated, and 60 % were downregulated. The top differentially expressed genes are listed in Additional file 1: Table S5. The top associated molecular functions predicted by IPA were cell death/survival (p = 1.29E−02), and cell proliferation/growth (p = 3.25E−03). The genes attributed to cell death/survival were killer cell immunoglobulin-like receptor, two domains, short cytoplasmic tail, 5 (KIR2DS5) 1.75-fold, p = 2.27E−02, ubiquitin specific peptidase 18 (USP18) 1.63-fold, p = 3.10E−03. The genes related to cell proliferation/growth were epithelial cell adhesion molecule (EPCAM) −2.06-fold, p = 9.93E−03, USP18, and hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 1 (HSD3B1) −1.64-fold, p = 2.73E−02.
It is of particular interest that vascular endothelial growth factor receptor-2 (VEGFR-2) was downregulated under hyperglycemia in a dose-dependent manner (−1.95-fold, p = 3.23E−04) for 16.5 mmol/L and (−3.0-fold, p = 5.3E−06) for 25 mmol/L and not improved by metformin treatment (Additional file 1: Table S3).
Metformin drives a gene expression profile consistent with pro-angiogenic action in CD34+ cells under hyperglycemia with hypoxia
We identified 1006 differentially expressed genes in cells under hyperglycemia exposed to hypoxia compared with hyperglycemia. 52 % of the genes were upregulated whereas 48 % were downregulated. The top 20 significantly differentially expressed genes are listed in Table 2. The most affected functions predicted by IPA were cell cycle (p = 1.95E−19), cellular movement (p = 2.03E−09), and cellular proliferation (p = 1.39E−07), which were predicted to be activated, whereas chemotaxis of vascular endothelial cells (p = 6.44E−03) was predicted to be inhibited. Downregulation of pro-angiogenic cytokines IL-8 (−2.12-fold, p = 3.55E−04) and HGF (−1.64-fold, p = 2.33E−03) was detected, while upregulation of angiogenic inhibitors TIMP1 (2.36-fold, p = 6.34E−06), TIMP3 (2.15-fold, p = 2.29E−02), and CXCL10 (1.89-fold, p = 2.04E−02) was indicated.
Under metformin combined with hyperglycemia–hypoxia, 317 genes were differentially expressed versus no metformin. Forty-two percent of the genes were upregulated whereas 58 % were downregulated. The 20 most differentially expressed genes are listed in Table 3. The most affected functions predicted by IPA were cellular movement (p = 3.19E−05), and DNA replication and repair (p = 1.41E−04). The main canonical pathways affected were mitochondrial dysfunction (p = 2.0E−03), triacylglycerol biosynthesis (p = 7.1E−03), MAPK signaling (p = 7.9E−03), Type 1 DM signaling (p = 9.5E−03), and IL-8 signaling (p = 2.3E−02) (Additional file 1: Table S6).
Furthermore, metformin downregulated the angiogenic inhibitors TIMP1 [30] (−1.68-fold, p = 3.90E−04), and CXCL10 [31] (−2.01-fold, p = 1.28E−02), whilst had no effect on expression of pro-angiogenic factors under hyperglycemia–hypoxia versus no metformin.
Confirmation of effects of metformin on gene expression in CD34+ cells using qRT-PCR
Seven pro-angiogenic factors CCL2, CCL5, HGF, IL-1a, IL-6, IL-8, selectin P (SELP) and two angiogenic inhibitors CXCL10 and TIMP1 with critical biological functions were validated by qRT-PCR (Additional file 1: Figure S3) and their results were compared to those obtained from the microarray experiments using the same RNA samples (Additional file 1: Figure S4). The qRT-PCR results were concordant with all gene expression changes indicated by the microarray data except TIMP1 which was justified later (Additional file 1: Table S7). Moreover, hyperglycemia and metformin resulted in an increase of the mRNA levels of HGF (1.7-fold, p = 0.001), and IL-6 (2.8-fold, p = 0.01) although this increase remained below normal level observed at euglycemia.
Discussion
To our knowledge, this is the first report describing the effect of a physiological concentration of metformin on the angiogenic potential of CD34+ cells. Metformin is a hypoglycemic agent found to have cardioprotective properties as borne by a large clinical trial [14]. It has been shown that metformin has a protective effect against free fatty acid induced apoptosis [32] and its administration in diabetic patients prior to stroke onset was associated with reduced neurological severity and improved acute-phase therapy outcomes [33]. Moreover, metformin’s pleiotropic effects and cardioprotective role beyond glucose-lowering effect has been recently highlighted, encouraging implementation in prospective studies [34]. A recent study has shown that metformin monotherapy lead to an improvement in multiple clinical parameters and a reduction in all-cause mortality and CVD events in Type 2 DM patients [35] however, the underlying mechanism is unclear.
In diabetic patients CD34+ cells displayed a vasoreparative dysfunction due to impaired paracrine function and reduced sensitivity to hypoxia [10]. Whilst, the outcome of trials involving administration of angiogenic cytokines for the purpose of therapeutic angiogenesis as an alternative treatment for ischemic heart disease have been disappointing [36–38].
Autologous CD34+ based stem cell therapies have been pioneered as an alternative treatment for CVD [39–42]. Therefore, we studied vasoreparative properties of CD34+ cells following incubation with physiological concentration of metformin, under conditions of hyperglycemia, and/or hypoxia as a model of intervention window during acute myocardial infarction. Our research focused on the paracrine secretion of selected pro-inflammatory, pro-angiogenic factors and angiogenic inhibitors, which effect was functionally studied using an in vitro tube formation assay. Furthermore, gene expression profiling of CD34+ cells, treated under the above mentioned conditions, was performed focusing on pro-angiogenic mechanisms.
Pro-angiogenic effect of metformin
We found that a physiological dose of metformin increased tube formation under the condition of hyperglycemia–hypoxia combined. This was associated with downregulation of the mRNA of the angiogenic inhibitors CXCL10 and TIMP1 observed by microarray analysis and further confirmed by qRT-PCR. The discrepancy in TIMP1 results obtained from microarray data and qRT-PCR could be due to non-concordance of transcripts used in the microarray probe set and qRT-PCR [43]. Conversely, in the absence of metformin, CXCL10 and TIMP1 gene expression was significantly upregulated under hyperglycemia–hypoxia versus hyperglycemia. Our findings are concordant with clinical/laboratory data that demonstrated a suppression in angiogenesis due to an increase in angiogenic inhibitors in patients with diabetes combined with an increase in CXCL10 and TIMP1 expression at the transcriptional and protein levels [44, 45].
Under conditions of euglycemia and euglycemia-hypoxia, metformin had no measured effect on angiogenesis as both tube formation and gene expression of CXCL10 and TIMP1 were unchanged. Therefore, it appears that metformin has no additional pro-angiogenic effect in the non-diabetic state. This finding is in keeping with recent clinical studies, which revealed that metformin exhibited no effect on several surrogate markers of CVD in non-diabetic patients nor led to beneficial clinical outcome [46, 47].
In support of enhanced angiogenesis by metformin, we found that although the concentration of VEGFA was progressively increased under hyperglycemia–hypoxia it was further enhanced following metformin.
We confirmed in our in vitro model, as it was shown in clinical studies, that hyperglycemia alone increased VEGFA but downregulated VEGFR-2 gene expression, neither of which were affected by metformin. Our findings are concordant with another study in which VEGFA expression was found to be increased in myocardial tissue of diabetic patients, whilst a decrease in VEGF receptors was documented leading to downregulation of VEGF signal transduction [48].
Thus, although metformin further increased angiogenic factor, VEGFA, we have shown that the cardioprotection offered was predominantly by reducing angiogenic inhibitors; CXCL10 and TIMP-1; rather than further increasing VEGFA alone. This means that the process of angiogenesis is under a fine balance of pro and anti-angiogenic factors to ensure tightly regulated process. Consequently, in the condition of diabetes combined with hypoxia it is not sufficient to further increase proangiogenic factors alone, but downregulation of inhibitors is required for altering the balance towards angiogenesis (Fig. 3).

Summary of the effect of metformin on CD34+ cells incubated with hyperglycemia–hypoxia. CD34+ cells were treated with hyperglycemia–hypoxia versus euglycemia (a), and hyperglycemia–hypoxia with metformin versus metformin untreated-condition (b). Green shading indicates downregulation, orange shading indicate upregulation and gray shading indicate unchanged gene expression. In vitro tube formation (angiogenesis), green shading indicates inhibition, whereas orange shading indicates activation
Pro-inflammatory effect of metformin
It has been assumed that mechanism behind CD34+ cells based therapy for CVD is mediated by paracrine function. Thus, we hypothesized that metformin improved the paracrine function of CD34+ cells. However, we found very low protein levels of IL-1β, IL-6, and TNF-α with non-significantly reduced levels of IL-8 expression in CD34+ cell CM in all studied conditions including metformin. These findings can be explained by downregulation of genes coding for inflammatory cytokines in CD34+ cells under metformin and euglycemia, euglycemia-hypoxia, and hyperglycemia.
We have shown that under euglycemia, metformin displayed an inhibitory effect on genes of pro-inflammatory factors CCL2, CCL5, CD226, IL-1α, IL-6, IL-8, and Integrin, beta 3 (ITGB3) whilst upregulating STEAP family member 4 (STEAP4). We thus believe that the anti-inflammatory action of metformin could be mediated by augmenting mRNA expression of STEAP4. This anti-inflammatory effect has been previously demonstrated in fibroblast-like synoviocytes by upregulation of STEAP4 leading to the suppression of IL-6 and IL-8 expression [49]. In concordance with our results another study in smooth muscle cells, ECs, or macrophages has shown that a physiological dose of metformin inhibited the expression of IL-1β, IL-6, and IL-8 [50].
Furthermore, the same mechanism of inhibition of pro-inflammatory cytokines was demonstrated under euglycemia-hypoxia with the increase in STEAP4 expression, while metformin had no additional effect on those genes.
These data suggest that metformin and/or hypoxia in CD34+ cells are beneficial in suppressing the inflammatory response. In fact, low oxygen tension in the range of 3–5 % (hypoxia) is the preferable environment for stem cells (including CD34+ cells) to reside quiescent in their niches and maintain pluripotency with no effect on proliferation [51].
In addition, we have established that TOB2 which acts as an anti-proliferative factor [52] was upregulated under hypoxic condition but was inhibited by metformin. TOB2 is a member of the mammalian BTG/TOB family of anti-proliferative proteins [53]. TOB2 were previously detected to be expressed in mouse embryonic stem cells that play a critical role in the maintenance of stem cells properties [52].
Similarly, under hyperglycemia and hyperglycemia–hypoxia our transcriptome analysis demonstrated downregulation of pro-inflammatory and pro-angiogenic cytokines, chemokines and their receptors with no additional effect of metformin. The failure to detect the suppressive effect of metformin on those cells is possibly due to already maximally suppressed cytokine gene expression.
We studied pro-inflammatory paracrine secretion by CD34+ cells without cell co-culture or cell expansion. This may be a limitation of our approach as we underestimate the interactions of CD34+ cells with other cells known to induce cytokine secretion in co-culture [54] or cytokine secretion from expanded CD34+ cells [55]. Specifically, the co-culture of CD34+ cells with HUVEC induced paracrine secretion of pro-inflammatory cytokines including IL-8, IL-6, CCL2, CCL3, and CCL4 and angiogenesis under hypoxia [54]. Also, the co-culture of CD34+ and CD34− cells lead to enhancement of tube formation and cell migration compared with the culture of CD34+ cells alone through the cell to cell interactions and paracrine effects [56]. Furthermore, it has been reported that macrophages migrate to hypoxic areas that stimulate expression of pro-angiogenic such as VEGFA and pro-inflammatory genes in an attempt to repair the damage [57]. Together these data demonstrate the significant role played by CD34− cells in interaction with CD34+ cells to improve tissue repair.
It is envisaged that the findings from our study will be validated in in vivo settings using suitable animal models with CXCL10 and/or TIMP1 overexpression. However, the current lack of appropriate animal models make it impossible at present to confirm the suggested mechanism in vivo. Moreover, it will be of importance if these findings can also be proven in CD34+ cells obtained from diabetic patients.
Conclusions
In conclusion, vascular protection by metformin is mediated by its dual effect to enhance the angiogenic potential of CD34+ cells by suppressing several angiogenic inhibitors including CXCL10 and TIMP1 whilst further increasing VEGFA secretion. Those angiogenic inhibitors are potential therapeutic targets for CVD interventions in diabetes.
Abbreviations
- C3:
-
complement 3
- CCL(number):
-
chemokine (C–C motif) ligand (number)
- CM:
-
conditioned medium
- CVD:
-
cardiovascular disease
- CXCL10:
-
chemokine (C–X–C motif) ligand 10
- DM:
-
diabetes mellitus
- EPCAM:
-
epithelial cell adhesion molecule
- HGF:
-
hepatocyte growth factor
- HSD3B1:
-
hydroxy-delta-5-steroid dehydrogenase, 3 bet- and steroid delta-isomerase 1
- HUVEC:
-
human umbilical vein endothelial cell
- IL-(number):
-
interleukin-(number)
- ITGB3:
-
integrin, beta 3
- KIR2DS5:
-
killer cell immunoglobulin-like receptor, two domains, short cytoplasmic tail, 5
- MI:
-
myocardial infarction
- MT2A:
-
metallothionein 2A
- SELP:
-
selectin P
- SPINK7:
-
serine peptidase inhibitor, Kazal type 7
- STEAP4:
-
STEAP family member 4
- TIMP1:
-
tissue inhibitor of metalloproteinase 1
- TOB2:
-
transducer of ERBB2, 2
- UCB:
-
umbilical cord blood
- USP18:
-
ubiquitin specific peptidase 18
- VEGFA:
-
vascular endothelial growth factor A
References
Organization. WH: WHO mortality database—January 2015 update. http://www.who.int/mediacentre/factsheets/fs317/en/.
Federation ID: IDF Diabetes Atlas 6th edition-2013. https://www.idf.org/sites/default/files/EN_6E_Atlas_Full_0pdf.
Ekezue BF, Laditka SB, Laditka JN, Studnicki J, Blanchette CM. Diabetes complications and adverse health outcomes after coronary revascularization. Diabetes Res Clin Pract. 2014;103(3):530–7.
Piccolo R, Galasso G, Iversen AZ, Eitel I, Dominguez-Rodriguez A, Gu YL, de Smet BJ, Mahmoud KD, Abreu-Gonzalez P, Trimarco B, et al. Effects of baseline coronary occlusion and diabetes mellitus in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am J Cardiol. 2014;114(8):1145–50.
Eelen G, de Zeeuw P, Simons M, Carmeliet P. Endothelial cell metabolism in normal and diseased vasculature. Circ Res. 2015;116(7):1231–44.
Roberts AC, Porter KE. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diab Vasc Dis Res. 2013;10(6):472–82.
Cogle CR, Wise E, Meacham AM, Zierold C, Traverse JH, Henry TD, Perin EC, Willerson JT, Ellis SG, Carlson M, et al. Detailed analysis of bone marrow from patients with ischemic heart disease and left ventricular dysfunction: BM CD34, CD11b, and clonogenic capacity as biomarkers for clinical outcomes. Circ Res. 2014;115(10):867–74.
Wang J, Zhang S, Rabinovich B, Bidaut L, Soghomonyan S, Alauddin MM, Bankson JA, Shpall E, Willerson JT, Gelovani JG, et al. Human CD34+ cells in experimental myocardial infarction: long-term survival, sustained functional improvement, and mechanism of action. Circ Res. 2010;106(12):1904–11.
Caballero S, Sengupta N, Afzal A, Chang KH, Li Calzi S, Guberski DL, Kern TS, Grant MB. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56(4):960–7.
Jarajapu YP, Hazra S, Segal M, Li Calzi S, Jadhao C, Qian K, Mitter SK, Raizada MK, Boulton ME, Grant MB. Vasoreparative dysfunction of CD34+ cells in diabetic individuals involves hypoxic desensitization and impaired autocrine/paracrine mechanisms. PLoS ONE. 2014;9(4):e93965.
Tan K, Lessieur E, Cutler A, Nerone P, Vasanji A, Asosingh K, Erzurum S, Anand-Apte B. Impaired function of circulating CD34(+) CD45(−) cells in patients with proliferative diabetic retinopathy. Exp Eye Res. 2010;91(2):229–37.
Daniele G, Guardado Mendoza R, Winnier D, Fiorentino TV, Pengou Z, Cornell J, Andreozzi F, Jenkinson C, Cersosimo E, Federici M, et al. The inflammatory status score including IL-6, TNF-α, osteopontin, fractalkine, MCP-1 and adiponectin underlies whole-body insulin resistance and hyperglycemia in type 2 diabetes mellitus. Acta Diabetol. 2014;51(1):123–31.
Lowe G, Woodward M, Hillis G, Rumley A, Li Q, Harrap S, Marre M, Hamet P, Patel A, Poulter N, et al. Circulating inflammatory markers and the risk of vascular complications and mortality in people with type 2 diabetes and cardiovascular disease or risk factors: the ADVANCE study. Diabetes. 2014;63(3):1115–23.
UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352:854–65.
Cheang WS, Tian XY, Wong WT, Lau CW, Lee SS, Chen ZY, Yao X, Wang N, Huang Y. Metformin protects endothelial function in diet-induced obese mice by inhibition of endoplasmic reticulum stress through 5′ adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor δ pathway. Arterioscler Thromb Vasc Biol. 2014;34(4):830–6.
de Aguiar LG, Bahia LR, Villela N, Laflor C, Sicuro F, Wiernsperger N, Bottino D, Bouskela E. Metformin improves endothelial vascular reactivity in first-degree relatives of type 2 diabetic patients with metabolic syndrome and normal glucose tolerance. Diabetes Care. 2006;29(5):1083–9.
de Jager J, Kooy A, Schalkwijk C, van der Kolk J, Lehert P, Bets D, Wulffele MG, Donker AJ, Stehouwer CD. Long-term effects of metformin on endothelial function in type 2 diabetes: a randomized controlled trial. J Intern Med. 2014;275(1):59–70.
Nimgaonkar MT, Roscoe RA, Persichetti J, Rybka WB, Winkelstein A, Ball ED. A unique population of CD34+ cells in cord blood. Stem Cells. 1995;13(2):158–66.
Ueda T, Yoshida M, Yoshino H, Kobayashi K, Kawahata M, Ebihara Y, Ito M, Asano S, Nakahata T, Tsuji K. Hematopoietic capability of CD34+ cord blood cells: a comparison with CD34+ adult bone marrow cells. Int J Hematol. 2001;73(4):457–62.
Altannavch TS, Roubalova K, Kucera P, Andel M. Effect of high glucose concentrations on expression of ELAM-1, VCAM-1 and ICAM-1 in HUVEC with and without cytokine activation. Physiol Res. 2004;53(1):77–82.
Bakhashab S, Lary S, Ahmed F, Schulten HJ, Bashir A, Ahmed FW, Al-Malki AL, Jamal HS, Gari MA, Weaver JU. Reference genes for expression studies in hypoxia and hyperglycemia models in human umbilical vein endothelial cells. G3 (Bethesda). 2014;4(11):2159–65.
Takami S, Yamashita S, Kihara S, Kameda-Takemura K, Matsuzawa Y. High concentration of glucose induces the expression of intercellular adhesion molecule-1 in human umbilical vein endothelial cells. Atherosclerosis. 1998;138(1):35–41.
http://www.fda.gov/ohrms/dockets/dailys/02/May02/053102/800471e6.pdf.
Thygesen K, Alpert JS, White HD, Joint ESCAAHAWHFTFftRoMI, Jaffe AS, Apple FS, Galvani M, Katus HA, Newby LK, Ravkilde J, et al. Universal definition of myocardial infarction. Circulation. 2007;116(22):2634–53.
Sahoo S, Klychko E, Thorne T, Misener S, Schultz KM, Millay M, Ito A, Liu T, Kamide C, Agrawal H, et al. Exosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activity. Circ Res. 2011;109(7):724–8.
Merdad A, Karim S, Schulten HJ, Dallol A, Buhmeida A, Al-Thubaity F, Gari MA, Chaudhary AG, Abuzenadah AM, Al-Qahtani MH. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer: MMP-9 as a potential biomarker for cancer invasion and metastasis. Anticancer Res. 2014;34(3):1355–66.
Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, Holloway AJ, Johnstone RW. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102(10):3697–702.
Raouf A, Zhao Y, To K, Stingl J, Delaney A, Barbara M, Iscove N, Jones S, McKinney S, Emerman J, et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell. 2008;3(1):109–18.
Yamasaki M, Nomura T, Sato F, Mimata H. Metallothionein is up-regulated under hypoxia and promotes the survival of human prostate cancer cells. Oncol Rep. 2007;18(5):1145–53.
Johnson MD, Kim HR, Chesler L, Tsao-Wu G, Bouck N, Polverini PJ. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase. J Cell Physiol. 1994;160(1):194–202.
Glaser J, Gonzalez R, Perreau VM, Cotman CW, Keirstead HS. Neutralization of the chemokine CXCL10 enhances tissue sparing and angiogenesis following spinal cord injury. J Neurosci Res. 2004;77(5):701–8.
Eriksson L, Nystrom T. Activation of AMP-activated protein kinase by metformin protects human coronary artery endothelial cells against diabetic lipoapoptosis. Cardiovasc Diabetol. 2014;13:152.
Mima Y, Kuwashiro T, Yasaka M, Tsurusaki Y, Nakamura A, Wakugawa Y, Okada Y. Impact of metformin on the severity and outcomes of acute ischemic stroke in patients with type 2 diabetes mellitus. J Stroke Cerebrovasc Dis. 2016;25(2):436–46.
Bromage DI, Yellon DM. The pleiotropic effects of metformin: time for prospective studies. Cardiovasc Diabetol. 2015;14:109.
Fung CS, Wan EY, Wong CK, Jiao F, Chan AK. Effect of metformin monotherapy on cardiovascular diseases and mortality: a retrospective cohort study on Chinese type 2 diabetes mellitus patients. Cardiovasc Diabetol. 2015;14:137.
Kastrup J, Jorgensen E, Ruck A, Tagil K, Glogar D, Ruzyllo W, Botker HE, Dudek D, Drvota V, Hesse B, et al. Direct intramyocardial plasmid vascular endothelial growth factor-A165 gene therapy in patients with stable severe angina pectoris A randomized double-blind placebo-controlled study: the Euroinject One trial. J Am Coll Cardiol. 2005;45(7):982–8.
Kusumanto YH, van Weel V, Mulder NH, Smit AJ, van den Dungen JJ, Hooymans JM, Sluiter WJ, Tio RA, Quax PH, Gans RO, et al. Treatment with intramuscular vascular endothelial growth factor gene compared with placebo for patients with diabetes mellitus and critical limb ischemia: a double-blind randomized trial. Hum Gene Ther. 2006;17(6):683–91.
Stewart DJ, Kutryk MJ, Fitchett D, Freeman M, Camack N, Su Y, Della Siega A, Bilodeau L, Burton JR, Proulx G, et al. VEGF gene therapy fails to improve perfusion of ischemic myocardium in patients with advanced coronary disease: results of the NORTHERN trial. Mol Ther. 2009;17(6):1109–15.
Assmus B, Schachinger V, Teupe C, Britten M, Lehmann R, Dobert N, Grunwald F, Aicher A, Urbich C, Martin H, et al. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction (TOPCARE-AMI). Circulation. 2002;106(24):3009–17.
Fernandez-Aviles F, San Roman JA, Garcia-Frade J, Fernandez ME, Penarrubia MJ, de la Fuente L, Gomez-Bueno M, Cantalapiedra A, Fernandez J, Gutierrez O, et al. Experimental and clinical regenerative capability of human bone marrow cells after myocardial infarction. Circ Res. 2004;95(7):742–8.
Schachinger V, Assmus B, Britten MB, Honold J, Lehmann R, Teupe C, Abolmaali ND, Vogl TJ, Hofmann WK, Martin H, et al. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction: final one-year results of the TOPCARE-AMI Trial. J Am Coll Cardiol. 2004;44(8):1690–9.
Strauer BE, Brehm M, Zeus T, Kostering M, Hernandez A, Sorg RV, Kogler G, Wernet P. Repair of infarcted myocardium by autologous intracoronary mononuclear bone marrow cell transplantation in humans. Circulation. 2002;106(15):1913–8.
Dallas PB, Gottardo NG, Firth MJ, Beesley AH, Hoffmann K, Terry PA, Freitas JR, Boag JM, Cummings AJ, Kees UR. Gene expression levels assessed by oligonucleotide microarray analysis and quantitative real-time RT-PCR—how well do they correlate? BMC Genom. 2005;6:59.
Chung AW, Hsiang YN, Matzke LA, McManus BM, van Breemen C, Okon EB. Reduced expression of vascular endothelial growth factor paralleled with the increased angiostatin expression resulting from the upregulated activities of matrix metalloproteinase-2 and -9 in human type 2 diabetic arterial vasculature. Circ Res. 2006;99(2):140–8.
Shimada A, Morimoto J, Kodama K, Suzuki R, Oikawa Y, Funae O, Kasuga A, Saruta T, Narumi S. Elevated serum IP-10 levels observed in type 1 diabetes. Diabetes Care. 2001;24(3):510–5.
Preiss D, Lloyd SM, Ford I, McMurray JJ, Holman RR, Welsh P, Fisher M, Packard CJ, Sattar N. Metformin for non-diabetic patients with coronary heart disease (the CAMERA study): a randomised controlled trial. Lancet Diabetes Endocrinol. 2014;2(2):116–24.
El Messaoudi S, Nederlof R, Zuurbier CJ, van Swieten HA, Pickkers P, Noyez L, Dieker HJ, Coenen MJ, Donders AR, Vos A, et al. Effect of metformin pretreatment on myocardial injury during coronary artery bypass surgery in patients without diabetes (MetCAB): a double-blind, randomised controlled trial. Lancet Diabetes Endocrinol. 2015;3(8):615–23.
Sasso FC, Torella D, Carbonara O, Ellison GM, Torella M, Scardone M, Marra C, Nasti R, Marfella R, Cozzolino D, et al. Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signaling in the myocardium of type 2 diabetic patients with chronic coronary heart disease. J Am Coll Cardiol. 2005;46(5):827–34.
Tanaka Y, Matsumoto I, Iwanami K, Inoue A, Minami R, Umeda N, Kanamori A, Ochiai N, Miyazawa K, Sugihara M, et al. Six-transmembrane epithelial antigen of prostate4 (STEAP4) is a tumor necrosis factor alpha-induced protein that regulates IL-6, IL-8, and cell proliferation in synovium from patients with rheumatoid arthritis. Mod Rheumatol. 2012;22(1):128–36.
Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, Schonbeck U, Libby P. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol. 2006;26(3):611–7.
Ezashi T, Das P, Roberts RM. Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci USA. 2005;102(13):4783–8.
Chen Y, Wang C, Wu J, Li L. BTG/Tob family members Tob1 and Tob2 inhibit proliferation of mouse embryonic stem cells via Id3 mRNA degradation. Biochem Biophys Res Commun. 2015;462(3):208–14.
Winkler GS. The mammalian anti-proliferative BTG/Tob protein family. J Cell Physiol. 2010;222(1):66–72.
Scheubel RJ, Holtz J, Friedrich I, Borgermann J, Kahrstedt S, Navarrete Santos A, Silber RE, Simm A. Paracrine effects of CD34 progenitor cells on angiogenic endothelial sprouting. Int J Cardiol. 2010;139(2):134–41.
Majka M, Janowska-Wieczorek A, Ratajczak J, Ehrenman K, Pietrzkowski Z, Kowalska MA, Gewirtz AM, Emerson SG, Ratajczak MZ. Numerous growth factors, cytokines, and chemokines are secreted by human CD34(+) cells, myeloblasts, erythroblasts, and megakaryoblasts and regulate normal hematopoiesis in an autocrine/paracrine manner. Blood. 2001;97(10):3075–85.
Kwon SM, Lee JH, Lee SH, Jung SY, Kim DY, Kang SH, Yoo SY, Hong JK, Park JH, Kim JH, et al. Cross talk with hematopoietic cells regulates the endothelial progenitor cell differentiation of CD34 positive cells. PLoS One. 2014;9(8):e106310.
Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, Lewis CE. Hypoxia-induced gene expression in human macrophages: implications for ischemic tissues and hypoxia-regulated gene therapy. Am J Pathol. 2003;163(4):1233–43.
Authors’ contributions
SB and JUW conceptualized the study. SB, FWA, MG, HJS, AB, SK, and JUW collected the cord blood from subjects, performed experiments and analyzed data. MHA, AMA, and MAG. provided scientific advice. SB, FA, HJS, SL, ALA and JUW wrote the manuscript. AGC and JUW corrected the final version of the manuscript. JUW is the guarantor of this work and had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to show great appreciation for Prof. Simi Ali, Institute of Cellular Medicine, Newcastle University, UK, Prof. Dianne Ford, Institute of Cell and Molecular Biosciences, Newcastle University, UK, and Prof. Stefan Przyborski, School of Biological and Biomedical Sciences, Durham University, UK, for their valuable comments on the manuscript. Additionally, the authors thank the midwives at Queen Elizabeth Hospital Gateshead, UK and Mrs. Manar M Ata, Microarray Service Unit at CEGMR, King Abdulaziz University, Saudi Arabia.
Competing interests
The authors declare that they have no competing interests.
Funding
This study was funded by the Diabetes Research and Wellness Foundation, UK (open funding 2011), Newcastle University summer scholarship, Joint Supervision Program, King Abdulaziz University, Jeddah 21589, Saudi Arabia and King Abdulaziz City for Science and Technology, Riyadh 11442, Saudi Arabia (GSP-32-68).
Author information
Authors and Affiliations
Corresponding author
Additional file
12933_2016_344_MOESM1_ESM.pdf
Additional file 1: Figure S1. Cell viability assay on HUVEC treated with sunitinib. Figure S2. Effect of metformin on euglycemia-hypoxia treated CD34+ cells. Figure S3. Validation of selected pro-angiogenic factors and angiogenic inhibitors in CD34+ cells by qRT-PCR. Figure S4. Heatmap of the pro-angiogenic and angiogenic inhibitors in CD34+ stem cells. Table S1. Effect of 3 h hypoxia on biological functions involved in CD34+ cells. Table S2. Top 30 differentially expressed genes in CD34+ cells induced by hyperglycemia compared to control. Table S3. Top biological functions involved in CD34+ cells cultured in hyperglycemia. Table S4. Top canonical pathways involved in CD34+ cells cultured in hyperglycemia. Table S5. Top 30 differentially expressed genes in CD34+ cells induced by hyperglycemia and treated with metformin compared to hyperglycemia. Table S6. Effect of metformin on canonical pathways involved in CD34+ cells cultured in combined hyperglycemia-hypoxia for 3 h. Table S7. Comparison of microarray and real-time PCR data on CD34+ cells treated with hypoxia, hyperglycemia and hyperglycemia-hypoxia in the presence and absence of metformin.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bakhashab, S., Ahmed, F.W., Schulten, HJ. et al. Metformin improves the angiogenic potential of human CD34+ cells co-incident with downregulating CXCL10 and TIMP1 gene expression and increasing VEGFA under hyperglycemia and hypoxia within a therapeutic window for myocardial infarction. Cardiovasc Diabetol 15, 27 (2016). https://doi.org/10.1186/s12933-016-0344-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-016-0344-2