
Abstract
Background
To investigate the long-term efficacy and safety of empagliflozin monotherapy compared with placebo and sitagliptin in drug-naïve patients with type 2 diabetes mellitus.
Methods
Of 899 patients randomized to receive empagliflozin 10 mg, empagliflozin 25 mg, placebo, or sitagliptin 100 mg once daily for 24 weeks, 615 continued in a double-blind extension trial for ≥52 weeks. Exploratory endpoints included changes from baseline in HbA1c, weight and blood pressure at week 76.
Results
Compared with placebo, adjusted mean changes from baseline in HbA1c at week 76 were −0.78 % (95 % CI −0.94, −0.63; p < 0.001) and −0.89 % (95 % CI −1.04, −0.73; p < 0.001) for empagliflozin 10 mg and 25 mg, respectively. Compared with placebo, adjusted mean changes from baseline in weight at week 76 were −1.8 kg (95 % CI −2.4, −1.3; p < 0.001) and −2.0 kg (95 % CI −2.6, −1.5; p < 0.001) for empagliflozin 10 mg and 25 mg, respectively. Empagliflozin led to reductions in systolic blood pressure (SBP) compared with placebo in the primary analysis but not in sensitivity analyses. Compared with sitagliptin, empagliflozin 25 mg reduced HbA1c and both empagliflozin doses reduced weight and SBP. Adverse events (AEs) were reported in 76.8, 78.0, 76.4 and 72.2 % of patients on empagliflozin 10 mg, empagliflozin 25 mg, placebo and sitagliptin, respectively. Confirmed hypoglycaemic AEs (glucose ≤3.9 mmol/l and/or requiring assistance) were reported in two patients (0.9 %) per treatment group.
Conclusions
Empagliflozin monotherapy for ≥76 weeks was well tolerated and led to sustained reductions in HbA1c and weight compared with placebo.
Trial registration: clinicaltrials.gov NCT01289990
Similar content being viewed by others


Background
Inhibition of the sodium glucose cotransporter 2 (SGLT2), located in the proximal tubule of the kidney, leads to increased urinary glucose excretion (UGE) and a reduction in plasma glucose levels in patients with type 2 diabetes mellitus (T2DM) [1–4]. This mechanism of action is associated with a low risk of hypoglycaemia, with additional benefits of weight loss and reductions in blood pressure [1, 5, 6].
Empagliflozin is a potent and selective SGLT2 inhibitor [7], which, when given as monotherapy or as add-on therapy for T2DM, has consistently reduced HbA1c, weight and systolic blood pressure (SBP) compared with placebo [8–16]. As well as reducing fasting plasma glucose (FPG), empagliflozin reduces post-prandial glucose in patients with T2DM [17].
In a Phase III, parallel-group, randomized, double-blind trial in drug-naïve patients with T2DM (EMPA-REG MONO™), empagliflozin 10 mg and 25 mg and the dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin 100 mg given as monotherapy for 24 weeks were well tolerated and improved glycaemic control. Adjusted mean differences versus placebo in change from baseline in haemoglobin A1c (HbA1c) at week 24 were −0.74 % for empagliflozin 10 mg and −0.85 % for empagliflozin 25 mg, with no significant difference in change from baseline in HbA1c between empagliflozin and sitagliptin. Treatment with empagliflozin also significantly reduced body weight and SBP compared with placebo and sitagliptin [9].
This 52-week extension to the above study, EMPA-REG EXTEND™ MONO, evaluated the long-term safety, tolerability and efficacy of empagliflozin 10 mg and 25 mg compared with placebo and sitagliptin as monotherapy in patients with T2DM.
Methods
Study design
In the initial 24-week study, drug-naïve patients with T2DM (no oral or injectable anti-diabetes therapy for ≥12 weeks prior to randomization) with insufficient glycaemic control despite a diet and exercise regimen (HbA1c ≥7 to ≤10 %, or HbA1c ≥7 to ≤9 % in Germany) and body mass index ≤45 kg/m2 were enrolled. Key exclusion criteria included uncontrolled hyperglycaemia (glucose concentration >13.3 mmol/l following an overnight fast, confirmed by a second measurement), an estimated glomerular filtration rate (eGFR) [Modification of Diet in Renal Disease (MDRD) formula] <50 ml/min/1.73 m2, indication of liver disease, and contraindications to sitagliptin according to the local label [9].
In the initial study, patients were randomized (1:1:1:1) to receive empagliflozin 10 mg, empagliflozin 25 mg, sitagliptin 100 mg, or placebo once daily for 24 weeks. Patients who completed 24 weeks’ treatment, who still did not contravene the exclusion criteria for the initial study, and who did not contravene additional exclusion criteria for the extension study, e.g., eGFR <30 ml/min at the last visit of the initial trial, could decide to continue their double-blind treatment for ≥52 weeks (i.e., a total treatment duration of ≥76 weeks). Patients remained on the treatments they received in the initial study, but were required to re-confirm their consent before starting the extension trial. The initial trial and the extension trial were registered with clinicaltrials.gov (NCT01177813 and NCT01289990, respectively) and were carried out in compliance with the protocols and the principles of the Declaration of Helsinki, and in accordance with the International Conference on Harmonization Harmonized Tripartite Guideline for Good Clinical Practice. The studies were approved by Institutional Review Boards, Independent Ethics Committees and Competent Authorities according to national and international regulations.
During the extension trial, patients continued to receive diet and exercise counselling based on local recommendations. Patients who received rescue medication during the initial 24-week study and were still receiving it at the start of the extension study were to continue their rescue medication throughout the extension study. Rescue medication could be initiated during the extension trial if a patient had a confirmed plasma glucose level >10 mmol/l after an overnight fast or HbA1c >8 %. The choice and dose of rescue medication were at the discretion of the investigator, except that DPP-4 inhibitors and glucagon-like peptide-1 analogues were not permitted. In cases of hypoglycaemia, dose reduction or discontinuation of rescue medication was to be initiated. If hyper- or hypoglycaemia could not be controlled, the patient was to be discontinued from the trial.
Endpoints and assessments
The primary efficacy endpoint of change from baseline in HbA1c at week 24 was analyzed in the initial trial [9]. No primary endpoint was defined for the extension study. Exploratory efficacy endpoints in the extension trial were change from baseline in HbA1c, FPG, body weight, SBP and diastolic blood pressure (DBP) at week 52 and week 76. Other exploratory endpoints were the percentage of patients who were treated in the extension trial and had HbA1c ≥7 % at baseline who reached HbA1c <7 % at week 76 and the use of rescue therapy over 76 weeks. Baseline was defined as the last observed measurement before the first administration of study drug in the initial trial.
Safety was assessed through the reporting of adverse events [AEs; coded using the Medical Dictionary for Drug Regulatory Activities (MedDRA), version 16.0]. AEs of special interest included confirmed hypoglycaemic events (plasma glucose ≤3.9 mmol/l and/or requiring assistance) and AEs consistent with urinary tract infection (UTI), genital infection and volume depletion, which were identified using prospectively defined search categories based on 77, 89 and 8 MedDRA preferred terms, respectively. Changes from baseline in clinical laboratory values at week 76 were also assessed.
Statistical analyses
No formal sample size calculation was performed for the extension trial; the extension trial was open to all patients who were eligible to participate.
Changes from baseline in HbA1c, FPG, weight, SBP and DBP at week 52 and 76 were analyzed using an analysis of covariance (ANCOVA) model in the full analysis set (FAS: patients who received ≥1 dose of study drug and had a baseline HbA1c measurement in the initial study) with baseline HbA1c and the baseline value of the endpoint in question as linear covariates, and baseline eGFR (MDRD), region and treatment as fixed effects. Data following initiation of rescue therapy were set to missing and missing data were imputed using the last observation carried forward (LOCF) approach. The percentage of patients reaching HbA1c <7 % at week 76 was assessed in patients from the FAS who were treated in the extension trial using a logistic regression model that included treatment, baseline eGFR, region and baseline HbA1c, with non-completers considered failures (patients who did not enter the extension trial were not considered non-completers).
Changes over time in HbA1c, FPG, weight, SBP and DBP were analyzed by means of sensitivity analyses using a restricted maximum likelihood-based mixed model repeated measures (MMRM) approach with baseline HbA1c and the baseline value of the endpoint in question as linear covariates, and baseline eGFR, region, treatment, visit, and visit by treatment interaction as fixed effects, based on observed cases (OC) in the FAS and in FAS-completers. The FAS-completers set comprised patients from the FAS who completed 76 ± 1 weeks’ treatment and had an HbA1c measurement at the week 76 visit. The use of rescue therapy was assessed in the FAS using logistic regression, including treatment as a factor and baseline HbA1c as a covariate.
Safety was assessed in the treated set (patients treated with ≥1 dose of study drug in the initial study) and analyses were descriptive, except for changes from baseline in lipid parameters, which were assessed using ANCOVA with the baseline value and baseline HbA1c as linear covariates, and baseline eGFR and treatment as fixed effects.
Results
Patient disposition and characteristics
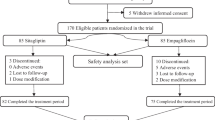
The FAS comprised 899 patients. The demographic and baseline characteristics of the FAS were balanced across treatment groups and are summarized in Table 1. Of these 899 patients, 615 (68.4 %) continued in the extension study (Fig. 1). The baseline characteristics of the patients who continued in the extension trial were comparable with the baseline characteristics of the overall patient population treated in the initial 24-week study.

Study flow
Efficacy
Reductions in HbA1c at week 76 were greater with both doses of empagliflozin compared with placebo. Differences of adjusted means versus placebo were −0.78 % [95 % confidence interval (CI) −0.94, −0.63] with empagliflozin 10 mg and −0.89 % (−1.04, −0.73) with empagliflozin 25 mg; p < 0.001 for both doses (Table 2). Compared with sitagliptin, adjusted mean changes from baseline in HbA1c at week 76 were greater for empagliflozin 25 mg (differences of adjusted means −0.22 %, 95 % CI −0.38, −0.07, p = 0.005), but not for empagliflozin 10 mg (Table 2). Adjusted mean HbA1c values over 76 weeks are presented in Fig. 2a. The results of sensitivity analyses based on MMRM OC analyses in the FAS and FAS-completers were consistent with those from the ANCOVA LOCF analysis in the FAS (Additional file 1). The proportion of patients treated in the extension trial who had HbA1c ≥7 % at baseline who reached HbA1c <7 % at week 76 was greater with both empagliflozin doses compared with placebo and with empagliflozin 10 mg compared with sitagliptin (Fig. 2b).

Glycaemic control a HbA1c over time [mixed model repeated measures (MMRM) in the full analysis set (FAS), observed cases (OC)]. b Patients with HbA1c ≥7 % at baseline who had HbA1c <7 % at week 76 (logistic regression in patients from the FAS treated in the extension trial using non-completers considered failures approach). c Change from baseline in FPG over time (MMRM in the FAS, OC). Empagliflozin 10 mg versus placebo odds ratio 4.17 (95 % CI 2.31, 7.51); empagliflozin 25 mg versus placebo odds ratio 3.96 (95 % CI 2.20, 7.14); sitagliptin versus placebo odds ratio 2.42 (95 % CI 1.32, 4.43). Empagliflozin 10 mg versus sitagliptin odds ratio 1.72 (95 % CI 1.04, 2.86); empagliflozin 25 mg versus sitagliptin odds ratio 1.64 (95 % CI 0.99, 2.72)
At week 76, reductions in FPG were greater for both empagliflozin doses compared with placebo or sitagliptin (Table 2). Differences of adjusted means versus placebo were −1.8 mmol/l (95 % CI −2.1, −1.4) with empagliflozin 10 mg and −1.9 mmol/l (95 % CI −2.3, −1.6) with empagliflozin 25 mg; p < 0.001 for both doses. Differences of adjusted means versus sitagliptin were −0.9 mmol/l (95 % CI −1.2, −0.5) with empagliflozin 10 mg and −1.0 mmol/l (95 % CI −1.4, −0.7) with empagliflozin 25 mg; p < 0.001 for both doses. Figure 2c shows the adjusted mean changes from baseline in FPG over the 76-week treatment period. The results of sensitivity analyses based on MMRM OC in the FAS and FAS-completers were consistent with findings from the ANCOVA LOCF analysis in the FAS (Additional file 1).
The proportions of patients who received rescue medication up to week 76 were 32.5 % for placebo versus 12.1 % for empagliflozin 10 mg [odds ratio (OR) 0.25, 95 % CI 0.15, 0.41, p < 0.001], 5.8 % for empagliflozin 25 mg (OR 0.11, 95 % CI 0.05, 0.20, p < 0.001) and 15.7 % for sitagliptin (OR 0.37, 95 % CI 0.23, 0.60, p < 0.001). When compared with sitagliptin, ORs were 0.67 (95 % CI 0.38, 1.18, p = 0.163) for empagliflozin 10 mg and 0.29 (95 % CI 0.14, 0.57, p < 0.001) for empagliflozin 25 mg.
At week 76, empagliflozin 10 mg and 25 mg resulted in a decrease in weight compared with placebo or sitagliptin (Table 2). Differences of adjusted means versus placebo were −1.8 kg (95 % CI −2.4, −1.3) with empagliflozin 10 mg and −2.0 kg (95 % CI −2.6, −1.5) with empagliflozin 25 mg; p < 0.001 for both doses. Differences of adjusted means versus sitagliptin were −2.3 kg (95 % CI −2.9, −1.8) with empagliflozin 10 mg and −2.6 kg (95 % CI −3.1, −2.0) with empagliflozin 25 mg; p < 0.001 for both doses. Adjusted mean changes from baseline in weight over the 76-week trial period are shown in Fig. 3. The results of sensitivity analyses based on MMRM OC (FAS and FAS-completers) were consistent with those from the ANCOVA LOCF analysis in the FAS (Additional Table 1).

Body weight Change from baseline in body weight over time [mixed model repeated measures (MMRM) in the full analysis set (FAS), observed cases (OC)]
Significant reductions in SBP were noted for empagliflozin 10 mg and 25 mg compared with placebo and sitagliptin at week 76 (Table 2). Differences of adjusted means versus placebo were −3.4 mmHg (95 % CI −5.5, −1.2) with empagliflozin 10 mg (p = 0.003) and −3.4 mmHg (95 % CI −5.6, −1.2) with empagliflozin 25 mg (p = 0.002). Differences of adjusted means versus sitagliptin were −3.7 mmHg (95 % CI −5.9, −1.6) with empagliflozin 10 mg and −3.8 mmHg (95 % CI −6.0, −1.6) with empagliflozin 25 mg; p = 0.001 for both doses. Adjusted mean changes from baseline in SBP up to week 76 are presented in Additional file 2. Sensitivity analyses based on MMRM OC in the FAS and FAS-completers showed no difference in adjusted mean change from baseline in SBP between empagliflozin 10 mg or empagliflozin 25 mg and placebo at week 76, but significant differences between both doses of empagliflozin and sitagliptin (Additional file 1).
At week 76, the reductions in DBP with empagliflozin 10 mg and 25 mg were not significantly different compared with placebo, but a significant decrease was noted for both doses when compared with sitagliptin (Table 2). Adjusted mean changes in DBP up to week 76 are presented in Additional file 3. Sensitivity analyses based on MMRM OC in the FAS and FAS-completers showed no difference in adjusted mean change from baseline in DBP for either empagliflozin dose compared with placebo or for empagliflozin 25 mg compared with sitagliptin, but showed a significant difference between empagliflozin 10 mg and sitagliptin at week 76 (Additional file 1).
No clinically meaningful changes in pulse rate were observed. Mean (standard deviation) changes from baseline in pulse rate were −0.2 (9.9), −0.5 (9.0), −1.0 (8.4) and −0.3 (10.1) beats per minute with placebo, empagliflozin 10 mg, empagliflozin 25 mg and sitagliptin, respectively.
The results of exploratory efficacy analyses at week 52 were consistent with those observed at week 76, except that DBP was significantly reduced with empagliflozin 25 mg compared with placebo but not significantly reduced with empagliflozin 10 mg compared with sitagliptin (Additional file 4).
Safety and tolerability
Median exposure was 17.3, 20.2, 20.2 and 18.1 months in the placebo, empagliflozin 10 mg, empagliflozin 25 mg and sitagliptin groups, respectively. Safety findings are presented in Table 3. The proportion of patients with ≥1 adverse event was similar across the treatment groups. A lower percentage of patients on empagliflozin 10 mg, empagliflozin 25 mg or sitagliptin had AEs leading to discontinuation (4.9, 4.0 and 4.9 %, respectively) compared with placebo (6.6 %). Two patients (0.9 %) per treatment group had confirmed hypoglycaemic AEs; one patient on empagliflozin 10 mg required assistance. Events consistent with UTI were reported in a similar proportion of patients in each treatment group (10.9, 9.4, 9.0, and 9.0 % on placebo, empagliflozin 10 mg, empagliflozin 25 mg, and sitagliptin, respectively) and in a higher proportion of female than male patients. All events consistent with UTI were mild or moderate in intensity except in one patient on empagliflozin 25 mg and one patient on sitagliptin. Events consistent with genital infection were reported in a higher proportion of patients on empagliflozin 10 and 25 mg (5.8 and 6.3 %, respectively) than placebo and sitagliptin (1.7 and 0.9 %, respectively). All events consistent with genital infection were mild or moderate in intensity. Events consistent with volume depletion were reported in 0.4, 2.7, 0.9 and 1.3 % of patients on placebo, empagliflozin 10 mg, empagliflozin 25 mg and sitagliptin, respectively. There was one death in the placebo group and one death in the sitagliptin group (sudden death), which was not regarded by the investigator as related to study drug.
Small increases in haematocrit and eGFR and small decreases in serum uric acid were observed in the empagliflozin groups (Additional file 5). Compared with placebo, there was an increase from baseline in low density lipoprotein (LDL) cholesterol and total cholesterol in patients treated with empagliflozin 25 mg and in high density lipoprotein (HDL) cholesterol in both empagliflozin dose groups. No differences versus placebo in change from baseline in triglycerides or LDL/HDL cholesterol ratio were noted for either empagliflozin dose (Table 4).
Discussion
This extension study showed that treatment with empagliflozin 10 mg or 25 mg or sitagliptin for 76 weeks led to sustained improvements in glycaemic control compared with placebo. When compared with sitagliptin, empagliflozin 25 mg reduced HbA1c, and both empagliflozin doses reduced FPG. Furthermore, empagliflozin 10 mg and 25 mg led to sustained weight loss compared with placebo or sitagliptin. The weight loss observed after 76 weeks’ treatment with empagliflozin is clinically meaningful as weight management remains a major challenge in the treatment of patients with T2DM [18] and weight gain with glucose-lowering medication is a concern for the majority of patients [19, 20]. A study of empagliflozin 25 mg given as add-on to metformin for 104 weeks found that nearly 90 % of the weight loss with empagliflozin was due to a reduction in fat mass, and that empagliflozin reduced both abdominal visceral adipose tissue and abdominal subcutaneous adipose tissue [13]. The initial weight loss seen with SGLT2 inhibitors may be due to the mild osmotic effects associated with UGE, but sustained weight loss is believed to result from loss of calories through UGE.
In previous Phase III studies, empagliflozin reduced SBP compared with placebo [8–12, 15, 21]. In this study, empagliflozin showed a reduction in SBP compared with placebo and sitagliptin at week 76 based on ANCOVA analyses with LOCF imputation, but no reductions with empagliflozin compared with placebo across the sensitivity analyses (based on MMRM, OC). This discrepant result may be explained by the higher rate of early discontinuation and the greater need for rescue therapy in the placebo group.
The sustained effects of empagliflozin on glycaemic control and weight in this extension trial were consistent with the results of a study in which patients received empagliflozin monotherapy for 90 weeks [22]. The effects of sitagliptin on glycaemic control and weight in this study were comparable with those observed in previous studies of sitagliptin monotherapy in drug-naïve patients with T2DM [23–25].
Recently published results of the EMPA-REG OUTCOME® trial have shown that in patients with type 2 diabetes and high cardiovascular risk (i.e., established cardiovascular disease), empagliflozin added to standard of care reduced the primary composite outcome of cardiovascular death, non-fatal myocardial infarction or non-fatal stroke (3-point major adverse cardiovascular events); cardiovascular death; hospitalisation for heart failure; and overall mortality compared to placebo [21, 26]. Although the mechanisms behind the observed effects of empagliflozin in this patient population are not yet understood, they may involve reductions in hyperglycaemia, blood pressure and weight as well as effects on plasma volume and sodium retention [27]; and reductions in arterial stiffness [28, 29].
Both doses of empagliflozin and sitagliptin were well tolerated. In accordance with its insulin-independent mode of action [30], and the results of previous studies, empagliflozin monotherapy was not associated with an increased risk of hypoglycaemia. In line with previous trials of SGLT2 inhibitors, events consistent with genital infection were reported in a greater proportion of patients treated with empagliflozin than with placebo, and were more common in female than male patients [31]. Consistent with previous studies of empagliflozin [9–12, 21], there was no higher risk of UTI in patients treated with empagliflozin in this study.
Strengths of the design of this study include the long duration, that treatment remained double-blind throughout the extension period and the inclusion of a sitagliptin group as an active comparator. Limitations of this study include that all endpoints, while pre-specified, were exploratory, with no primary endpoint defined for the extension study, and the amount of missing data. Only 68 % of patients randomized in the initial 24-week study entered the extension period; however, this is within the range for extension studies of other SGLT2 inhibitors [32–34]. Data obtained after initiation of rescue therapy (5.8–32.5 % across treatment groups) were set to missing and imputed. Overall, 51.4 % of HbA1c data at week 76 were imputed using a LOCF approach. The methods for handling missing data were analyzed by means of sensitivity analyses, which revealed consistent results regarding improvements in glycaemic control and weight loss with empagliflozin. A further limitation of this study was that the results may not be generalizable to all patients with T2DM, as for example, approximately two-thirds of the patients in the trial were Asian.
In conclusion, results from this extension study indicate that empagliflozin 10 mg and 25 mg given as monotherapy to drug-naïve patients with T2DM leads to sustained improvements in glycaemic control and reductions in weight compared with placebo and sustained reductions in HbA1c (for empagliflozin 25 mg), weight and SBP compared with sitagliptin over 76 weeks.
Abbreviations
- AE:
-
adverse events
- ANCOVA:
-
analysis of covariance
- CI:
-
confidence interval
- DBP:
-
diastolic blood pressure
- DPP-4:
-
dipeptidyl peptidase-4
- FAS:
-
full analysis set
- FPG:
-
fasting plasma glucose
- eGFR:
-
estimated glomerular filtration rate
- HbA1c:
-
haemoglobin A1c
- HDL:
-
high density lipoprotein
- LDL:
-
low density lipoprotein
- LOCF:
-
last observation carried forward
- MDRD:
-
modification of diet in renal disease formula
- MedDRA:
-
Medical Dictionary for Drug Regulatory Activities
- MMRM:
-
mixed model repeated measures
- OC:
-
observed cases
- OR:
-
odds ratio
- SGLT2:
-
sodium glucose cotransporter 2
- SE:
-
standard error
- SBP:
-
systolic blood pressure
- T2DM:
-
type 2 diabetes mellitus
- UGE:
-
urinary glucose excretion
- UTI:
-
urinary tract infection
References
DeFronzo RA, Davidson JA, del Prato S. The role of the kidneys in glucose homeostasis: a new path towards normalizing glycaemia. Diabetes Obes Metab. 2012;14:5–14.
Gallo LA, Wright EM, Vallon V. Probing SGLT2 as a therapeutic target for diabetes: basic physiology and consequences. Diab Vasc Dis Res. 2015;12:78–89.
Liu XY, Zhang N, Chen R, Zhao JG, Yu P. Efficacy and safety of sodium-glucose cotransporter 2 inhibitors in type 2 diabetes: a meta-analysis of randomized controlled trials for 1 to 2 years. J Diabetes Complicat. 2015;29:1295–303.
Michel MC, Mayoux E, Vallon V. A comprehensive review of the pharmacodynamics of the SGLT2 inhibitor empagliflozin in animals and humans. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:801–16.
Baker WL, Smyth LR, Riche DM, Bourret EM, Chamberlin KW, White WB. Effects of sodium-glucose co-transporter 2 inhibitors on blood pressure: a systematic review and meta-analysis. J Am Soc Hypertens. 2014;8(262–75):e9.
Hedrington MS, Davis SN. The role of empagliflozin in the management of type 2 diabetes by patient profile. Ther Clin Risk Manag. 2015;11:739–49.
Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14:83–90.
Haering H-U, Merker L, Seewaldt-Becker E, Weimer M, Meinicke T, Woerle HJ, et al. Empagliflozin as add-on to metformin plus sulfonylurea in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2013;36:3396–404.
Roden M, Weng J, Eilbracht J, Delafont B, Kim G, Woerle HJ, et al. Empagliflozin monotherapy in drug-naive patients with type 2 diabetes: a randomised, 24-week, double-blind, placebo-controlled, parallel group, trial with sitagliptin as active comparator. Lancet Diabetes Endocrinol. 2013;1:208–19.
Kovacs CS, Seshiah V, Swallow R, Jones R, Rattunde H, Woerle HJ, et al. Empagliflozin improves glycaemic and weight control as add-on therapy to pioglitazone or pioglitazone plus metformin in patients with type 2 diabetes: a 24-week, randomized, placebo-controlled trial. Diabetes Obes Metab. 2014;16:147–58.
Haering H-U, Merker L, Seewaldt-Becker E, Weimer M, Meinicke T, Broedl UC, et al. Empagliflozin as add-on to metformin in patients with type 2 diabetes: a 24-week, randomized, double-blind, placebo-controlled trial. Diabetes Care. 2014;37:1650–9.
Barnett AH, Mithal A, Manassie J, Jones R, Rattunde H, Woerle HJ, et al. Efficacy and safety of empagliflozin added to existing antidiabetes treatment in patients with type 2 diabetes and chronic kidney disease: a randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2:369–84.
Ridderstrale M, Andersen KR, Zeller C, Kim G, Woerle HJ, Broedl UC, et al. Comparison of empagliflozin and glimepiride as add-on to metformin in patients with type 2 diabetes: a 104-week randomised, active-controlled, double-blind, phase 3 trial. Lancet Diabetes Endocrinol. 2014;2:691–700.
Rosenstock J, Jelaska A, Frappin G, Salsali A, Kim G, Woerle HJ, et al. Improved glucose control with weight loss, lower insulin doses, and no increased hypoglycemia with empagliflozin added to titrated multiple daily injections of insulin in obese inadequately controlled type 2 diabetes. Diabetes Care. 2014;37:1815–23.
Tikkanen I, Narko K, Zeller C, Green A, Salsali A, Broedl UC, et al. Empagliflozin reduces blood pressure in patients with type 2 diabetes and hypertension. Diabetes Care. 2015;38:420–8.
Rosenstock J, Jelaska A, Zeller C, Kim G, Broedl UC, Woerle HJ. Impact of empagliflozin added on to basal insulin in type 2 diabetes inadequately controlled on basal insulin: a 78-week randomized, double-blind, placebo-controlled trial. Diabetes Obes Metab. 2015;17:936–48.
Nishimura R, Tanaka Y, Koiwai K, Inoue K, Hach T, Salsali A, Lund SS, Broedl UC. Effect of empagliflozin monotherapy on postprandial glucose and 24-hour glucose variability in Japanese patients with type 2 diabetes mellitus: a randomized, double-blind, placebo-controlled, 4-week study. Cardiovasc Diabetol. 2015;14:11.
Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, et al. Management of hyperglycaemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2015;38:140–9.
Lund A, Knop FK. Worry vs. knowledge about treatment-associated hypoglycaemia and weight gain in type 2 diabetic patients on metformin and/or sulphonylurea. Curr Med Res Opin. 2012;28:731–6.
Hauber AB, Mohamed AF, Johnson FR, Falvey H. Treatment preferences and medication adherence of people with type 2 diabetes using oral glucose-lowering agents. Diabet Med. 2009;26:416–24.
Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–28.
Ferrannini E, Berk A, Hantel S, Pinnetti S, Hach T, Woerle HJ, et al. Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care. 2013;36:4015–21.
Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE, et al. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care. 2006;29:2632–7.
Raz I, Hanefeld M, Xu L, Caria C, Williams-Herman D, Khatami H, et al. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia. 2006;49:2564–71.
Aschner P, Katzeff HL, Guo H, Sunga S, Williams-Herman D, Kaufman KD, et al. Efficacy and safety of monotherapy of sitagliptin compared with metformin in patients with type 2 diabetes. Diabetes Obes Metab. 2010;12:252–61.
Zinman B, Inzucchi SE, Lachin JM, Wanner C, Ferrari R, Fitchett D, et al. Rationale, design, and baseline characteristics of a randomized, placebo-controlled cardiovascular outcome trial of empagliflozin (EMPA-REG OUTCOME™). Cardiovasc Diabetol. 2014;13:102.
Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. 2014;129:587–97.
Chilton R, Tikkanen I, Cannon CP, Crowe S, Woerle HJ, Broedl UC, Johansen OE. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes Metab. 2015;17:1180–93.
Lin B, Koibuchi N, Hasegawa Y, Sueta D, Toyama K, Uekawa K, et al. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc Diabetol. 2014;13:148.
Ferrannini E, Muscelli E, Frascerra S, Baldi S, Mari A, Heise T, et al. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest. 2014;124:499–508.
Hasan FM, Alsahli M, Gerich JE. SGLT2 inhibitors in the treatment of type 2 diabetes. Diabetes Res Clin Pract. 2014;104:297–322.
Bailey CJ, Gross JL, Hennicken D, Iqbal N, Mansfield TA, List JF. Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med. 2013;11:43.
Bolinder J, Ljunggren Ö, Johansson L, Wilding J, Langkilde AM, Sjöström CD, et al. Dapagliflozin maintains glycaemic control while reducing weight and body fat mass over 2 years in patients with type 2 diabetes mellitus inadequately controlled on metformin. Diabetes Obes Metab. 2014;16:159–69.
Stenlöf K, Cefalu WT, Kim KA, Jodar E, Alba M, Edwards R, et al. Long-term efficacy and safety of canagliflozin monotherapy in patients with type 2 diabetes inadequately controlled with diet and exercise: findings from the 52-week CANTATA-M study. Curr Med Res Opin. 2014;30:163–75.
Authors’ contributions
MR and LM contributed to the acquisition and interpretation of data and writing of the manuscript. AVC, FR, AS, GK, PS, HJW and UCB contributed to the study design, interpretation of data and writing of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The studies reported here were funded by Boehringer Ingelheim and Eli Lilly and Company. Boehringer Ingelheim’s involvement included study design, data collection and data analysis. Eli Lilly and Company’s involvement was limited to co-funding of the study. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Julie Fleming and Wendy Morris of Fleishman-Hillard Group, Ltd, London, UK, during the preparation of this manuscript. The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development and have approved the final version.
Competing interests
MR has served on scientific advisory boards and/or received speaker’s honoraria for AstraZeneca, Boehringer Ingelheim, Eli Lilly and Company, Genentech and Novo Nordisk. He has been involved with clinical trial research for Boehringer Ingelheim, Novartis, Poxel and Sanofi-Aventis. LM has been a consultant and/or speaker for AstraZeneca, Boehringer Ingelheim, Bristol–Myers Squibb, Eli Lilly and Company, Merck Sharp & Dohme and Novo Nordisk without any direct financial benefit. AVC, FR, AS, GK, PS, HJW and UCB are all employees of Boehringer Ingelheim.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional files
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Roden, M., Merker, L., Christiansen, A.V. et al. Safety, tolerability and effects on cardiometabolic risk factors of empagliflozin monotherapy in drug-naïve patients with type 2 diabetes: a double-blind extension of a Phase III randomized controlled trial. Cardiovasc Diabetol 14, 154 (2015). https://doi.org/10.1186/s12933-015-0314-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-015-0314-0