
Abstract
Background
Acute hyperglycemia co-presenting with myocardial infarction (in diabetic and non-diabetic individuals) is often associated with a poor prognosis. Although acute hyperglycemia induces oxidative stress that can lead to dysregulation of the ubiquitin–proteasome system (UPS), it is unclear whether increased/decreased UPS is detrimental with ischemia–reperfusion under such conditions. As our earlier data implicated the UPS in cardiac damage, we hypothesized that its inhibition results in cardioprotection with ischemia–reperfusion performed under conditions that simulate acute hyperglycemia.
Methods
Ex vivo rat heart perfusions were performed with Krebs–Henseleit buffer containing 33 mM glucose vs. controls (11 mM glucose) for 60 min stabilization, followed by 20 min global ischemia and 60 min reperfusion ± 5 µM lactacystin and 10 µM MG-132, respectively. The UPS inhibitors were added during the first 20 min of the reperfusion phase and various cardiac functional parameters evaluated. In parallel experiments, infarct sizes were assessed following 20 min regional ischemia and 120 min reperfusion ± each of the respective UPS inhibitors (added during reperfusion). Heart tissues were collected and analyzed for markers of oxidative stress, UPS activation, inflammation and autophagy.
Results
The proteasome inhibitor doses and treatment duration here employed resulted in partial UPS inhibition during the reperfusion phase. Both lactacystin and MG-132 administration resulted in cardioprotection in our experimental system, with MG-132 showing a greater effect. The proteasome inhibitors also enhanced cardiac superoxide dismutase protein levels (SOD1, SOD2), attenuated pro-inflammatory effects and caused an upregulation of autophagic markers.
Conclusions
This study established that partial proteasome inhibition elicits cardioprotection in hearts exposed to ischemia–reperfusion with acute simulated hyperglycemia. These data reveal that protease inhibition triggered three major protective effects, i.e. (a) enhancing myocardial anti-oxidant defenses, (b) attenuating inflammation, and (c) increasing the autophagic response. Thus the UPS emerges as a unique therapeutic target for the treatment of ischemic heart disease under such conditions.
Similar content being viewed by others

Background
The link between hyperglycemia and increased morbidity/mortality with heart diseases such as acute myocardial infarction (AMI) is receiving growing attention [1–3]. Hyperglycemia can manifest as a more chronic condition (typically found with diabetes) or as a stress-induced state, e.g. in diabetic and non-diabetic persons challenged by an AMI. Chronic hyperglycemia may sometimes be linked to the development of macrovascular complications [4], with AMI the chief contributor to cardiovascular mortality with diabetes [5–7]. Acute, stress-induced hyperglycemia is associated with increased in-hospital deaths, congestive heart failure, and cardiogenic shock [8–10]. The presentation of hyperglycemia in non-diabetic individuals likely represents a combination of undiagnosed diabetes, impaired glucose tolerance [11, 12], and a robust response to acute stress [8, 9].
Hyperglycemia-induced oxidative stress can lead to the formation of misfolded or damaged proteins that are usually eliminated by the ubiquitin–proteasome system (UPS), the major role player responsible for non-lysosomal protein degradation [13, 14]. However, a dysfunctional UPS is linked with a pro-inflammatory milieu and impaired cardiac function in diabetic mice [15] and diabetic patients presenting with unstable angina [16]. Although altered UPS functioning is implicated with diminished cardiac function, it remains unclear whether greater or lesser UPS activation is detrimental with ischemia–reperfusion under such conditions. This conundrum also applies for UPS regulation in response to ischemia–reperfusion per se, i.e. under normoglycemic conditions. For example, some found that oxidative stress, inflammation and myocardial reperfusion injury can be blunted by proteasomal inhibitor treatment [15, 16]. Furthermore, UPS inhibition leads to a reduction in ventricular tachyarrhythmias [17] and preserved cardiac function following myocardial ischemia under baseline glucose conditions. Conversely, others established that UPS inactivation (due to oxidative damage occurring with ischemia–reperfusion) is a robust contributor to the damaging effects elicited by an AMI [18]. Such discordant findings may be due to different experimental protocols/models utilized and due to variations in UPS inhibitor class, dosages and length of administration [19, 20]. With the paucity of studies investigating UPS regulation with ischemia–reperfusion under acute hyperglycemic conditions, efforts to gain greater insight into this intriguing question are essential as it will contribute to (a) the delineation of underlying mechanisms, and (b) the development of novel cardioprotective agents.
As our earlier findings implicated increased UPS activation in hyperglycemia-mediated cardiac damage [21] we hypothesized that its inhibition during early reperfusion provides cardioprotection following ischemia–reperfusion under high glucose conditions. We employed an established rat model of ex vivo isolated heart perfusions subjected to ischemia–reperfusion and exposed to simulated acute hyperglycemia as before [21, 22]. To further strengthen these data two different UPS inhibitor classes were used, i.e. lactacystin (2-(acetylamino)-3-[({3-hydroxy-2-[1-hydroxy-2-methylpropyl]-4-methyl-5-oxopyrrolidin-2-yl}carbonyl)sulfanyl]propanoic acid) and MG-132 (N-(benzyloxycarbonyl) leucinylleucinylleucinal Z-Leu-Leu-Leu-al). The current study demonstrates that proteasome inhibition elicits cardioprotection in hearts exposed to ischemia–reperfusion with acute simulated hyperglycemia by upregulating anti-oxidant defenses, enhancing anti-inflammatory effects and increasing autophagy.
Methods
Animals and ethics statement
All animals were treated in accordance with the Guide for the Care and Use of Laboratory Animals of the National Academy of Sciences (NIH publication No. 85-23, revised 1996). This study was carried out with the approval of the Animal Ethics Committee of Stellenbosch University.
Ex vivo perfusion protocol
Male Wistar rats weighing 180–220 g were used for this study. Rats were anesthetized (pentobarbitone, 100 mg/kg i.p.) and hearts rapidly excised and perfused in a modified Langendorff model as described before [21, 22]. Briefly, Krebs–Henseleit buffer containing (in mM) 11 glucose, 118 NaCl, 4.7 KCl, 1.2 MgSO4·7H2O, 2.5 CaCl2·2H2O, 1.2 KH2PO4, 25 NaHCO3 was equilibrated with 95% O2–5% CO2 (37°C, pH 7.4) at a constant pressure (100 cm). Buffer was not recirculated and the hearts were allowed to beat at their natural rate. During perfusion, a latex balloon attached to a pressure transducer (Stratham MLT 0380/D, ADInstruments Inc, Bella Vista, NSW, Australia) compatible with the PowerLab System ML410/W (ADInstruments Inc, Bella Vista, NSW, Australia) was inserted into the left ventricle via the mitral valve and inflated to produce a systolic pressure of 80–120 mmHg and a diastolic pressure of 4–12 mmHg. The temperature of the heart was maintained at 37°C by suspending it in a heated water jacket.
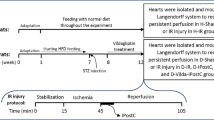
The protocol was divided into two parts, i.e. perfusions (a) with global ischemia and (b) regional ischemia, followed by reperfusion for cardiac functional assessments and infarct size determination, respectively. For each of these, hearts were randomly distributed into six experimental groups for perfusions: baseline control (11 mM glucose) ± either 5 μM lactacystin or 10 μM MG-132; and high glucose (33 mM glucose) ± either 5 μM lactacystin or 10 μM MG-132 (n = 8 rats were used for each of the experimental groups indicated). The respective UPS inhibitors (lactacystin, MG-132) were dissolved in the buffer and added for the first 20 min of reperfusion.
For our inhibition studies we aimed to achieve so-called “non-toxic proteasome inhibition’’, i.e. only partial attenuation of chymotrypsin-like activity as high and sustained doses can result in severe toxic effects with detrimental outcomes [23]. Lactacystin is a natural compound that binds mainly to the β5 proteasomal subunit (responsible for chymotrypsin-like activity) of the 20S proteasome [24], leading to irreversible inhibition of chymotrypsin-like activity (reviewed in [25]). However, the trypsin- and caspase-like activities are blunted to a lesser extent in this instance [26]. We also employed the reversible aldehyde inhibitor MG-132 that specifically binds to proteasomal subunit β5 of the 20S proteasome (reviewed in [25]). Proteasome inhibitor doses were selected to be in the range of what was employed in previously published studies [27, 28]. High glucose perfusions were used to simulate acute hyperglycemia and since ex vivo Langendorff perfusions are typically performed with 11 mM glucose at baseline, the 33 mM dose would be representative of a threefold elevation of glucose levels (above normal) within the clinical setting. Additional experiments were also performed in order to rule out the effects of osmotic pressure on heart function. Here rat hearts were perfused with 22 mM mannitol plus 11 mM glucose (total molarity = 33 mM) and subjected to ischemia–reperfusion as described above.
Ex-vivo global ischemia and reperfusion
The protocol included a 60 min stabilization period, 20 min of global ischemia, followed by 60 min of reperfusion. Contractile parameters assessed throughout the experiment included heart rate (HR), left ventricular developed pressure (LVDP), end diastolic pressure (EDP), rate-pressure product (RPP = HR × LVDP), and coronary flow. The percentage recovery for LVDP and RPP was also calculated where reperfusion data points were expressed as a percentage of pre-ischemic values. Coronary flow was measured by collection of the effluent at regular timed intervals. Both left and right ventricular tissues were immediately collected after the 60 min reperfusion period with tissues freeze-clamped in liquid nitrogen with pre-cooled Wollenberger tongs, whereafter it was stored at −80°C for further molecular and biochemical analyses.
Ex-vivo regional ischemia and reperfusion
To further strengthen our Langendorff functional data, we also evaluated the effects of lactacystin and MG-132 by infarct size determination. This was performed as described before [21, 22], and we employed regional ischemia with a reperfusion time of 2 h. Here a 3/0 silk suture was placed on the proximal portion of the left anterior descending coronary artery and passing the ends through a plastic tube. For induction of regional ischemia the ends were tightened by pressing the plastic tube against the surface of the heart (above the artery) for 20 min. The snare was released during the reperfusion period. The efficacy of ischemia was confirmed by regional cyanosis and a substantial decrease in coronary flow.
After completion of each regional ischemia–reperfusion experiment the snare was re-tightened and 2.5% Evans blue dye (in Krebs buffer) was perfused through the hearts for identification of the area at risk of ischemia. Hearts were subsequently removed from the Langendorff apparatus, blotted dry, suspended (using suture) within 50 ml plastic tubes and frozen at −20°C for 3 days. Frozen hearts were thereafter sliced into 2 mm transverse sections and incubated with 1% 2,3,5-triphenyl tetrazolium chloride in phosphate-buffered saline for 20 min at 37°C. Slices were then fixed in 10% formalin for 24 h at room temperature before being placed between glass plates for scanning. The infarct size and the area-at-risk were determined using Image J software (v1.46p, NIH, USA) and infarct size was expressed as a percentage of the area-at-risk.
Measurement of intracellular E3 ligases, oxidative stress, inflammation and autophagic markers
Protein isolation was performed as previously described [29]. Briefly, collected heart tissues were homogenized with modified RIPA buffer, the supernatant centrifuged twice at 4,300g for 10 min at 4°C then stored at −80°C until further use. Protein expression was determined by SDS-PAGE as described previously by our laboratory [21, 29], and Western blotting performed with representative markers for: (a) E3 ligases; Muscle RING Finger 1 (MURF1; Abcam, Cambridge, MA, USA) and muscle atrophy F-box (MAFbx; Santa Cruz Biotechnologies, Santa Cruz, CA, USA); (b) oxidative stress: superoxide dismutase 1 and 2 (SOD1, SOD2; Santa Cruz Biotechnologies, Santa Cruz, CA, USA); (c) inflammation: tumor necrosis factor-alpha (TNF-α; Sigma-Aldrich, St. Louis, MO, USA), and inhibitor of nuclear factor kappa-B kinase subunit alpha (IκBα; Sigma-Aldrich, St. Louis, MO, USA); and (d) autophagy: microtubule‐associated protein 1 light chain 3 II (LC3-II; Cell Signaling, Danvers, MA, USA), and p62 (Cell Signaling, Danvers, MA, USA). Western blots were quantified by densitometric analysis and β-actin (Cell Signaling, Danvers, MA, USA) employed as a loading control as described before [21, 22].
Proteasome activity experiments
Heart tissues were cut into small slices and homogenized in 1 ml of Tris–HCl buffer (pH 7.4) using an IKA Ultra Turrax T25 homogenizer (IKA Labortechnik, Staufen, Germany) and incubated on ice for 10 min before centrifugation at 9,000g for 15 min to remove cell debris. The supernatant was employed for protein quantification using the Bradford assay as described previously [30].
The Proteasome-Glo™ 3-Substrate System (Promega, Madison, WI, USA) consists of three homogeneous bioluminescent assays that separately measure the three proteolytic activities associated with the proteasome, i.e. chymotrypsin-like (LLVY), trypsin-like (LSTR), and caspase-like (LLE) activities. The three assays differ in their ability to detect different protease activities based on their substrate components, i.e. the luminogenic substrates provided for the LLVY, LSTR, and LLE are Suc-LLVY-aminoluciferin, Z-LRR-aminoluciferin, and Z-nLPnLD-aminoluciferin, respectively. Each substrate is added to a buffer system optimized for proteasome and luciferase activities to make a Proteasome-Glo™ Reagent for a particular catalytic activity. The individual Proteasome-Glo™ Reagent is added to test samples in an “add-mix-measure” format, resulting in proteasome-induced cleavage of each particular substrate. Substrate cleavage generates a “glow-type” luminescent signal produced by the luciferase reaction that is proportional to proteasome activity.
Assays were performed with ~50 µg of protein lysate [in 25 mM Tris–HCl (pH 7.5)] together with the respective substrate—incubated together for 30 min at 37°C. Aminomethylcoumarin and β-naphthylamine luminescence was subsequently measured using a luminometer microplate reader (BMG Labtech, Ortenberg, Germany) and data were normalized to protein concentrations.
Statistical analysis
Data are presented as mean ± standard error of mean (SEM). Statistical analysis was performed by the Mann–Whitney t-test, or one-way analysis of variance (ANOVA) followed by the Tukey–Kramer post hoc test (GraphPad Prism v5, San Diego, CA, USA). Values were considered significant when p < 0.05.
Results
To rule out the possibility of attributing the effects of hyperglycemia on heart function to changes in osmolarity, separate perfusion experiments were completed with 11 mM glucose plus 22 mM mannitol (total molarity 33 mM). These data demonstrate no differences in LVDP recovery and dP/dtmax for the osmotic control group vs. hearts perfused under baseline glucose (11 mM) conditions (Fig. 1a, b). By contrast, hearts exposed to high glucose displayed lower LVDP recovery compared to the osmotic control group (p < 0.05) with no differences for dP/dtmax (Fig. 1c, d). High glucose perfusions also resulted in greater infarct sizes compared to both control groups (p < 0.01) (Fig. 1e).

High glucose-induced cardiac contractile dysfunction independent of osmotic pressure. Isolated rat hearts were perfused under high glucose conditions (33 mM glucose) vs. controls (11 mM glucose) for 60 min and thereafter subjected to 20 min of global ischemia, followed by 60 min of reperfusion. In parallel experiments, groups were similarly perfused under hyperosmotic conditions (22 mM mannitol + 11 mM glucose; 33 mM). a Left ventricular developed pressure (LVDP) (% recovery) under baseline conditions vs. mannitol control, b maximum ventricular contraction (dP/dtmax) under baseline glucose conditions vs. mannitol control, c LVDP (% recovery) under high glucose conditions vs. mannitol control, d dP/dtmax under high glucose conditions vs. mannitol control, and e Infarct size/area at risk (%) under baseline, high glucose and hyperosmotic conditions after subjecting rat hearts to regional ischemia as described before. Values are expressed as mean ± SEM (n = 6). *p < 0.05, **p < 0.01 vs. respective controls.
We next determined the effects of high glucose exposure on post-ischemic UPS activities. Here trypsin-, chymotrypsin- and caspase-like activities were significantly upregulated (Fig. 2). The effects of lactacystin and MG-132 were determined and both compounds—as expected—significantly lowered myocardial chymotrypsin-like activity (Fig. 2). To further validate the degree of UPS inhibition we also assessed peptide levels of E3 ligases (MURF1 and MAFbx) involved in the final step of the ubiquitination process. Here MAFbx expression was significantly increased under high glucose conditions, while both lactacystin and MG-132 administration decreased MURF1 and MAFbx expression following ischemia–reperfusion under high glucose conditions (Fig. 3).

Lactacystin and MG-132 administration attenuate proteasomal activities during ischemia–reperfusion. Isolated rat hearts were perfused under high glucose (33 mM) conditions vs. controls (11 mM) and subjected to global ischemia and reperfusion as earlier described. For UPS inhibition, either 5 μM lactacystin or 10 μM MG-132 was added for the first 20 min of the reperfusion period. The respective proteasomal activities evaluated: a, d trypsin-like (LSTR), b, e chymotrypsin-like (LLVY), c, f caspase-like (LLE). Values are expressed as mean ± SEM (n = 8). *p < 0.05, **p < 0.01, ***p < 0.001 vs. respective controls.

Decreased expression of E3 ubiquitin ligases during ischemia–reperfusion. Isolated rat hearts were perfused under high glucose conditions (33 mM glucose) vs. controls (11 mM glucose) for 60 min and subsequently subjected to 20 min of global ischemia, followed by 60 min of reperfusion ± either 5 μM lactacystin or 10 μM MG-132 treatment for the first 20 min of reperfusion. Western blot analysis was completed for: a muscle RING finger 1 (MuRF1) and b muscle atrophy F-box (MAFbx) with lactacystin treatment; c MURF1 and d MAFbx with MG-132 administration. We employed β-actin as loading control. Values are expressed as mean ± SEM (n ≥ 4). *p < 0.05, **p < 0.01 vs. respective controls.
We next evaluated functional parameters in our experimental system and found that high glucose exposure blunted cardiac contractile function vs. matched controls following ischemia–reperfusion (Figs. 4, 5). Acute lactacystin administration during the reperfusion phase significantly improved LVDP recovery at baseline (Fig. 4a) and also under high glucose conditions (from 10 ± 2.3% to 36 ± 4%; p < 0.01 vs. untreated high glucose) (Fig. 4c). RPP data exhibited a similar trend although lactacystin had no effect under baseline glucose conditions compared to the untreated group (Fig. 4b). MG-132 elicited robust cardioprotection (LVDP, RPP) at baseline and in response to high glucose exposure (Fig. 5). Moreover, both lactacystin and MG-132 decreased end-diastolic pressure in hearts exposed to high glucose conditions, although there were no differences for the control groups (Table 1). Proteasome inhibitor treatment also enhanced coronary flow although this was only statistically significant for MG-132 (p < 0.05 vs. untreated high glucose) (Table 1). Our comparative analyses show that MG-132 administration resulted in greater cardioprotection compared to lactacystin (Fig. 6). The regional ischemia experiments further support these findings by demonstrating that lactacystin blunted the high glucose-induced increase in the infarct size/area-at-risk from 66.4 ± 3.4% to 48 ± 3.9% (p < 0.05 vs. high glucose untreated) (Fig. 7). Likewise, MG-132 treatment decreased infarct sizes under baseline and high glucose perfusion conditions. Here MG-132 significantly attenuated the infarct size/area-at-risk to 40.3 ± 2.5% and 49.7 ± 7.4% vs. 56.3 ± 2.8% and 73.5 ± 4.9% at baseline and high glucose perfusions, respectively. There were no differences in the area-at-risk for any of the experimental groups investigated (data not shown).

Lactacystin administration improves contractile function during ischemia–reperfusion. Isolated rat hearts were perfused under high glucose conditions (33 mM glucose) vs. controls (11 mM glucose) for 60 min and thereafter subjected to 20 min of global ischemia, followed by 60 min of reperfusion. Lactacystin (5 μM) was added to treatment groups during the first 20 min of reperfusion. a Left ventricular developed pressure (LVDP) (% recovery) at baseline glucose levels (11 mM), and c with high glucose (33 mM). Rate pressure product (RPP) (% recovery) at baseline glucose levels (b), and d under high glucose conditions. Values are expressed as mean ± SEM (n = 8).*p < 0.05, **p < 0.01 vs. respective controls.

MG-132 administration improves contractile function during ischemia–reperfusion. Isolated rat hearts were perfused under high glucose conditions (33 mM glucose) vs. controls (11 mM glucose) for 60 min and thereafter subjected to 20 min of global ischemia, followed by 60 min of reperfusion. MG-132 (10 μM) was added to treatment groups during the first 20 min of reperfusion. a Left ventricular developed pressure (LVDP) (% recovery) at baseline glucose levels (11 mM), and c with high glucose (33 mM). Rate pressure product (RPP) (% recovery) at baseline glucose levels (b), and d under high glucose conditions. Values are expressed as mean ± SEM (n = 8). ***p < 0.001 vs. respective controls.

Comparative analysis of UPS inhibition on contractile function during ischemia–reperfusion. Isolated rat hearts were perfused under high glucose conditions (33 mM glucose) vs. controls (11 mM glucose) for 60 min and subsequently subjected to 20 min of global ischemia, followed by 60 min of reperfusion. Either 5 μM lactacystin or 10 μM MG-132 was added during the first 20 min of reperfusion. a Left ventricular developed pressure (LVDP) (% recovery) at baseline glucose levels (11 mM), and b with high glucose (33 mM). Rate pressure product (RPP) (% recovery) at baseline glucose levels (c), and d under high glucose conditions. Values are expressed as mean ± SEM (n = 8). *p < 0.05, **p < 0.01, ***p < 0.001 vs. respective controls.

UPS inhibition attenuates infarct size. Isolated rat hearts were perfused under high glucose conditions (33 mM) vs. controls (11 mM) for 60 min and thereafter subjected to regional ischemia as earlier described. UPS inhibitors (5 μM lactacystin or 10 μM MG-132) were administered for the first 20 min of the 2-h reperfusion period. a Infarct size/area at risk (%) following lactacystin administration and b infarct size/area at risk (%) following MG-132 treatment under baseline vs. high glucose perfusion conditions. Values are expressed as mean ± SEM (n = 8). *p < 0.05, **p < 0.01, ***p < 0.001 vs. respective controls.
For the current study, high glucose exposure diminished myocardial SOD1, but not SOD2 expression levels following ischemia–reperfusion (Fig. 8). Myocardial TNF-α expression was also significantly elevated while IkBα levels remained unaltered (vs. baseline glucose conditions) following ischemia–reperfusion in the presence of high glucose (Fig. 9). Due to the interplay between the UPS and autophagy we also investigated the effects of lactacystin (more specific inhibitor vs. MG-132) in this context. LC3-II protein levels were not altered following ischemia–reperfusion under high glucose conditions (Fig. 10). However, lactacystin treatment resulted in a robust elevation in cardiac LC3-II expression (p < 0.001 vs. high glucose untreated). The expression of p62 was not significantly altered in response to high glucose but was significantly increased after lactacystin administration (Fig. 10).

UPS inhibition enhances anti-oxidant defenses during ischemia–reperfusion. Isolated rat hearts were perfused under high glucose conditions (33 mM) vs. controls (11 mM) for 60 min and subjected to global ischemia as earlier described ± addition of 5 μM lactacystin or 10 μM MG-132 during the first 20 min of reperfusion. Western blot analysis for: a SOD1 and b SOD2 with lactacystin treatment; c SOD1 and d SOD2 with MG-132 administration. We employed β-actin as loading control. Values are expressed as mean ± SEM (n ≥ 4). *p < 0.05, **p < 0.01, ***p < 0.001 vs. respective controls.

Decreased expression of inflammatory markers following UPS inhibition with ischemia–reperfusion. Isolated rat hearts were perfused under high glucose conditions (33 mM) vs. controls (11 mM) and subjected to global ischemia and reperfusion ± 5 μM lactacystin or 10 μM MG-132 treatment for the first 20 min of the reperfusion period. Western blot analysis for: a TNF-α and b IkBα with lactacystin treatment; c TNF-α and d IkBα with MG-132 administration. We employed β-actin as loading control. Values are expressed as mean ± SEM (n ≥ 4). *p < 0.05, **p < 0.01, ***p < 0.001 vs. respective controls.

Increased expression of autophagy markers following UPS inhibition with ischemia–reperfusion. Isolated rat hearts were perfused under simulated acute hyperglycemia conditions (33 mM glucose) vs. controls (11 mM glucose) for 60 min and thereafter subjected to 20 min of global ischemia, followed by 60 min of reperfusion. 5 μM lactacystin was added for the first 20 min of reperfusion. Western blot analysis for: a LC3-II and b p62 protein expression levels are shown with β-actin as loading control. Values are expressed as mean ± SEM (n ≥ 4). *p < 0.05, ***p < 0.001 vs. respective controls.
Discussion
This study demonstrates that both lactacystin and MG-132 administration elicited cardioprotection during ischemia–reperfusion performed under acute high glucose conditions. Here initial experiments demonstrated a robust increase in post-ischemic myocardial UPS activities together with elevated protein expression of an E3 ligase (MAFbx). Moreover, the proteasome inhibitor doses employed and the treatment duration resulted in partial UPS impairment together with cardioprotective effects.
Previous studies found decreased proteasome function following ischemia–reperfusion, although the underlying mechanisms remain unclear (reviewed in [31]). This likely occurs due to higher free radical generation with ischemia–reperfusion, thereby leading to direct oxidative modification and subsequent attenuation of proteasome function [18, 31]. In agreement, we detected increased proteasome activities in rat hearts subjected to ischemia–reperfusion performed under baseline glucose conditions vs. pre-ischemic hearts (data not shown). However, with hyperglycemia excess oxidative stress triggers higher UPS activity to help degrade and remove damaged proteins. For example, we recently reported increased cardiac superoxide levels together with decreased SOD activity following ischemia–reperfusion performed under high glucose perfusion conditions [21, 22]. These findings are consistent with other studies demonstrating higher UPS activity in diabetic mouse hearts [15], streptozotocin-infarcted rat hearts and diabetic patients suffering from unstable angina [16]. What about the functional effects of proteasome inhibition with ischemia–reperfusion under hyperglycemic conditions? This study shows—for the first time as far as we are aware—that both lactacystin and MG-132 administration triggered cardioprotective effects with ischemia–reperfusion performed under such conditions. This is in agreement with previous findings showing that high glucose levels trigger myocardial oxidative stress and apoptosis in parallel with impaired contractile function [22].
The effects observed under baseline glucose conditions are consistent with earlier research establishing that proteasome inhibition results in cardioprotection with ischemia–reperfusion [17, 32–34]. Although both beneficial and detrimental effects were observed with proteasome inhibition, the collective data indicate that cardioprotection with ischemia–reperfusion occurs only with acute drug administration [31]. Moreover, the outcomes of UPS inhibition are strongly dose- and time-dependent with lower, non-toxic doses likely upregulating anti-oxidant defenses and exerting anti-inflammatory effects, while higher doses and chronic treatment may result in damaging consequences on protein turnover and cell death [31]. This may also depend on distinct intracellular sub-compartments, with some suggesting that beneficial effects may be due to the inhibition of proteasomal activity within the non-cardiomyocyte compartment where it may exert anti-inflammatory effects [35]. In support, others found that moderate cardiomyocyte-restricted proteasome inhibition exacerbated ischemia–reperfusion injury in mouse hearts [36]. Further studies are, however, required to investigate these intriguing possibilities.
The comparative analyses performed establish that MG-132 administration lead to greater cardioprotection compared to lactacystin. We are unclear regarding the underlying mechanism(s) whereby MG-132 resulted in such a striking improvement in cardiac function. As indicated before, proteasome inhibitor dose and treatment duration are crucial parameters that may affect functional outcomes and we therefore cannot exclude the possibility that lower lactacystin dosages could potentially result in enhanced cardioprotection. Lactacystin is also considered a more specific inhibitor than MG-132 and it is therefore possible that the superiority of MG-132 may be due to non-UPS-related effects. For example, MG-132 can exert additional effects on calpains and cathepsins [37] and also protect cardiomyocytes from injury through the inhibition of NF-kB and downregulation of transforming growth factor beta 1 (TGFβ1) [38]. As MG-132 significantly augmented coronary flow, it is possible that it may also exert advantageous effects on cardiac endothelial cells. In support, an in vitro study found that low-dose proteasome inhibition upregulated endothelial nitric oxide synthase expression and activity together with improved endothelial function [39]. This may therefore lead to increased oxygen supply to the myocardium resulting in improved post-ischemic function [40].
To gain a better understanding of the mechanisms whereby proteasome inhibitors elicit cardioprotection, we assessed whether it exhibits anti-oxidant and/or anti-inflammatory properties in our experimental system. It is well established that myocardial ischemia–reperfusion is associated with increased oxidative stress resulting in cell death [41, 42]. For example, we recently reported that several markers of myocardial oxidative stress were significantly increased in our experimental model [21, 22]. Of note, high glucose availability leads to oxidative stress due to an imbalance in terms of free radical production and intracellular anti-oxidant defenses [43–45]. For the current study high glucose exposure diminished myocardial SOD1, but not SOD2 expression levels following ischemia–reperfusion. Both lactacystin and MG-132 administration resulted in higher cardiac SOD1 and SOD2 protein levels and this is likely a key mechanism to detoxify increased free radical levels. Although we are unclear regarding the transcriptional modulators implicated in this process, the basic leucine zipper transcription factor, nuclear factor-erythroid 2-related factor 2 (Nrf2), is a strong candidate as it is a known UPS target [46, 47]. It is likely that UPS inhibition in our experimental system attenuates Nrf2 ubiquitination and subsequent degradation, thereby increasing its stability and translocation to the nucleus for activation of target anti-oxidant genes [48–50].
The effects of proteasome inhibition on myocardial inflammation were investigated by evaluating TNF-α peptide levels and NFkβ activation. For the latter, we determined myocardial IkBα levels—a UPS target—that when bound to NFκβ in the cytosol prevents its translocation to the nucleus and the subsequent activation of target genes such as TNF-α. Here TNF-α expression was significantly elevated while IkBα levels remained unaltered following ischemia–reperfusion in the presence of high glucose. Lactacystin administration blunted the TNF-α spike under baseline and high glucose conditions, while this effect was not observed with MG-132. However, IkBα levels were significantly higher following lactacystin and MG-132 administration. As lactacystin is a more potent proteasome inhibitor than MG-132, this suggests that its cardioprotective effects are more strongly linked to the attenuation of UPS-mediated induction of inflammatory pathways. In support, a disproportionately elevated UPS can lead to NFκβ activation, thereby resulting in downstream transcriptional effects and the establishment of a pro-inflammatory milieu with hyperglycemia and/or ischemia [16]. Thus these data establish that the strong pro-inflammatory effect triggered in response to ischemia–reperfusion and hyperglycemia can be blunted by partial proteasome inhibition.
Due to the interplay between the UPS and autophagy we also investigated the effects of lactacystin (more specific inhibitor vs. MG-132) in this context. LC3-II levels increased while there was also a significant decrease in p62 expression, indicating greater autophagic flux. Here p62 can shuttle ubiquitinated proteins not removed by the UPS to the autophagic clearance system [51], thus serving as an adapter that is able to interact with both the UPS and autophagic machinery to eventually help with the degradation of misfolded and damaged proteins [52]. These data are consistent with others demonstrating that low dose proteasome inhibitor treatment (reducing chymotrypsin-like activity to ~46%) increased myocardial autophagy [53]. Lower p62 levels would indicate greater clearance and uptake of damaged proteins by cardiac autophagosomes. Thus with UPS inhibition there is an upregulation of autophagy following ischemia–reperfusion under high glucose conditions, likely an adaptive mechanism that assists with the removal of misfolded and damaged proteins. This is in agreement with others reporting that experimentally-induced upregulation of autophagy was linked to improved heart function in diabetic mice [54].
Limitations of study
For the proteasome activity assays we did not employ proteasome inhibitors and the data here presented may also contain contributions from non-proteasomal proteases. Thus it is possible that the degree of proteasomal inhibition could be an underestimation, although this should not affect our relative comparisons.
Conclusions
This study established that partial proteasome inhibition elicits cardioprotection in hearts exposed to ischemia–reperfusion with acute simulated hyperglycemia. Our findings also show three major protective effects, i.e. (a) enhancing myocardial anti-oxidant defences, (b) attenuating inflammation, and (c) increasing the autophagic response. These data are encouraging as it could lead to the development of novel therapeutic interventions that may help treat individuals burdened with hyperglycemia and suffering associated cardiovascular complications such as an AMI.
Abbreviations
- µg:
-
microgram
- µl:
-
microliter
- µM:
-
micromolar
- °C:
-
degree Celsius
- AMI:
-
acute myocardial infarction
- ANOVA:
-
one way analysis of variance
- CA:
-
California
- CaCl2 :
-
calcium chloride
- CMRG:
-
Cardio-Metabolic Research Group
- CO2 :
-
carbon dioxide
- dP/dtmax :
-
maximum velocity of contraction
- EDP:
-
end diastolic pressure
- g :
-
gravitational acceleration
- g:
-
gram
- HR:
-
heart rate
- h:
-
hour
- IκBα:
-
inhibitor of nuclear factor kappa-B kinase subunit alpha
- i.p.:
-
intraperitoneal
- Inc.:
-
incorporation
- KH2PO4 :
-
potassium dihydrogen phosphate
- LC3-II:
-
microtubule‐associated protein 1 light chain 3 II
- LLE:
-
caspase-like proteasome activity
- LLVY:
-
chymotrypsin-like proteasome activity
- LSTR:
-
trypsin-like proteasome activity
- LVDP:
-
left ventricular developed pressure
- MA:
-
Massachusetts
- MAFbx:
-
muscle atrophy F-box
- mg/kg:
-
milligrams per kilogram
- MgCl2 :
-
magnesium chloride
- MgSO4 :
-
magnesium sulphate
- min:
-
minute(s)
- mmHg:
-
millimetres of mercury
- mM:
-
millimolar
- MO:
-
Missouri
- MURF1:
-
muscle RING finger1
- n:
-
number
- NaCl:
-
sodium chloride
- NaHCO3 :
-
sodium hydrogen carbonate
- NFkβ:
-
nuclear factor kappa beta
- NIH:
-
National Institutes of Health
- Nrf2:
-
basic leucine zipper transcription factor, nuclear factor-erythroid 2-related factor 2
- NSW:
-
New South Wales
- O2 :
-
oxygen
- RIPA:
-
radioimmunoprecipitation assay
- RPP:
-
rate pressure product
- SDS-PAGE:
-
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEM:
-
standard error of the mean
- SOD1/2:
-
superoxide dismutase 1/2
- TGFβ1:
-
transforming growth factor beta 1
- TNF-α:
-
tumor necrosis factor-alpha
- UPS:
-
ubiquitin proteasome system
- USA:
-
United States of America
- vs.:
-
versus
- WI:
-
Wisconsin
References
Soedamah-Muthu S, Fuller J, Mulnier HE, Raleigh VS, Lawrenson R, Colhoun HM (2006) High risk of cardiovascular disease in a cohort study using the General Practice Research Database. Diabetes Care 29:798–804
Gardner A (2014) The benefits of tight glycemic control in critical illness: sweeter than assumed? Indian J Crit Care Med 18(12):807
Martín-Timón I, Sevillano-Collantes C, Segura-Galindo A, Cañizo-Gómez FJ (2014) Type 2 diabetes and cardiovascular disease: have all risk factors the same strength? World J Diabetes 5(4):444–470
Marcovecchio ML, Lucantoni M, Chiarelli F (2011) Role of chronic and cute hyperglycemia in the development of diabetes complications. Diabetes Technol Ther 13:389–394
United Kingdom Prospective Diabetes Study (UKPDS) Group (1998) Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with T2DM (UKPDS 33). Lancet 352:837–845
Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
Kannel WB, Hjortland M, Castelli WP (1974) Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol 34:29–34
Capes SE, Hunt D, Malmberg K, Gerstein HC (2000) Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet 355:773–778
Marfella R, Di Filippo C, Portoghese M, Ferraraccio F, Rizzo MR, Siniscalchi M et al (2009) Tight glycemic control reduces heart inflammation and remodeling during acute myocardial infarction in hyperglycemic patients. J Am Coll Cardiol 53(16):1425–1436
Sleiman I, Morandi A, Sabatini T, Ranhoff A, Ricci A, Rozzini R et al (2008) Hyperglycemia as a predictor of in-hospital mortality in elderly patients without diabetes mellitus admitted to a sub-intensive care unit. J Am Geriatr Soc 56(6):1106–1110
Norhammar A, Tenerz A, Nilsson G, Hamsten A, Efendíc S, Rydén L et al (2002) Glucose metabolism in patients with acute myocardial infarction and no previous diagnosis of diabetes mellitus: a prospective study. Lancet 359:2140–2144
Sawin G, Shaughnessy AF (2010) Glucose control in hospitalized patients. Am Fam Physician 81(9):1121–1124
Powell SR (2006) The ubiquitin–proteasome system in cardiac physiology and pathology. Am J Physiol Heart Circ Physiol 291:H1–H19
Kriegenburg F, Ellgaard L, Hartmann-Petersen R (2012) Molecular chaperones in targeting misfolded proteins for ubiquitin-dependent degradation. FEBS J 279(4):532–542
Wang Y, Sun W, Du B, Miao X, Bai Y, Xin Y et al (2013) Therapeutic effect of MG-132 on diabetic cardiomyopathy is associated with its suppression of proteasomal activities: roles of Nrf2 and NF-κB. Am J Physiol Heart Circ Physiol 304(4):H567–H578
Marfella R, Di C, Portoghese M, Siniscalchi M, Martis S, Ferraraccio F et al (2009) The ubiquitin–proteasome system contributes to the inflammatory injury in ischemic diabetic myocardium : the role of glycemic control. Cardiovasc Pathol 18(6):332–345
Huang S, Patterson E, Yu X, Garrett MW, De Aos I, Kem DC (2008) Proteasome inhibition 1 h following ischemia protects GRK2 and prevents malignant ventricular tachyarrhythmias and SCD in a model of myocardial infarction. Am J Physiol Heart Circ Physiol 294(3):H1298–H1303
Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B et al (2001) Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem 276(32):30057–30063
Zolk O, Schenke C, Sarikas A (2006) The ubiquitin–proteasome system: focus on the heart. Cardiovasc Res 70(3):410–421
Calise J, Powell SR (2013) The ubiquitin proteasome system and myocardial ischemia. Am J Physiol Heart Circ Physiol 304(3):H337–H349
Mapanga R, Rajamani U, Dlamini N, Zungu-Edmondson M, Kelly-Laubscher R, Shafiullah M et al (2012) Oleanolic acid: a novel cardioprotective agent that blunts hyperglycemia-induced contractile dysfunction. PLoS One 7(10):e47322
Mapanga RF, Joseph D, Symington B, Garson K-L, Kimar C, Kelly-Laubscher R et al (2014) Detrimental effects of acute hyperglycaemia on the rat heart. Acta Physiol (Oxf) 210:546–564
Harris F, Pierpoint L (2012) Photodynamic therapy based on 5-aminolevulinic acid and its use as an antimicrobial agent. Med Res Rev 32(6):1292–1327
Fenteany G, Standaert R, Lane W, Choi S, Corey E, Schreiber S (1995) Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 268:726–731
Borissenko L, Groll M (2007) 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev 107(3):687–717
Craiu A, Gaczynska M, Akopian T, Gramm CF, Fenteany G, Goldberg AL et al (1997) Lactacystin and clasto-lactacystin beta-lactone modify multiple proteasome beta-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J Biol Chem 272(20):13437–13445
Divald A, Powell SR (2006) Proteasome mediates removal of proteins oxidized during myocardial ischemia. Free Radic Biol Med 40:156–164
Powell S, Wang P, Katzeff H, Shringarpure R, Teoh C, Khaliulin I et al (2005) Oxidized and ubiquitinated proteins may predict recovery of postischemic cardiac function: essential role of the proteasome. Antioxid Redox Signal 7:538–546
Rajamani U, Joseph D, Roux S, Essop MF (2011) The hexosamine biosynthetic pathway can mediate myocardial apoptosis in a rat model of diet-induced insulin resistance. Acta Physiol (Oxf) 202(2):151–157
Rajamani U, Essop MF (2010) Hyperglycemia-mediated activation of the hexosamine biosynthetic pathway results in myocardial apoptosis. Am J Physiol Cell Physiol 299(1):C139–C147
Yu X, Kem DC (2010) Proteasome inhibition during myocardial infarction. Cardiovasc Res 85:312–320
Campbell B, Adams J, Shin Y, Lefer A (1999) Cardioprotective effects of a novel proteasome inhibitor following ischemia and reperfusion in the isolated perfused rat heart. J Mol Cell Cardiol 31:467–476
Pye J, Ardeshirpour F, McCain A, Bellinger DA, Merricks E, Adams J et al (2003) Proteasome inhibition ablates activation of NF-kappa B in myocardial reperfusion and reduces reperfusion injury. Am J Physiol Heart Circ Physiol 284(3):H919–H926
Stansfield WE, Moss NC, Willis MS, Tang R, Selzman CH (2007) Proteasome inhibition attenuates infarct size and preserves cardiac function in a murine model of myocardial ischemia–reperfusion injury. Ann Thorac Surg 84(1):120–125
Wang X, Robbins J (2014) Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol 71:16–24
Tian Z, Zheng H, Li J, Li Y, Su H, Wang X (2012) Genetically induced moderate inhibition of the proteasome in cardiomyocytes exacerbates myocardial ischemia–reperfusion injury in Mice. Circ Res 111(5):532–542
Kisselev AF, Goldberg AL (2001) Proteasome inhibitors: from research tools to drug candidates. Chem Biol 8(8):739–758
Ma Y, Chen B, Liu D, Yang Y, Xiong Z, Zeng J et al (2011) MG132 treatment attenuates cardiac remodeling and dysfunction following aortic banding in rats via the NF-κβ/TGFβ1 pathway. Biochem Pharmacol 81(10):1228–1236
Stangl K, Günther C, Frank T, Lorenz M, Meiners S, Röpke T et al (2002) Inhibition of the ubiquitin–proteasome pathway induces differential heat-shock protein response in cardiomyocytes and renders early cardiac protection. Biochem Biophys Res Commun 291:542–549
Kassiotis C, Rajabi M, Taegtmeyer H (2008) Metabolic reserve of the heart: the forgotten link between contraction and coronary flow. Prog Cardiovasc Dis 51(1):74–88
Buja LM, Entman ML (1998) Modes of myocardial cell injury and cell death in ischemic heart disease. Circulation 98(14):1355–1357
Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D (2004) Protein kinase C delta activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J Biol Chem 279(46):47985–47991
Brownlee M (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54:1615–1625
Nourooz-Zadeh J, Rahimi A, Tajaddini-Sarmadi J, Tritschler H, Rosen P, Halliwell B et al (1997) Relationships between plasma measures of oxidative stress and metabolic control in NIDDM. Diabetologia 40:647–653
Rösen P, Du X, Tschöpe D (1998) Role of oxygen derived radicals for vascular dysfunction in the diabetic heart: prevention with alpha tocopherol? Mol Cell Biochem 188:103–111
Holtzclaw WD, Dinkova-Kostova AT, Talalay P (2004) Protection against electrophile and oxidative stress by induction of phase 2 genes: the quest for the elusive sensor that responds to inducers. Adv Enzyme Regul 44:335–367
Kobayashi A, Ohta T, Yamamoto M (2000) Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol 2004(378):273–286
McMahon M, Itoh K, Yamamoto M, Hayes JD (2003) Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem 278(24):21592–21600
Kobayashi A, Kang M, Okawa H, Zenke Y, Chiba T, Igarashi K et al (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24(16):7130–7139
Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, Hannink M (2005) Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem 280(34):30091–30099
Willis MS, Townley-Tilson WHD, Kang EY, Homeister JW, Patterson C (2010) Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res 106(3):463–478
Korolchuk VI, Menzies FM, Rubinsztein DC (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584(7):1393–1398
Zheng Q, Su H, Tian Z, Wang X (2011) Proteasome malfunction activates macroautophagy in the heart. Am J Cardiovasc Dis 1(3):214–226
Wang B, Yang Q, Sun YY, Xing YF, Bin WY, Lu XT et al (2014) Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J Cell Mol Med 18(8):1599–1611
Authors’ contributions
BA performed experiments, analyzed the data, and wrote the manuscript. RFM conceived the idea, performed experiments, analyzed the data, and wrote the manuscript. MFE conceived the idea, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgments
This work was supported by the South African National Research Foundation and Stellenbosch University (to MFE).
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Adams, B., Mapanga, R.F. & Essop, M.F. Partial inhibition of the ubiquitin–proteasome system ameliorates cardiac dysfunction following ischemia–reperfusion in the presence of high glucose. Cardiovasc Diabetol 14, 94 (2015). https://doi.org/10.1186/s12933-015-0258-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-015-0258-4