
Abstract
Patients with type 2 diabetes have a several-fold increased risk of developing cardiovascular disease when compared with nondiabetic controls. Myocardial infarction and stroke are responsible for 75% of all death in patients with diabetes, who present a 2-4× increased incidence of death from coronary artery disease. Patients with diabetes are considered for cardiovascular disease secondary prevention because their risk level is similar to that reported in patients without diabetes who have already suffered a myocardial infarction. More recently, with a better risk factors control, mainly in intensive LDL cholesterol targets with statins, a significant decrease in acute cardiovascular events was observed in population with diabetes. Together with other major risk factors, type 2 diabetes must be considered as an important cause of cardiovascular disease.
Glucagon like peptide-1 receptor agonists represent a novel class of anti-hyperglycemic agents that have a cardiac-friendly profile, preserve neuronal cells and inhibit neuronal degeneration, an anti-inflammatory effect in liver protecting it against steatosis, increase insulin sensitivity, promote weight loss, and increase satiety or anorexia.
This review is intended to rationally compile the multifactorial cardiovascular effects of glucagon-like peptide-1 receptor agonists available for the treatment of patients with type 2 diabetes.
Similar content being viewed by others


Introduction
When compared with nondiabetic controls, T2D patients have a several-fold increased risk of developing cardiovascular disease (CVD) [1]. At least, 68% of people >65 years of age with diabetes die of some form of CVD. Among adults with diabetes, CVD death rates are 2-4× higher than the rates for adults without diabetes [2]. Patients with T2D are considered for CVD secondary prevention because their risk level is similar to that reported in nondiabetic patients who have already suffered a MI [3]. More recently, with a better risk factors control, mainly in intensive LDL cholesterol targets with statins, a significant decrease in acute cardiovascular events was observed in diabetic population. Together with other major risk factors, T2D must be considered as an important cause of CVD. Indeed, from a cardiological point of view, “diabetes is a cardiovascular disease” [4]. Unfortunately, despite significant advances in anti-glycemic therapies, macrovascular complications are still the most common cause of death in T2D patients [5].
The observation that intrajejunal glucose promotes greater insulin release than intravenous glucose administration was reported 50 years ago by McIntyre et al. [6] Perley and Kipnis estimated the intestinal component accounts for 50%-70% of the total insulin secreted after an oral glucose load. The `incretin effect’ was coined by Creuzfeld and Ebert in 1985 to designate this phenomenon [7]. Nauck et al. [8] demonstrated that the incretin effect is impaired in patients with T2D. Recently, An et al. observed that the improvement in the glycemic control potentiates insulin secretion induced by oral glucose ingestion but does not change the insulin secretion after iv glucose infusion indirectly demonstrating that the incretin effect is dependent of glucose/food absorption by the intestines [9].
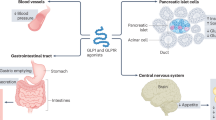
Glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are the two factors that account for most of the incretin effect [10]. GLP-1 and GIP are both secreted in response to food ingestion. GLP-1 is mainly produced in the enteroendocrine L cells located in the distal intestine exerting its effects through binding to the GLP-1 receptors (GLP-1R) expressed in the pancreas, heart, blood vessels, gastrointestinal tract (GIT), kidney, lung, breast, and central nervous system [11],[12]. In the pancreatic ß-cell GLP-1, on elevated glucose concentrations, leads to stimulation of insulin secretion (Figure 1) [13].

GLP-1 is released from the small intestine after meal ingestion and enhance glucose-stimulated insulin secretion ( incretin action ). DPP-4 rapidly converts GLP-1 and GIP to their inactive metabolites in vivo. Inhibition of DPP-4 activity prevents GLP-1 and GIP degradation, thereby enhancing incretin action (Adapted from Abrahamson MJ. The increting effect of GLP-1. Diabetes, Cardiovascular Disease and Stroke: Mechanisms and Risk Reduction. http://www.medscape.org/viewarticle/557239 and reference [14]).
Because the majority of the α-cells do not express GLP-1R, and also because GLP-1 inhibits glucagon secretion even in T1D patients with low insulin pancreatic reserve, the mechanism by which GLP-1 decreases glucagon secretion is still a matter of debate [15]. Recently, De Marinis et al. reported that GLP-1-induced suppression of glucagon release is dependent of protein kinase A (PKA) and independent of glucose or paracrine effects mediated by insulin or by somatostatin acting on somatostatin receptor subtype-2 [16],[17].
Furthermore, GLP-1 decreases gastrointestinal motility extending the entry of nutrients to be absorbed by the GIT [18]. This effect seems to be very important for the normalization of postprandial glucose (PPG) elevations, and potentially even more important than the insulinotropic effects of GLP-1 for maintaining PPG homeostasis [19]. Many reports showed that GLP-1 effects involve neural modulation and peripheral effects, which increase satiety, resting energy expenditure and lower plasma concentrations of free fatty acids [20]-[22].
Either GLP-1 or GIP potentiates glucose-dependent insulin response protecting α-cells against cytokine-induced apoptosis or glucose [23]. All of these GLP-1 effects (Figure 2) are potentially beneficial for patients with T2D, and have been sought exploited clinically with the development of GLP-1R agonists.

Pleitropic effects of GLP-1 or GLP-1R agonists (Adapted from references [[24]] ).
The phenomenon of induction of glucose sensitivity observed in glucose-resistant ß-cells is called glucose competence, i.e. the magnitude of the insulin secretion response to a rise in glucose concentration. More recently, Cornu et al. demonstrated in KO mice that the regulation of ß-cell number and function by GLP-1 depends on the cAMP/PKA mediated-induction of insulin-like growth factor-1 receptor (IGF-1R) expression and the increased activity of insulin-like growth factor 2 (IGF-2)/IGF-1R by an autocrine mode of action [25].
As depicted in Figure 1, dipeptidylpeptidase-4 (DPP-4) is an ubiquitously expressed transmembrane protein that removes N-terminal dipeptides from various substrate hormones, chemokines, neuropeptides and growth factors. DPP-4 inhibitors (DPP-4i) block degradation of GLP-1 and GIP, extend their insulinotropic effect, and improve glycemia [26].
Although the SAVOR and EXAMINE studies were delineated to verify cardiovascular safety of two DPP-4i, namely saxagliptin and alogliptin, respectively, both are considered to be the first controlled clinical trials to address the effect of GLP-1 system on CVD. In the SAVOR study DPP-4 inhibition with saxagliptin did not increase or decrease the rate of ischemic events, though the rate of hospitalization for heart failure was increased [27]. On the other hand, EXAMINE study showed that T2D patients, who had had a recent acute coronary syndrome, the rates of major adverse cardiovascular events were not increased with the DPP-4 inhibitor alogliptin as compared with placebo [28].
GLP-1R agonists represents a novel class of anti-hyperglycemic agent that improve health and survival of ß-cells (improvement in postprandial hyperglycemia), suppress glucagon (improvement in fasting hyperglycemia), improve insulin resistance (modest effect) and influence energy intake (augment satiety signal) with minimal, if at all, any side effects (weight neutral and non-hypoglycemic). The incretins address most of the proposed pathophysiologic mechanisms involved in the development of T2D (Figure 2). Besides the ß-cell dysfunction, incretin deficiency is now considered among the key factors in the pathophysiology of T2D. So, exogenous GLP-1R agonists have been more recently considered as a good choice for treating patients with diabetes [29].
In this brief review, we shall discuss recent important evidences of the potential clinical benefit of the incretin effect on cardiovascular system as well as the biological mechanisms, which underlie these effects.
GLP-1R distribution in the cardiovascular system
GLP-1R is a member of the class B1 family of G protein-coupled receptor for which there are only predicted structures [30]. GLP-1R has been detected by reverse transcriptase-polymerase chain reaction in cardiac and vascular tissues isolated from both human and animal models [11],[31], and their mRNA transcripts were demonstrated in the human heart [32].
GLP-1R protein has also been detected in human coronary artery endothelial cells (HCAEC) and human umbilical vein endothelial cells (HUVEC) [33],[34]. Ban et al. detected GLP-1R protein expression by immunohistochemistry in mouse coronary endothelial and smooth muscle cells [35].
Many reports using polyclonal antisera were unable to detect GLP-1R protein in ventricular cardiomyocytes leaded to the revaluation of those earlier reports. Because of lack of sensitivity and problems with non-specificity of commercially available GLP-1R antisera detected multiple nonspecific immunoreactive proteins even in tissue extracts from Glp1r-/- mice. Although Glp1r mRNA transcripts are detected in whole heart extracts such methods do not distinguish between vascular smooth muscle cells or atrial cardiomyocytes, there were technical difficulties in isolating pure cardiomyocyte populations from the ventricle without contamination of atrial cardiomyocytes isolated from neonatal mouse [36].
Cardiac GLP-1R signaling pathways
There is considerable overlap between pathways induced by the GLP-1R activation. As shown in Figure 3, GLP-1R couples to adenylyl cyclase leading to the activation of cAMP. Using a GLP-1 conjugated to tetramethyl rhodamine to monitor the internalization of the receptor, Kuna et al. [37] observed that after internalization, GLP-1R/ligand complex could be co-localized with adenylate cyclase in the endosome. Indeed, cAMP levels can be elicited by GLP-1 treatment of mouse cardiomyocytes without increasing intracellular Ca2+ or inducing contractility [38].

The involvement of cAMP/PKA/CREB pathway on decrease myocyte apoptosis has been unveiled by experiments prepared in cultured mouse cardiomyocytes. This function is mediated via the activation of the cAMP/PKA/CREB (cAMP-responsive element binding protein) and the transactivation of the EGF-R (epidermal growth factor receptor) leading to the activation of phosphatidylinositol-3 kinase (PI3K), protein kinase Cζ(PKCζ), Akt-protein kinase B (AKT/PKB), extracellular regulated kinase (ERK1/2 or mitogen-activated protein kinase [MAPK]) signaling pathways and to the up-regulation of the expression of the cell cycle regulator cyclinD1 (Figure 3). The increase in Akt and ERK phosphorylations lead to cardiomyocyte growth and activation of glucose metabolism [24],[39]-[41].
The main mechanism of cardioprotective effects of GLP-1R agonists against oxidative stress-induced injury in H9c2 cells (cardiomyoblasts from Rattus norvegicus) and in cardiomyocytes is related to the scavenging of reactive oxygen species, by increasing the concentrations of endogenous antioxidant defenses and inhibiting cardiomyocyte apoptosis [42]. This cytoprotective effect is mediated by PI3K and partially dependent on ERK1/2 and seems to be glucose-independent. A protective role of retinoid X receptor is also related to hypoxia/reoxygenation injury in those cells [43]. Moreover, GLP-1 also inhibits palmitate- and ceramide-induced phosphatidylserine exposure and DNA fragmentation [44].
Effects of GLP-1R activation in the heart
The observation that GLP-1 decreases contractility in primary culture of adult rat cardiomyocytes, despite increasing cAMP levels, [38] has also been reported in isolated rat hearts [45]. Studies in dogs demonstrated an increased myocardial glucose uptake during a hyperinsulinemic-euglycemic clamp [46].
Moreover, cardiometabolic effects of GLP-1 are attenuated in obesity and T2D, via mechanisms that may involve impaired p38-MAPK signaling. Using a swine experimental model, Moberly et al. confirmed and extended the observation, where GLP-1 significantly increased myocardial glucose uptake under basal conditions in lean humans, but this effect was impaired in T2D [47]. GLP-1 did not increase myocardial oxygen consumption or blood flow in humans or in swine, increasing p38-MAPK activity in lean, but not obese cardiac tissue [48].
Experimental studies in animals suggested that incretins may preserve cardiomyocyte viability, increase metabolic efficiency, inhibit the structural, and functional remodeling after MI [49]. Shannon’s group was the first to demonstrate that infusion of GLP-1(7-36) (1.5 pmol/kg/min) for 72 h in patients with left ventricular dysfunction (LVD) after MI improved global and regional left ventricular wall motion scores reducing hospital stay and in-hospital mortality [50],[51].
GLP-1-mediated control of heart rate (HR) and blood pressure (BP) is complex and species specific. Synthetic human GLP-1 administered into the jugular vein of male rats acutely increased systolic and diastolic BP, as well as HR, which returned to basal levels 25 min after GLP-1 administration. The pre-treatment with propranolol or phentolamine did not prevent the increase in BP and HR. In rodents, these increases involve a dual pathways originating from both central nervous system and periphery with intact neural vagus transmission [52].
Intravenous infusion of GLP-1 for 48 h in healthy human subjects increased muscle sympathetic nerve activity but had no effect on BP, norepinephrine plasma concentration, or the sympathetic/parasympathetic balance as estimated by the HR variability, suggesting that the increase in sympathetic drive is at least partially compensated by an increase in the parasympathetic activity [53]. On the other hand, acute subcutaneous (SC) injection of GLP-1 increases HR and BP transiently in healthy human subjects, returning to normal ranges 50-60 min after injection [54]. A mean increase in HR of 1.86 beats/min was reported in a meta-analysis including available data from randomized controlled trials testing GLP-1 agonists against placebo in patients with T2D [53].
In the cardiovascular system, incretins have been recently associated to the increase of endogenous antioxidant defenses, inhibition of cardiomyocyte apoptosis, attenuation of endothelial inflammation and dysfunction [55].
GLP-1R activation and artery wall function
GLP-1 has pleiotropic effects on the cardiovascular system. A GLP-1R agonist (liraglutide) increased endothelial nitric oxide synthase phosphorylation and nitric oxide (NO) production by the 5-AMP-activated protein kinase (AMPK)-dependent pathway and subsequent NO production in cultured HCAEC [56],[57]. GLP-1 protects the cardiac microvessels against oxidative stress, apoptosis, and the resultant microvascular barrier dysfunction in diabetes, contributing to improvement of cardiac function and cardiac glucose metabolism. The protective effects of GLP-1 are dependent on downstream inhibition of Rho through a cAMP/PKA-mediated pathway [58].
As shown by Batchuluun at al., metformin and liraglutide improve high glucose-induced oxidative stress via inhibition of PKC-NADPH oxidase pathway in human aortic endothelial cells. All these effects were even more pronounced when both drugs were used together, suggesting a potential clinical benefit of this drug combination in decreasing the endothelial damage induced by hyperglycemia [59].
In humans, Kelly et al. showed that exenatide therapy for that 3-mo in patients with obesity and pre-diabetes had similar effects on microvascular endothelial function, markers of inflammation, oxidative stress, and vascular activation, as metformin. They assumed that improvements in endothelial function with GLP-1R agonists might be limited to the postprandial setting, particularly following the consumption of a high-fat meal [60].
GLP-1 enhanced acetylcholine-induced forearm blood flow but had no effect on blood flow induced by sodium nitro-prusside in healthy human subjects [61]. Patients with T2D with stable CAD presented an improvement in endothelial function expressed by an increase in flow-mediated vasodilation of the brachial artery, independent of changes in systolic and diastolic blood pressure during a hyperinsulinemic clamp in response to GLP-1 [35]. However, GLP- 1-mediated improvements in blood flow were considerably attenuated after a 2-month period of better glycemic control [62].
In obese patients, GLP-1 infusion significantly increased excretion of sodium (by 60%), calcium (by 60%) and chloride (by 44%) and significantly decreased excretion of H+ (by 75%). Besides these renoprotective qualities, these changes suggest that GLP-1 may have renoprotective qualities [63]. Moreover, Kim et al. demonstrated that cardiac GLP-1R expression is localized in cardiac atria and that GLP-1R activation promotes secretion of atrial natriuretic peptide (ANP) and increases BP [64].
In animal and cell models, GLP-1 has been shown to impact the development and/or progression of atherosclerotic plaques. Since GLP-1R has been localized by immunocytochemistry in mouse aortic smooth muscle cells, endothelial cells, monocytes, and macrophages, both direct and indirect actions of GLP-1 may contribute to the potential reduction of atherogenesis. Continuous infusion of exendin-4 in nondiabetic C57BL/6 and ApoE-/- mice reduced monocyte adhesion to aortic endothelial cells and atherosclerotic lesion size after 40 days of treatment. The inflammatory markers monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis factor α(TNF- α), were reduced by treatment with exendin-4 in response to lipopolysaccharide in cultured peritoneal macrophages harvested from mice [65]. However, circulating GLP-1 was found to be positively associated with total coronary load in humans in a fully adjusted model [OR: 2.53 (95% CI: 1.12-6.08; p = 0.03)], [66] but further studies are required to confirm these observations.
Continuous infusion of exendin-4 for 4 wk. in C57BL/6 mice reduced neointimal formation after endothelial denudation of the femoral artery [67]. Nagashima et al. [68] reported that continuous GLP-1 infusion reduced foam cell formation and the development of atherosclerotic lesions in ApoE-/- mice. Recently, the same group demonstrated similar effects after infusion of liraglutide in apoE-/- mice [69]. In addition to these findings on atherosclerotic plaque formation, GLP-1R agonists prevented the increase in plasminogen activator inhibitor type-1 (PAI-1) and vascular cell adhesion molecule-1 (VCAM-1) gene expression in response to TNF- α or hyperglycemia in HUVEC through PKA pathway [24],[70].
GLP-1R agonists effects on cardiovascular events
The first pieces of evidence of the potential clinical benefit of GLP-1 agonists have emerged recently, pointing to a decrease of major adverse cardiovascular and cerebrovascular events (MACCE) including stroke, MI, cardiac mortality, acute coronary syndrome, and revascularization procedures. A retrospective medical database analysis of T2D patients (n = 39,275) treated with exenatide or other glucose-lowering therapies (n = 381,218) indicated that the GLP-1 agonist might reduce in 19% the incidence of MACCE and in 12% cardiovascular hospitalizations [71]. Despite the absence of adverse cardiovascular effects has been confirmed, this benefit was not verified in an integrated analysis of 12 controlled, randomized, short-term clinical trials (12-52 weeks) comparing exenatide with placebo or insulin [72].
In both diabetic and non-diabetic patients presenting class II/IV heart failure, GLP-1 infusion led to an improvement of left ventricular (LV) ejection fraction, myocardial ventilation oxygen consumption, 6-min walk distance, and quality of life [50]. A study with infusion of exenatide in T2D patients with chronic heart failure reduced the pulmonary capillary wedge pressure and increased both inotropism and chronotropism [73]. These favorable results require further clinical trials in order to elucidate whether these effects will result or not in reduced mortality. In a study involving patients with CAD and preserved LV function, who were scheduled to undergo coronary artery bypass grafting (CABG), were randomized to receive standard therapy or treatment with GLP-1 (1.5 pmol/kg/min) as a continuous infusion beginning 12 h before CABG and continuing for 48 h after the procedure. The control group required greater use of inotropic and vasoactive infusions during the 48 h period after CABG to achieve the same hemodynamic results observed in the group receiving GLP-1. There were also more frequent arrhythmias requiring anti-arrhythmic agents in the control group [50].
In a non-randomized pilot study, Nikolaidis et al. investigated the safety and efficacy of a 72-h infusion of native GLP-1 combined to background therapy in 10 patients with acute MI and LV ejection fraction <40% (mean 29 ± 2%) after successful primary angioplasty, compared to 11 control patients. GLP-1 treatment was safe and elicited a significant improvement in LV ejection fraction (to a mean of 39 ± 2%) [74].
In a study involving 14 patients with normal ejection fraction and CAD awaiting revascularization, normal saline and native GLP-1 were infused on two different occasions from 30 min before until 30 min after a dobutamine stress echocardiography for evaluation of the global LV function; native GLP-1 significantly improved LV function at peak stress and at 30 min into recovery, predominantly in ischemic segments [75].
Fifty-eight patients with ST-segment-elevation MI and thrombolysis were randomized to receive either saline (n = 40) or exenatide (n = 18, 10 μg SC and 10 μg as an IV bolus 5 min before the onset of reperfusion plus 10 μg SC twice daily on the following 2 days) to evaluate whether this GLP-1R agonist could reduce the area of necrosis; exenatide administration significantly decreased release of CK-MB and troponin and reduced infarct size as evaluated by cardiac magnetic resonance after 1 month [14].
Differences between GLP-1R agonists
GLP-1R agonists already in the market and others in development present the same mode of action and, having the same pleiotropic effects of native GLP-1, the differences in clinical profiles between them are related to differences in pharmacokinetics, structure and size of each formulation. The most remarkable clinically Important pharmacokinetic characteristic is the difference between the short-acting and continuous-acting compounds (Table 1) [13]. Indeed, continuous exenatide exposure once weekly elicited a greater response than did short-acting exenatide, improving glycemic and lipids controls and lipoprotein metabolism, and decreasing systemic inflammation. Indeed, as shown by Hermansen et al. liraglutide treatment in patients with T2D significantly reduced postprandial excursions of triglyceride and apo B48 after a fat-rich meal, independently of gastric emptying. Results indicate that liraglutide’s potential to reduce CVD risk via improvement of postprandial lipemia.
Meloni et al. calculated the absolute benefit increase of using exenatide once week, GLP-1R agonist, vs an oral glucose-lowering medication or insulin glargina to achieve ADA-recommended goals. Exenatide once a week assisted more patients in reaching the majority of ADA-recommended therapeutic goals than treatment with sitagliptin, pioglitazone, or insulin glargine.
Conclusions
Mechanistic and preliminary clinical evidence have consistently pointed toward beneficial effects of GLP-1 analogues and GLP-1R agonists on cardiovascular diseases in T2D patients. Indeed, a search at http://www.clinicaltrials.gov (as of, Sep ‘14), employing cardiovascular disease and GLP-1 as keywords, returns 112 records comprising 42% completed, 39% recruiting, 8% not yet recruiting, 6% active, but not yet recruiting, 4% terminated, and 1% withdraw studies. Among those, 12% studies has results and 88% has no results available, which indicates towards an increasing interest in the cardiovascular effects of incretin-based therapies. The actual evaluation of cardiovascular benefits will be achieved with the completion of the Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial, which enrolled 9,340 T2D patients randomized for treatment with liraglutide or placebo for an estimated period of 5 years (ClinicalTrials.gov Identifier: NCT01179048).
Tables 2 and 3 depict the most important results of GLP-1 and GLP-1R agonists from animal and human studies mentioned in the text.
The potential cardiovascular benefits expected from this new therapeutic approach to obtain glycemic control in T2D arises a possibility to change the excessive cardiovascular burden related to this disease in both developed and developing countries worldwide.
References
Booth GL, Kapral MK, Fung K, Tu JV: Recent trends in cardiovascular complications among men and women with and without diabetes. Diabetes Care. 2006, 29 (1): 32-7. 10.2337/diacare.29.01.06.dc05-0776.
Haffner SJ, Cassells H: Hyperglycemia as a cardiovascular risk factor. Am J Med. 2003, 115 (Suppl 8A): 6S-11S. 10.1016/j.amjmed.2003.09.009.
Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M: Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998, 339 (4): 229-34. 10.1056/NEJM199807233390404.
Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, Mitch W, Smith SC, Sowers JR: Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation. 1999, 100 (10): 1134-46. 10.1161/01.CIR.100.10.1134.
Perk J, De Backer G, Gohlke H, Graham I, Reiner Z, Verschuren WM, Albus C, Benlian P, Boysen G, Cifkova R, Deaton C, Ebrahim S, Fisher M, Germano G, Hobbs R, Hoes A, Karadeniz S, Mezzani A, Prescott E, Ryden L, Scherer M, Syvänne M, Scholte Op Reimer WJ, Vrints C, Wood D, Zamorano JL, Zannad F: European Guidelines on cardiovascular disease prevention in clinical practice (version 2012): The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Atherosclerosis. 2012, 223 (1): 1-68. 10.1016/j.atherosclerosis.2012.05.007.
McIntyre N, Holsworth DC, Turner DS: New interpretation of oral glucose tolerance. Lancet. 1964, 2: 20-21. 10.1016/S0140-6736(64)90011-X.
Creutzfeldt W, Ebert R: New developments in the incretin concept. Diabetologia. 1985, 28: 565-73. 10.1007/BF00281990.
Nauck M, Stockmann F, Ebert R, Creutzfeldt W: Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986, 29 (1): 46-52. 10.1007/BF02427280.
An Z, Prigeon RL, D’Alessio DA: Improved glycemic control enhances the incretin effect in patients with type 2 diabetes. J Clin Endocrinol Metab. 2013, 98 (12): 4702-4708. 10.1210/jc.2013-1199.
Kieffer TJ, Habener JF: The glucagon-like peptides. Endocr Rev. 1999, 20 (6): 876-913. 10.1210/edrv.20.6.0385.
Wei Y, Mojsov S: Tissue-specific expression of the human receptor for glucagon-like peptide-I: brain, heart and pancreatic forms have the same deduced amino acid sequences. FEBS Lett. 1995, 358: 219-24. 10.1016/0014-5793(94)01430-9.
Körner M, Stöckli M, Waser B, Reubi JC: GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J Nucl Med. 2007, 48: 736-43. 10.2967/jnumed.106.038679.
Lund A, Knop FK, Vilsbøll T: Glucagon-like peptide-1 receptor agonists for the treatment of type 2 diabetes: differences and similarities. Eur J Intern Med. 2014, 25 (5): 407-14. 10.1016/j.ejim.2014.03.005.
Baggio LL, Drucker DJ: Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007, 132 (6): 2131-57. 10.1053/j.gastro.2007.03.054.
Godoy-Matos AF: The role of glucagon on type 2 diabetes at a glance. Diabetol Metab Syndr. 2014, 6 (1): 91-10.1186/1758-5996-6-91.
De Marinis YZ, Salehi A, Ward CE, Zhang Q, Abdulkader F, Bengtsson M, Braha O, Braun M, Ramracheya R, Amisten S, Habib AM, Moritoh Y, Zang E, Reimann F, Rosengren AH, Shibasaki T, Gribble F, Renström E, Seino S, Eliasson L, Rorsman P: GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab. 2010, 11: 543-553. 10.1016/j.cmet.2010.04.007.
De Heer J, Rasmussen C, Coy DH, Holst JJ: Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia. 2008, 51: 2263-2270. 10.1007/s00125-008-1149-y.
Little TJ, Pilichiewicz AN, Russo A, Phillips L, Jones KL, Nauck MA, Wishart J, Horowitz M, Feinle-Bisset C: Effects of intravenous glucagon-like peptide-1 on gastric emptying and intragastric distribution in healthy subjects: relationships with postprandial glycemic and insulinemic responses. J Clin Endocrinol Metab. 2006, 91 (5): 1916-23. 10.1210/jc.2005-2220.
Flint A, Raben A, Astrup A, Holst JJ: Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998, 101 (3): 515-20. 10.1172/JCI990.
Dailey MJ, Moran TH: Glucagon-like peptide 1 and appetite. Trends Endocrinol Metab. 2013, 24: 85-91. 10.1016/j.tem.2012.11.008.
ørskov C, Poulsen SS, Møller M, Holst JJ: Glucagon-like peptide I receptors in the subfornical organ and the area postrema are accessible to circulating glucagon-like peptide I. Diabetes. 1996, 45: 832-5. 10.2337/diab.45.6.832.
Pannacciulli N, Bunt JC, Koska J, Bogardus C, Krakoff J: Higher fasting plasma concentrations of glucagon-like peptide 1 are associated with higher resting energy expenditure and fat oxidation rates in humans. Am J Clin Nutr. 2006, 84: 556-60.
Komatsu M, Takei M, Ishii H, Sato Y: Glucose-stimulated insulin secretion: a newer perspective. J Diabetes Investig. 2013, 4 (6): 511-6. 10.1111/jdi.12094.
Sussman MA, Völkers M, Fischer K, Bailey B, Cottage CT, Din S, Gude N, Avitabile D, Alvarez R, Sundararaman B, Quijada P, Mason M, Konstandin MH, Malhowski A, Cheng Z, Khan M, McGregor M: Myocardial AKT: the omnipresent nexus. Physiol Rev. 2011, 91: 1023-1070. 10.1152/physrev.00024.2010.
Cornu M, Modi H, Kawamori D, Kulkarni RN, Joffraud M, Thorens B: Glucagon-like peptide-1 increases beta-cell glucose competence and proliferation by translational induction of insulin-like growth factor-1 receptor expression. J Biol Chem. 2010, 285 (14): 10538-45. 10.1074/jbc.M109.091116.
Aroor AR, Sowers JR, Jia G, DeMarco VG: Pleiotropic Effects of the Dipeptidylpeptidase-4 Inhibitors on the Cardiovascular System. Am J Physiol Heart Circ Physiol. 2014, 307 (4): H477-H492. 10.1152/ajpheart.00209.2014.
Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I: SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013, 369 (14): 1317-26. 10.1056/NEJMoa1307684.
White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Cushman WC, Zannad F: EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013, 369 (14): 1327-35. 10.1056/NEJMoa1305889.
Gupta V: Pleiotropic effects of incretins. Indian J Endocrinol Metab. 2012, 16 (Suppl1): S47-S56. 10.4103/2230-8210.94259.
Kirkpatrick A, Heo J, Abrol R, Goddard WA: Predicted structure of agonist-bound glucagon-like peptide 1 receptor, a class B G protein-coupled receptor. Proc Natl Acad Sci U S A. 2012, 109 (49): 19988-93. 10.1073/pnas.1218051109.
Bullock BP, Heller RS, Habener JF: Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology. 1996, 137 (7): 2968-2978.
Ban K, Kim KH, Cho CK, Sauvé M, Diamandis EP, Backx PH, Drucker DJ, Husain M: Glucagon-like peptide (GLP)-1(9-36)amide-mediated cytoprotection is blocked by exendin(9-39) yet does not require the known GLP-1 receptor. Endocrinology. 2010, 151: 1520-1531. 10.1210/en.2009-1197.
Nyström T, Gutniak MK, Zhang Q, Zhang F, Holst JJ, Ahrén B, Sjöholm A: Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am J Physiol Endocrinol Metab. 2004, 287: E1209-E1215. 10.1152/ajpendo.00237.2004.
Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S: Glucagon-like peptide-1 (GLP-1) inhibits advanced glycation end product (AGE)-induced up-regulation of VCAM-1 mRNA levels in endothelial cells by suppressing AGE receptor (RAGE) expression. Biochem Biophys Res Commun. 2010, 391: 1405-1408. 10.1016/j.bbrc.2009.12.075.
Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M: Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and-independent pathways. Circulation. 2008, 117: 2340-2350. 10.1161/CIRCULATIONAHA.107.739938.
Ussher JR, Drucker DJ: Cardiovascular actions of incretin-based therapies. Circ Res. 2014, 114 (11): 1788-803. 10.1161/CIRCRESAHA.114.301958.
Kuna RS, Girada SB, Asalla S, Vallentyne J, Maddika S, Patterson JT, Smiley DL, DiMarchi RD, Mitra P: Glucagon-like peptide-1 receptor-mediated endosomal cAMP generation promotes glucose-stimulated insulin secretion in pancreatic ncreatic. Am J Physiol Endocrinol Metab. 2013, 305 (2): E161-70. 10.1152/ajpendo.00551.2012.
Vila Petroff MG, Egan JM, Wang X, Sollott SJ: Glucagon- like peptide-1 increases cAMP but fails to augment contraction in adult rat cardiac myocytes. Circ Res. 2001, 89: 445-452. 10.1161/hh1701.095716.
Gigoux V, Fourmy D: Acting on hormone receptors with minimal side effect on cell proliferation: a timely challenge illustrated with GLP-1R and GPER. Front Endocrinol. 2013, 4: 50-10.3389/fendo.2013.00050.
Xiao YF, Nikolskaya A, Jaye DA, Sigg DC: Glucagon-like peptide-1 enhances cardiac L-type Ca2+ currents via activation of the cAMP-dependent protein kinase A pathway. Cardiovasc Diabetol. 2011, 10: 6-10.1186/1475-2840-10-6.
Rose BA, Force T, Wang Y: Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010, 90: 1507-1546. 10.1152/physrev.00054.2009.
Chang G, Zhang D, Yu H, Zhang P, Wang Y, Zheng A, Qin S: Cardioprotective effects of exenatide against oxidative stress-induced injury. Int J Mol Med. 2013, 32 (5): 1011-20.
Shan PR, Xu WW, Huang ZQ, Pu J, Huang WJ: Protective role of retinoid X receptor in H9c2 cardiomyocytes from hypoxia/reoxygenation injury in rats. World J Emerg Med. 2014, 5 (2): 122-7. 10.5847/wjem.j.issn.1920-8642.2014.02.008.
Ravassa S, Zudaire A, Carr RD, Díez J: Antiapoptotic effects of GLP-1 in murine HL-1 cardiomyocytes. Am J Physiol Heart Circ Physiol. 2011, 300: H1361-H1372. 10.1152/ajpheart.00885.2010.
Nikolaidis LA, Elahi D, Hentosz T, Doverspike A, Huerbin R, Zourelias L, Stolarski C, Shen YT, Shannon RP: Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation. 2004, 110: 955-961. 10.1161/01.CIR.0000139339.85840.DD.
Moberly SP, Mather KJ, Berwick ZC, Owen MK, Goodwill AG, Casalini ED, Hutchins GD, Green MA, Ng Y, Considine RV, Perry KM, Chisholm RL, Tune JD: Impaired cardiometabolic responses to glucagon-like peptide 1 in obesity and type 2 diabetes mellitus. Basic Res Cardiol. 2013, 108 (4): 365-10.1007/s00395-013-0365-x.
Moberly SP, Berwick ZC, Kohr M, Svendsen M, Mather KJ, Tune JD: Intracoronary glucagon-like peptide 1 preferentially augments glucose uptake in ischemic myocardium independent of changes in coronary flow. Exp Biol Med. 2012, 237 (3): 334-42. 10.1258/ebm.2011.011288.
Zhao TC: Glucagon-like peptide-1 (GLP-1) and protective effects in cardiovascular disease: a new therapeutic approach for myocardial protection. Cardiovasc Diabetol. 2013, 12: 90-10.1186/1475-2840-12-90.
Sokos GG, Nikolaidis LA, Mankad S, Elahi D, Shannon RP: Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. J Card Fail. 2006, 12 (9): 694-699. 10.1016/j.cardfail.2006.08.211.
Sokos GG, Bolukoglu H, German J, Hentosz T, Magovern GJ, Maher TD, Dean DA, Bailey SH, Marrone G, Benckart DH, Elahi D, Shannon RP: Effect of glucagon-like peptide-1 (GLP-1) on glycemic control and left ventricular function in patients undergoing coronary artery bypass grafting. Am J Cardiol. 2007, 100 (5): 824-829. 10.1016/j.amjcard.2007.05.022.
Barragán JM, Rodríguez RE, Blázquez E: Changes in arterial blood pressure and heart rate induced by glucagon-like peptide-1-(7-36 amide) in rats. Am J Physiol. 1994, 266 (3): E459-E466.
Robinson LE, Holt TA, Rees K, Randeva HS, O’Hare JP: Effects of exenatide and liraglutide on heart rate, blood pressure and body weight: systematic review and meta-analysis.BMJ Open 2013, 3(1). doi:10.1136/bmjopen-2012-001986.,
Edwards CM, Todd JF, Ghatei MA, Bloom SR: Subcutaneous glucagon-like peptide-1 (7-36) amide is insulinotropic and can cause hypoglycaemia in fasted healthy subjects. Clin Sci. 1998, 95 (6): 719-724. 10.1042/CS19980050.
Ceriello A, Novials A, Canivell S, La Sala L, Pujadas G, Esposito K, Testa R, Bucciarelli L, Rondinelli M, Genovese S: Simultaneous GLP-1 and insulin administration acutely enhances their vasodilatory, antiinflammatory, and antioxidant action in type 2 diabetes. Diabetes Care. 2014, 37 (7): 1938-43. 10.2337/dc13-2618.
Erdogdu O, Nathanson D, Sjöholm A, Nyström T, Zhang Q: Exendin-4 stimulates proliferation of human coronary artery endothelial cells through eNOS-, PKA- and PI3K/Akt-dependent pathways and requires GLP-1 receptor. Mol Cell Endocrinol. 2010, 325: 26-35. 10.1016/j.mce.2010.04.022.
Wang D, Luo P, Wang Y, Li W, Wang C, Sun D, Zhang R, Su T, Ma X, Zeng C, Wang H, Ren J, Cao F: Glucagon-like peptide-1 protects against cardiac microvascular injury in diabetes via a cAMP/PKA/Rho-dependent mechanism. Diabetes. 2013, 62 (5): 1697-708. 10.2337/db12-1025.
Batchuluun B, Inoguchi T, Sonoda N, Sasaki S, Inoue T, Fujimura Y, Miura D, Takayanagi R: Metformin and liraglutide ameliorate high glucose-induced oxidative stress via inhibition of PKC-NAD(P)H oxidase pathway in human aortic endothelial cells. Atherosclerosis. 2014, 232 (1): 156-64. 10.1016/j.atherosclerosis.2013.10.025.
Kelly AS, Richard M, Bergenstal RM, Gonzalez-Campoy JM, Harold Katz H, Bank AJ: Effects of Exenatide vs. Metformin on endothelial function in obese patients with pre-diabetes: a randomized trial. Cardiovasc Diabetol. 2012, 11: 64-10.1186/1475-2840-11-64.
Basu A, Charkoudian N, Schrage W, Rizza RA, Basu R, Joyner MJ: Beneficial effects of GLP-1 on endothelial function in humans: dampening by glyburide but not by glimepiride. Am J Physiol Endocrinol Metab. 2007, 293: E1289-E1295. 10.1152/ajpendo.00373.2007.
Ceriello A, Esposito K, Testa R, Bonfigli AR, Marra M, Giugliano D: The possible protective role of glucagon-like peptide 1 on endothelium during the meal and evidence for an “endothelial resistance” to glucagon-like peptide 1 in diabetes. Diabetes Care. 2011, 34: 697-702. 10.2337/dc10-1949.
Gutzwiller JP, Tschopp S, Bock A, Zehnder CE, Huber AR, Kreyenbuehl M, Gutmann H, Drewe J, Henzen C, Goeke B, Beglinger C: Glucagon-like peptide 1 induces natriuresis in healthy subjects and in insulin-resistant obese men. J Clin Endocrinol Metab. 2004, 89 (6): 3055-61. 10.1210/jc.2003-031403.
Kim M, Platt MJ, Shibasaki T, Quaggin SE, Backx PH, Seino S, Simpson JA, Drucker DJ: GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat Med. 2013, 19 (5): 567-75. 10.1038/nm.3128.
Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, Fujitani Y, Hirose T, Kawamori R, Watada H: Inhibition of monocyte adhesion to endothelial cells and attenuation of atherosclerotic lesion by a glucagon-like peptide-1 receptor agonist, exendin-4. Diabetes. 2010, 59: 1030-1037. 10.2337/db09-1694.
Piotrowski K, Becker M, Zugwurst J, Biller-Friedmann I, Spoettl G, Greif M, Leber AW, Becker A, Laubender RP, Lebherz C, Goeke B, Marx N, Parhofer KG, Lehrke M: Circulating concentrations of GLP-1 are associated with coronary atherosclerosis in humans. Cardiovasc Diabetol. 2013, 12: 117-10.1186/1475-2840-12-117.
Goto H, Nomiyama T, Mita T, Yasunari E, Azuma K, Komiya K, Arakawa M, Jin WL, Kanazawa A, Kawamori R, Fujitani Y, Hirose T, Watada H: Exendin-4, a glucagon-like peptide-1 receptor agonist, reduces intimal thickening after vascular injury. Biochem Biophys Res Commun. 2011, 405: 79-84. 10.1016/j.bbrc.2010.12.131.
Nagashima M, Watanabe T, Terasaki M, Tomoyasu M, Nohtomi K, Kim-Kaneyama J, Miyazaki A, Hirano T: Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia. 2011, 54: 2649-2659. 10.1007/s00125-011-2241-2.
Tashiro Y, Sato K, Watanabe T, Nohtomi K, Terasaki M, Nagashima M, Hirano T: A glucagon-like peptide-1 analog liraglutide suppresses macrophage foam cell formation and atherosclerosis. Peptides. 2014, S0196-9781 (13): 00431-2.
Liu H, Dear AE, Knudsen LB, Simpson RW: A long-acting glucagon-like peptide-1 analogue attenuates induction of plasminogen activator inhibitor type-1 and vascular adhesion molecules. J Endocrinol. 2009, 201: 59-66. 10.1677/JOE-08-0468.
Best JH, Hoogwerf BJ, Herman WH, Pelletier EM, Smith DB, Wenten M, Hussein MA: Risk of cardiovascular disease events in patients with type 2 diabetes prescribed the glucagon-like peptide 1 (GLP-1) receptor agonist exenatide twice daily or other glucose-lowering therapies: a retrospective analysis of the LifeLink database. Diabetes Care. 2011, 34 (1): 90-5. 10.2337/dc10-1393.
Ratner R, Han J, Nicewarner D, Yushmanova I, Hoogwerf BJ, Shen L: Cardiovascular safety of exenatide BID: an integrated analysis from controlled clinical trials in participants with type 2 diabetes. Cardiovasc Diabetol. 2011, 10: 22-10.1186/1475-2840-10-22.
Nathanson D, Ullman B, Löfström U, Hedman A, Frick M, Sjöholm A, Nyström T: Effects of intravenous exenatide in type 2 diabetic patients with congestive heart failure: a double-blind, randomised controlled clinical trial of efficacy and safety. Diabetologia. 2012, 55 (4): 926-35. 10.1007/s00125-011-2440-x.
Nikolaidis LA, Mankad S, Sokos GG, Miske G, Shah A, Elahi D, Shannon RP: Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation. 2004, 109 (8): 962-5. 10.1161/01.CIR.0000120505.91348.58.
Woo JS, Kim W, Ha SJ, Kim JB, Kim SJ, Kim WS, Seon HJ, Kim KS: Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler Thromb Vasc Biol. 2013, 33 (9): 2252-60. 10.1161/ATVBAHA.113.301586.
Hermansen K, Bækdal TA, Düring M, Pietraszek A, Mortensen LS, Jørgensen H, Flint A: Liraglutide suppresses postprandial triglyceride and apolipoprotein B48 elevations after a fat-rich meal in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, cross-over trial. Diab Obesity Metabol. 2013, 15: 1040-1048. 10.1111/dom.12133.
Meloni AR, DeYoung MB, Han J, Best JH, Grimm M: Treatment of patients with type 2 diabetes with exenatide once weekly versus oral glucose-lowering medications or insulin glargine: achievement of glycemic and cardiovascular goals. Cardiovasc Diabetol. 2013, 12: 48-10.1186/1475-2840-12-48.
Zhao T, Parikh P, Bhashyam S, Bolukoglu H, Poornima I, Shen YT, Shannon RP: Direct effects of glucagon-like peptide-1 on myocardial contractility and glucose uptake in normal and postischemic isolated rat hearts. J Pharmacol Exp Ther. 2006, 317: 1106-1113. 10.1124/jpet.106.100982.
Hattori Y, Jojima T, Tomizawa A, Satoh H, Hattori S, Kasai K, Hayashi T: A glucagon-like peptide-1 (GLP-1) analogue, liraglutide, upregulates nitric oxide production and exerts anti-inflammatory action in endothelial cells. Diabetologia. 2010, 53 (10): 2256-63. 10.1007/s00125-010-1831-8.
Read PA, Khan FZ, Dutka DP: Cardioprotection against ischaemia induced by dobutamine stress using glucagon-like peptide-1 in patients with coronary artery disease. Heart. 2012, 98 (5): 408-13. 10.1136/hrt.2010.219345.
Chiquette E, Toth PP, Ramirez G, Cobble M, Chilton R: Treatment with exenatide once weekly or twice daily for 30 weeks is associated with changes in several cardiovascular risk markers. Vasc Health Risk Manag. 2012, 8: 621-9.
Acknowledgement
The author wishes to thank Daniel Soares Freire, MD, PhD for providing medical writing and editorial assistance on behalf of Springer Healthcare. This manuscript was prepared according to the International Society for Medical Publication Professionals’ Good Publication Practice for Communicating Company-Sponsored Medical Research: the GPP2 Guidelines. Funding to support the preparation of this manuscript was provided by Novo Nordisk Inc. Prof. Sposito was supported by a fellowship grant of productivity in research from Brazilian National Research Council (CNPq) (grant number 300313/2007-1).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests that would influence the content of this review.
Authors’ contributions
ACS and FKS participated in the concept and drafting of the manuscript and ACS made the critical review for intellectual content and final approval. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Saraiva, F.K., Sposito, A.C. Cardiovascular effects of Glucagon-like peptide 1 (GLP-1) receptor agonists. Cardiovasc Diabetol 13, 142 (2014). https://doi.org/10.1186/s12933-014-0142-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-014-0142-7