
Abstract
Background
Chronic obstructive pulmonary disease (COPD) is associated with elevated ATP levels in the extracellular space. Once released, ATP serves as danger signal modulating immune responses by activating purinergic receptors. Accordingly, purinergic signalling has been implicated in respiratory inflammation associated with cigarette smoke exposure. However, the role of P2X4-signalling has not been fully elucidated yet.
Methods
Here, we analysed the P2X4 mRNA expression in COPD patients as well as cigarette smoke-exposed mice. Furthermore, P2X4-signalling was blocked by either using a specific antagonist or genetic depletion of P2rx4 in mice applied to an acute and prolonged model of cigarette smoke exposure. Finally, we inhibited P2X4-signalling in macrophages derived from THP-1 before stimulation with cigarette smoke extract.
Results
COPD patients exhibited an increased P2X4 mRNA expression in cells isolated from the bronchoalveolar lavage fluid and peripheral mononuclear cells. Similarly, P2rx4 expression was elevated in lung tissue of mice exposed to cigarette smoke. Blocking P2X4-signalling in mice alleviated cigarette smoke induced airway inflammation as well as lung parenchyma destruction. Additionally, human macrophages derived from THP-1 cells released reduced concentrations of proinflammatory cytokines in response to cigarette smoke extract stimulation when P2X4 was inhibited.
Conclusion
Taken together, we provide evidence that P2X4-signalling promotes innate immunity in the immunopathologic responses induced by cigarette smoke exposure.
Similar content being viewed by others


Background
About two decades ago, adenosine 5′-triphosphate (ATP) has been indicated to play a role in chronic obstructive airway diseases for the first time [1]. Released into extracellular space ATP serves as damage-associated molecular pattern (DAMP), which drives inflammation by facilitating recruitment as well as activation of various immune cell types via binding to purinergic receptors [2, 3]. Consequently, elevated levels of extracellular ATP in the bronchoalveolar lavage fluid (BALF) have been reported in chronic inflammatory diseases of the airways including asthma and chronic obstructive pulmonary disease (COPD) [4, 5]. Accordingly, mice exposed to cigarette smoke exhibited increased ATP concentrations in the BALF compared to air controls [6]. The increased ATP release upon inflammatory conditions in the airways has been attributed to epithelial cells and activated immune cells such as macrophages and neutrophils [2, 7].
According to their functional properties, the P2 receptors are classified into the ionotropic P2X and the metabotropic P2Y receptors [8]. Members of both subclasses have been implicated in COPD pathogenesis. For instance, P2X7 activation has been shown to promote pulmonary inflammation via IL-1ß mediated release of proinflammatory cytokines, chemokines and proteases [9, 10]. Accordingly, genetic ablation of P2rx7 resulted in reduced IL-1ß production and alleviated cigarette smoke (CS)-induced airway inflammation in mice [11]. P2X7 shares high similarities in terms of sequences, expression patterns and physiological functions with P2X4 [12]. Interestingly, several studies suggest an interaction between these two P2X receptors [13,14,15,16]. However, although P2X7 has been implicated in CS-induced airway inflammation a potential role of P2X4 has not been elucidated yet.
During the last two decades much progress has been made in understanding the complex interplay of extracellular ATP and the pathophysiological processes in pulmonary inflammation [17]. However, many aspects about the role of purinergic signalling in chronic lung disorders like COPD remain elusive. Therefore, the aim of this study was to investigate the potential role of P2X4 in CS-induced airway inflammation as well as airway remodelling. Here we demonstrate that P2X4-signalling contributes to CS-induced airway inflammation by promoting innate immunity.
Methods
Human subjects
Individuals with COPD and healthy never-smokers (Additional file 1: Tables S1 and S2) were recruited at University Hospital Freiburg, Germany. Patients and healthy volunteers gave their written informed consent to use the obtained biologic samples for research. The analysis of the samples was approved by the local ethics committee (EC Nr. 06.2006 & 10.2006).
Mice
C57BL/6N (6–8 weeks old) mice were purchased from Charles River (Sulzfeld, Germany). The P2rx4-deficient (MGI: 3665297) mouse was originally designed in Francois Rassendren’s lab at the Centre National de la Recherche Scientifiqu in the Department of Molecular Pharmacology as previously described [18]. P2rx4-deficient animals were crossed back in C57BL/6 N background and bred under specific pathogen-free (SPF) conditions in the animal facility of Freiburg University. Male, 8–10 weeks old mice were used for all the experiments. For experiments including solely wild type mice, the treated animals and the corresponding controls were littermates. Experiments on P2rx4-deficiency included P2rx4-deficient mice and wild type controls from separate nests. All mice experiments were approved by the local ethics committee [G-12/106, G-14/108] and executed according to national law.
Murine models of cigarette smoke-induced airway inflammation and P2X4 inhibition
A whole-body exposure chamber containing the mice was mechanically ventilated (7025 rodent ventilators, Ugo Basile, Biological Research Instruments, Comerio, Italy) with either the smoke from five commercial Marlboro Red cigarettes (10 mg of tar, 10 mg of carbon monoxide and 0.9 mg nicotine) diluted 1:10 with air or air alone for 20 min. Mice were exposed to CS of five subsequent lit cigarettes for 20 min daily on three consecutive days in an acute or on each of five consecutive days per week for four months in a chronic approach.
Intratracheal (i.t.) administration of 80 µl PBS containing 10 µM 5-(3-Bromophenyl)-1,3-dihydro-2H-benzofuro-[3,2-e]-1,4-diazepin-2-one dissolved in DMSO (Tocris, Bristol, UK) or 80 µl PBS with 0.05% DMSO as vehicle control was performed 30 min prior to every smoke exposure. The amount of 10 µM chosen was based on preliminary dosing experiments (data not shown) according to results from an ATP-induced current inhibition assay applied with transfected HEK293 cells [19]. To minimize cage effects mice within the same cage were randomly allocated to the different treatment groups of the corresponding experiment.
Bronchoalveolar lavage fluid (BALF)
First, the trachea of sacrificed mice was penetrated with a cannula. Subsequently, the lung was flushed with 0.75 ml PBS (Gibco, Thermo Fisher Scientific, Germany) supplemented with 0.5 mM EDTA (Sigma-Aldrich, Taufkirchen, Germany) four times. Every first flush was collected in a separate tube and the corresponding supernatant was later used for cytokine measurements via enzyme-linked immunosorbent assay (ELISA). BAL cells from individual mouse were pooled and used for differential cell counting via flow cytometry.
Histology
After fixing the lungs with 10% formalin (Merck, Darmstadt, Germany) for 48 h, they were embedded in paraffin. The prior cut 7 µm lung sections were dewaxed and rehydrated using xylene and a series of decreasing alcohol concentrations. Finally, hematoxylin and eosin were applied to stain lung sections (Carl Roth, Karlsruhe, Germany).
Forty histological fields were evaluated both vertically and horizontally to determine the average interalveolar distance (mean linear intercept: Lm). Counting was performed independently by two members of the lab. The mean Lm and postfixation lung volume were used to calculate the internal surface area of the lungs (ISA) [20].
Flow cytometry
First, BALF cells were washed in PBS containing 0.5% BSA and 0.01% NaN3 and counted. To avoid nonspecific binding cells were incubated with an unlabeled anti-CD16/32 antibody (AB_467133) before adding a mix containing anti-Gr-1 FITC − (AB_465314), anti-F4/80 PE − (AB_465923), anti-Cd11c APC − (AB_469346), anti-CD3e PE-Cy7 (AB_469572) -labeled antibodies (eBioscience, San Diego, CA). Flow cytometry was performed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo v10 (TreeStar, Ashland, OR) software. Different cell types were identified with the aid of differential marker expression (CD3/B220 + CD11c − Lymphocytes, F4/80 + CD11c + macrophages and Gr-1 + CD3/B220 − CD11c − neutrophils) and via forward and side scatter positioning.
Quantitative real-time polymerase chain reaction (qPCR)
Murine lungs were incubated in RNAlater Stabilization Reagent (QIAGEN GmbH, Hilden, Germany) at 2–8° C overnight and frozen for long-term storage (− 20 °C). Total RNA from human cells or murine tissue was isolated using QIAzol lysis reagent (QIAGEN GmbH, Hilden, Germany). Chloroform was added to the lysates for separation. The aqueous phase containing the RNA was transferred before isopropanol was added for precipitation. The RNA pellet was washed two times with 70% Ethanol for the subsequent two washing steps, and eventually dissolved in nuclease-free water. 28S and 18S rRNA bands were evaluated after gel electrophoresis to confirm RNA integrity. After eliminating remaining genomic DNA via gDNA eliminator column (QIAGEN GmbH, Hilden, Germany), first strand cDNA synthesis kit (Thermo Fisher Scientific GmbH, Germany) was used to synthesize cDNA. The qPCR mixes containing Takyon mastermix (Eurogentec, Köln, Germany) were applied to a LightCycler 480 (Roche, Mannheim, Germany). Primer sequences are available in the Additional file 1. Gene expression was calculated using the following formula:
Cigarette smoke extract stimulation of macrophages derived from monocyte-like THP-1 cells
THP-1 cells were obtained from German Collection of Microorganisms and Cell Cultures GmbH (Braunschweig, Germany). Undifferentiated THP-1 cells were cultivated in RPMI-1640 medium (Gibco, Thermo Fisher Scientific, Germany) supplemented with 1% penicillin/streptomycin and 10% heat-inactivated fetal calf serum (FCS, Biocell Laboratories, Rancho Dominguez, CA) in 150 cm [2] culture flask (Corning Life Sciences, NY, USA) till a density of 3 × 107 was reached. After adjusting to 1.5 × 106 cells/well in a 6-well plate (Greiner Bio-One, Frickenhausen, Germany), cells were incubated with 100 nM phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, Taufkirchen, Germany) for three days to induce macrophage-like differentiation. Twenty-four hours before stimulation, the cells were detached by using 5 ml accutase (Sigma-Aldrich, Taufkirchen, Germany) for 45 min at 37 °C and adjusted to 2.5 × 105 cells/well in a 24-well-plate (Greiner Bio-One, Frickenhausen, Germany). After letting the cells adhere for 2 h, the medium was removed and replaced with FCS-free medium for conditioning.
The 100% cigarette smoke extract (CSE) was prepared by infusing CS of one cigarette (Marlboro Red, 10 mg of tar, 10 mg of carbon monoxide and 0,9 mg nicotine) into 10 ml pre-warmed 37 °C RPMI medium using a 50 ml syringe, followed by sterile filtration through a 0.22 μm cellulose acetate sterilizing system (Corning Life Sciences, NY, USA). The CSE was used immediately after preparation. A 5% CSE stimulation is associated with smoking slightly less than one pack of cigarettes per day. To block P2X4-signalling, 10 µM 5-(3-Bromophenyl)-1,3-dihydro-2H-benzofuro-[3,2-e]-1,4-diazepin-2-one (5-BDBD) or vehicle (PBS with 0.05% DMSO) were added to the cell culture 30 min prior to CSE stimulation.
Measurement of mediator concentrations
To determine the concentrations of inflammatory mediators including CXCL1/KC, MCP-1, IL-1ß, IL-6, neutrophil elastase, MMP-9 and TNFa, sandwich ELISAs was applied. Two replicates for standard as well as supernatant samples were measured and supernatant samples were diluted in the solvent of the standard protein if necessary. All ELISA kits were purchased from R&D Systems (Wiesbaden-Nordenstadt, Germany) and the procedure was performed according to the corresponding manufacturer’s kit manuals.
Statistical analysis
Statistical tests were performed using Graphpad Prism v9 (GraphPad Software, San Diego, CA) and Microsoft Excel. All data was analysed for homoscedasticity using Levene’s and Brown-Forsythe test. The P2RX4 expression in human cells and P2rx4 expression in murine lung tissue were analysed using the unpaired t-test with Welch’s correction to adjust for unequal variance. The data concerning antagonist treatment were analysed Welch’s analysis of variance (ANOVA) with Dunnett-T3 post hoc test to correct for multiple comparisons. Data concerning P2rx4-deficincy were analysed using two-way ANOVA with Tukey’s post hoc test to correct for multiple comparisons. P ≤ 0.05 was considered statistically significant.
Results
COPD and cigarette smoke exposure are associated with an enhanced P2RX4 expression
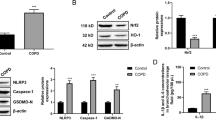
To investigate whether obstructive pulmonary disease (COPD) is associated with an altered P2RX4 expression, the P2RX4 expression in BALF cells as well as blood mononuclear cells (MNCs) of COPD patients was compared to never-smoking healthy individuals. Interestingly, COPD-patients showed an elevated P2RX4 in both, BALF cells and MNCs (Fig. 1a, b). Similarly, lung tissue (Fig. 1c) as well as BALF cells (Additional file 1: Fig. S1), isolated from mice exposed to CS for three consecutive days, exhibited an increased P2rx4 expression compared to the air-control animals.

P2RX4 expression in murine lung tissue, human BALF and peripheral blood mononuclear cells. Relative P2rx4 (murine)/P2RX4 (human) expression determined via quantitative PCR in a) BALF cells and b) peripheral blood mononuclear cells obtained from COPD patients as well as in c) murine lung tissue isolated after cigarette smoke (CS) exposure for three consecutive days compared to non-smoking (Air/NS) individuals. P2X4 mRNA expression was quantified relative to ß2 microglobulin (ß2m) levels. mice: Air n = 9, CS n = 17; human BALF: n = 15 in both groups; human blood mononuclear cells (MNCs): never smokers n = 22, COPD n = 26. Each symbol represents individual subject. Graphs include mean with standard deviation unpaired t-Test with Welch’s correction was applied for group comparison. P is depicted in the corresponding graph and was considered statistical significant when ≤ 0.5
In case of stratification by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) classification into “GOLD III and IV” and “GOLD I and II” COPD patients, P2RX4 expression was increased in BALF cells and blood MNCs of GOLD III and IV COPD patients while COPD patients classified as GOLD I and II showed no significant difference compared to never-smokers (Additional file 1: Fig. S2). Of note, the active smokers among the GOLD I and GOLD II COPD patients tended to exhibit a higher P2RX4 expression compared to GOLD I and GOLD II ex-smokers. However, due to the exploratory character of this human study there are differences in known confounding factors including age and gender ratio between the COPD patients and the never-smoking healthy subjects (Additional file 1: Tables S1, S2).
Blocking P2X4-signalling alleviates cigarette smoke-induced airway inflammation
To evaluate the role of P2X4 in CS-induced airway inflammation mice were treated with the selective P2X4 antagonist 5-BDBD i.t. prior daily CS exposure. Consequently, 5-BDBD administration caused an alleviated CS-induced acute airway inflammation compared to vehicle instillation in C57BL/6 N mice. This was evidenced by the reduction in numbers of BALF neutrophils, macrophages and lymphocytes (Fig. 2a) as well as concentrations of the proinflammatory cytokines CXCL1/KC, MCP-1, IL-1ß and IL-6 in the BALF supernatant (Fig. 2b).

Inhibition of P2RX4-mediated signalling alleviates CS-induced airway inflammation in mice. Differential cell counts as well as concentrations of proinflammatory cytokines (CXCL1/KC, MCP-1, IL-1ß, IL-6) in the BALF of mice a, b treated with 10 µM 5-BDBD 30 min prior each daily smoke exposure, and c, d deficient for P2rx4 (P2rx4−/−) after cigarette smoke (CS) exposure for three consecutive days compared to untreated (Vehicle/CS) or wild type (P2rx4+/+) animals, respectively. Data is presented as individual symbols (n = 6–7) and mean + SD. Treatment data was analysed applying Welch’s ANOVA corrected for multiple comparison using Dunett’s T3 test. Deficiency data was analysed applying two-way ANOVA corrected for multiple comparison using Tukey test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
For further confirmation, P2rx4-deficient mice were subjected to CS exposure on three consecutive days. Similar to 5-BDBD treatment, P2rx4-deficiency was associated with decreased differential cell numbers in the BALF compared to wild type control animals (Fig. 2c). Likewise, P2rx4-deficient mice exhibited reduced BALF concentrations of CXCL1/KC, MCP-1, IL-1ß and IL-6 (Fig. 2d). Interestingly, P2rx4 deficiency was also associated with diminished IL-17 concentrations (Additional file 1: Fig. S3A). Altogether, the results suggested that P2X4-signalling promotes acute CS-induced airway inflammation.
P2rx4-deficiency dampens cigarette smoke-driven emphysema development
Besides proinflammatory cytokines, P2rx4-deficiency was associated with a diminished release neutrophil elastase and MMP-9 (Additional file 1: Fig. S3B) after acute smoke exposure. Among other tissue-degrading proteases, MMP-9 and neutrophil elastase have been implicated in airway remodelling and emphysema development during COPD [21]. The reduction of these mediators in the BALF of P2rx4-deficient mice suggested that P2X4-signalling drives airway remodelling by facilitating protease release.
To evaluate the role of P2X4 in airway remodelling and emphysema development P2rx4-deficient mice were exposed to CS five days a week for four months. Analogous to the acute approach, P2rx4-deficiency was associated with diminished neutrophil, macrophage, and lymphocyte numbers (Fig. 3a) as well as concentrations proinflammatory cytokines (Additional file 1: Fig. S4), neutrophil elastase, and MMP9 (Fig. 3b) in the BALF compared to wild type controls. Furthermore, P2rx4-deficient mice were partially protected from lung parenchyma destruction evidenced by a reduced mean linear intercept (Lm) and increased mean internal surface area (ISA) (Fig. 3c).

P2rx4-deficiency is associated with reduced airway inflammation and remodelling in response to regular cigarette smoke exposure for 4 months. a Differential cell count, and b concentrations of proteases in the BALF (ELA2, MMP-9) of P2rx4-deficient mice (P2rx4−/−) after exposure with the smoke of 5 cigarettes per day, five days a week for four months compared to wild type animals (P2rx4+/+). c Exemplary hematoxylin & eosin stained paraffin lung section (40 × magnification, 100 µm scale bar) used to quantify lung parenchyma destruction via calculating the linear mean intercept (Lm) and mean internal surface area (ISA). Mean Lm and ISA ± SDs are depicted under the corresponding histology pictures. BALF data is presented as individual symbols (n = 5) and mean + SD. Two-way ANOVA corrected for multiple comparison using Tukey test was applied for group comparison. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
Inhibiting P2X4-signalling alleviates the release of proinflammatory cytokines by monocyte-derived macrophages in response to cigarette smoke extract stimulation
Airway inflammation as well as remodelling is driven by chronic pulmonary inflammation perpetuated by immune cells infiltrating the lung. Myeloid cells and lymphocytes have been shown to play a crucial role in COPD pathogenesis and both cell types, also express high P2RX4 levels [22,23,24]. Since P2RX4 is expressed in alveolar macrophages and shows an increased expression in MNCs isolated from COPD patients (Fig. 1b), we suggested monocyte-derived cells to play a pivotal role in P2X4-mediated effects in acute cigarette smoke-induced airway inflammation.
To investigate the role of P2X4-signalling in myeloid cells, we assessed the secretion of proinflammatory cytokines by 5-BDBD treated human non-polarized (M0) macrophages derived from P2X4-expressing THP-1 cells [25, 26] in response to CSE stimulation. Indeed, 5-BDBD treated THP-1 derived macrophages secreted less TNFα, IL-1ß, MCP-1 and IL-6 compared to cells not pre-incubated with 5-BDBD (Fig. 4).

Blocking P2X4-signaling in THP-1 derived macrophages alleviates pro-inflammatory cytokine release in response to cigarette smoke extract stimulation. Concentrations of IL-1ß, TNFα, IL-6 and MCP-1 in the supernatant of THP-1 derived non-polarized macrophages treated with 10 µM of the specific P2X4-antagonist 5-BDBD 30 min prior to stimulation with 5% cigarette smoke extract compared to non-treated cells. Data is presented as individual symbols for 6 wells from three different experiments and mean + SD. Welch’s ANOVA corrected for multiple comparison using Dunett’s T3 was used for group comparison. *P < 0.05, **P < 0.01
Discussion
It has been clearly shown that purinergic signalling plays a role in airway inflammation in mice and in humans [2]. However, the particular contribution of the different receptor subtypes to the pathological mechanisms related to the inflammatory response have not been fully elucidated, yet. A previous study has implicated that P2X7 drives CS-induced airway inflammation by modulating IL-1ß maturation and release [11]. Although P2X4 closely correlates with P2X7, [12,13,14,15,16] the role of P2X4 in CS-driven pulmonary inflammation remains unclear.
In the present study, we demonstrate that P2rx4 expression in the lungs of CS-exposed was increased. Furthermore, BALF cells and peripheral MNCs isolated from COPD patients, exhibit elevated P2RX4 levels compared to healthy never smokers. Of note, whereas GOLD III and IV COPD patients show a remarkable increase in P2RX4 levels, GOLD I and II COPD patients exhibit no significant P2RX4 induction. Interestingly, active smoking tends to yield a higher P2RX4 expression the BALF cells and blood MNCs compared to ex-smokers in the BALF cells and blood MNCs isolated from GOLD I and GOLD II patients. However, due to the pronounced differences in two known confounding factors, age and gender, between the groups as well as the small sample size, the results might be biased and we consider these human data rather exploratory. In order to draw a solid inference a larger number of volunteers with similar proportions of confounding factors is needed. Nevertheless, these data suggest that CS-exposure in mice as well as humans enhances P2RX4 expression.
The inhibition as well as disruption of P2X4-signalling resulted in an alleviated CS-induced airway inflammation and proved functional relevance of the increased P2rx4-expression observed in CS-exposed mice. This is in line with previous studies reporting a suppressive effect of 5-BDBD treatment as well as P2rx4-deficiency in allergen-driven models of pulmonary inflammation [27, 28]. Besides a reduced airway inflammation, P2rx4-deficient mice showed less parenchymal destruction after four months of CS exposure accompanied by diminished neutrophil elastase as well as MMP9 BALF levels compared to wild type animals. Both proteases have been implicated in matrix remodelling and airway destruction in COPD [21]. These data suggest P2X4-signalling to contribute to CS-driven airway inflammation and airway remodelling.
In the airways, IL-1ß is considered a master regulator in immune defence against inhaled noxious including cigarette smoke [29]. BALF and sputum of COPD patients exhibit an increased IL-1ß production compared to never-smokers [30, 31]. Accordingly, mice overexpressing human IL-1ß show pulmonary inflammation and an increased secretion of CXCL1/KC, CXCL2/ MIP-2, MMP-9 and MMP-12 [9]. Interestingly, previous studies demonstrated that P2X4 regulates ATP-driven inflammasome activation and subsequent IL-1ß release in several organs [32]. Therefore, we suggested P2X4-signalling to contribute to CS-induced airway inflammation by promoting ATP-driven IL-1ß maturation and subsequent release. Support for this hypothesis comes from the reduced IL-1ß, CXCL1/KC and MMP-9 concentrations observed in the BALF of 5-BDBD treated as well as P2rx4-deficient mice in response to CS-exposure.
Among other cell types activated monocytes and macrophages are considered a major source of IL-1ß. In addition, myeloid cells play a crucial role in COPD pathogenesis and represent the predominant cell type in the BALF of smokers [22, 33]. Of note, monocytes and macrophages have been reported to exhibit a high P2RX4 expression according to www.proteinatlas.org/ and literature [23, 34]. We demonstrate that P2RX4 expression, is considerably increased in COPD patients. In addition, non-polarized macrophages derived from the monocyte-like, human THP-1 cells pre-incubated with 5-BDBD released reduced amounts of not only IL-1ß, but other proinflammatory cytokines including IL-6, MCP-1 and TNFα in response to CSE stimulation.
It has been shown that P2X4 is preferentially localized in lysosomes of macrophages [35]. The intra-endolysosomal Ca2+ release triggered by activated P2X4 leads to calmodulin recruitment and the subsequent formation of P2X4-calmodulin complexes at endolysosomal membranes, which promote vesicle fusion [36]. Calcium/calmodulin binds to calcium-sensitive proteins like calmodulin dependent kinase II delta (CAMK2D), which have been implicated in modulating NF-κB signalling [37]. Active NF-κB, in turn, promotes the transcription of proIL-1ß, proIL-18 and Nlrp3, all associated with inflammasome activation [38]. Therefore, we suggest that P2X4 promotes CS-induced airway inflammation by driving inflammasome activation, IL-1ß release and NF-κB activation in monocytic cells. However, P2X4-signalling in other cell types including hematopoietic and structural cells may also contribute to the noxious effects in the lung induced by CS. For instance, P2X4-signalling in airway epithelial cells as well as alveolar type II cells has been associated with an augmented mucin and surfactant secretion, and alveolar fluid transport [39, 40].
Besides P2X4, P2X7 has also been implicated in an ATP-dependent NF-κB activation [41,42,43]. Additionally, former studies suggest a physical interaction of P2X4 and P2X7 in myeloid cells [44]. Functionally, this interaction has been associated with ATP-induced IL-1ß release [15, 45]. Furthermore, both P2X4 and P2X7 have been implicated in NALP3 inflammasome activation [46,47,48]. Weinhold et al. propose compensatory mechanism of the expression of P2X4 and P2X7, thus knocking down the expression of one increases the expression of the other [16]. In contrast, several studies report that knocking out either P2X4 or P2X7 reduces the expression of the other one as well [14, 28]. While bone marrow derived macrophages (BMDMs) derived from wild type mice exhibit an increased P2rx4 and P2rx7 expression, P2rx4-deficient BMDMs fail to induce P2rx7 in response to CSE stimulation (Additional file 1: Figs. S5, S6). Therefore, we suppose that P2X4-signalling modulates P2RX7 expression in monocytic cells, which might augment suppressive effects associated with PX4-inhibition and P2rx4-deficiency.
Conclusion
Taken together, our data demonstrate a clear role of P2X4 in cigarette smoke induced airway inflammation and cigarette smoke driven airway remodelling. The blocking of P2X4-signalling exerts inhibitory effects on immune cell lung recruitment and the subsequent release of proinflammatory cytokines and mediators including IL-1ß as well as proteases. This suggests that P2X4-signalling facilitates innate immunity in the lung and highlight the potential role of P2X4 in the course of immunopathologic responses induced by cigarette smoke. Therefore, the implications of these observations add another piece to the puzzle concerning the basic understanding of purinergic signalling in airway diseases.
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 5-BDBD:
-
5-(3-Bromophenyl)-1,3-dihydro-2H-benzofuro-[3,2-e]-1,4-diazepin-2-one
- BALF:
-
Bronchoalveolar lavage fluid
- CS:
-
Cigarette smoke
- CSE:
-
Cigarette smoke extract
- ISA:
-
Internal surface area
- Lm:
-
Mean linear intercept
- P2rx4/P2RX4:
-
Purinergic receptor P2X 4 (gene)
- P2X4:
-
Purinergic receptor P2X 4 (protein)
References
Pelleg A, Schulman ES. Adenosine 5′-triphosphate axis in obstructive airway diseases. Am J Ther. 2002;9:454–64. https://doi.org/10.1097/00045391-200209000-00014.
Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509:310–7. https://doi.org/10.1038/nature13085.
Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16:177–92. https://doi.org/10.1038/nri.2016.4.
Idzko M, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med. 2007;13:913–9. https://doi.org/10.1038/nm1617.
Lommatzsch M, et al. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:928–34. https://doi.org/10.1164/rccm.200910-1506OC.
Cicko S, et al. Purinergic receptor inhibition prevents the development of smoke-induced lung injury and emphysema. J Immunol. 2010;185:688–97. https://doi.org/10.4049/jimmunol.0904042.
Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367:2322–33. https://doi.org/10.1056/NEJMra1205750.
Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11:201–12. https://doi.org/10.1038/nri2938.
Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32:311–8. https://doi.org/10.1165/rcmb.2004-0309OC.
Mortaz E, Adcock IM, Shafei H, Masjedi MR, Folkerts G. Role of P2X7 receptors in release of IL-1beta: a possible mediator of pulmonary inflammation. Tanaffos. 2012;11:6–11.
Lucattelli M, et al. P2X7 receptor signaling in the pathogenesis of smoke-induced lung inflammation and emphysema. Am J Respir Cell Mol Biol. 2011;44:423–9. https://doi.org/10.1165/rcmb.2010-0038OC.
Kopp R, Krautloher A, Ramirez-Fernandez A, Nicke A. P2X7 interactions and signaling—making head or tail of it. Front Mol Neurosci. 2019. https://doi.org/10.3389/fnmol.2019.00183.
Antonio LS, et al. P2X4 receptors interact with both P2X2 and P2X7 receptors in the form of homotrimers. Br J Pharmacol. 2011;163:1069–77. https://doi.org/10.1111/j.1476-5381.2011.01303.x.
Craigie E, Birch RE, Unwin RJ, Wildman SS. The relationship between P2X4 and P2X7: a physiologically important interaction? Front Physiol. 2013;4:216. https://doi.org/10.3389/fphys.2013.00216.
Perez-Flores G, et al. The P2X7/P2X4 interaction shapes the purinergic response in murine macrophages. Biochem Biophys Res Commun. 2015;467:484–90. https://doi.org/10.1016/j.bbrc.2015.10.025.
Weinhold K, Krause-Buchholz U, Rodel G, Kasper M, Barth K. Interaction and interrelation of P2X7 and P2X4 receptor complexes in mouse lung epithelial cells. Cell Mol Life Sci. 2010;67:2631–42. https://doi.org/10.1007/s00018-010-0355-1.
Le TT, et al. Purinergic signaling in pulmonary inflammation. Front Immunol. 2019;10:1633. https://doi.org/10.3389/fimmu.2019.01633.
Sim JA, et al. Altered hippocampal synaptic potentiation in P2X4 knock-out mice. J Neurosci. 2006;26:9006–9. https://doi.org/10.1523/JNEUROSCI.2370-06.2006.
Coddou C, Sandoval R, Hevia MJ, Stojilkovic SS. Characterization of the antagonist actions of 5-BDBD at the rat P2X4 receptor. Neurosci Lett. 2019;690:219–24. https://doi.org/10.1016/j.neulet.2018.10.047.
Hasleton PS. The internal surface area of the adult human lung. J Anat. 1972;112:391–400.
Pandey KC, De S, Mishra PK. Role of proteases in chronic obstructive pulmonary disease. Front Pharmacol. 2017;8:512. https://doi.org/10.3389/fphar.2017.00512.
Ni L, Dong C. Roles of myeloid and lymphoid cells in the pathogenesis of chronic obstructive pulmonary disease. Front Immunol. 2018;9:1431. https://doi.org/10.3389/fimmu.2018.01431.
Wang L, Jacobsen SE, Bengtsson A, Erlinge D. P2 receptor mRNA expression profiles in human lymphocytes, monocytes and CD34+ stem and progenitor cells. BMC Immunol. 2004;5:16. https://doi.org/10.1186/1471-2172-5-16.
Thul PJ, et al. A subcellular map of the human proteome. Science. 2017. https://doi.org/10.1126/science.aal3321.
Layhadi JA, Fountain SJ. P2X4 receptor-dependent Ca(2+) influx in model human monocytes and macrophages. Int J Mol Sci. 2017. https://doi.org/10.3390/ijms18112261.
Stokes L, Surprenant A. Dynamic regulation of the P2X4 receptor in alveolar macrophages by phagocytosis and classical activation. Eur J Immunol. 2009;39:986–95. https://doi.org/10.1002/eji.200838818.
Chen H, et al. Effect of P2X4R on airway inflammation and airway remodeling in allergic airway challenge in mice. Mol Med Rep. 2016;13:697–704. https://doi.org/10.3892/mmr.2015.4622.
Zech A, et al. P2rx4 deficiency in mice alleviates allergen-induced airway inflammation. Oncotarget. 2016;7:80288–97. https://doi.org/10.18632/oncotarget.13375.
Osei ET, Brandsma CA, Timens W, Heijink IH, Hackett TL. Current perspectives on the role of interleukin-1 signalling in the pathogenesis of asthma and COPD. Eur Respir J. 2020. https://doi.org/10.1183/13993003.00563-2019.
Ekberg-Jansson A, et al. Neutrophil-associated activation markers in healthy smokers relates to a fall in DL(CO) and to emphysematous changes on high resolution CT. Respir Med. 2001;95:363–73. https://doi.org/10.1053/rmed.2001.1050.
Pauwels NS, et al. Role of IL-1alpha and the Nlrp3/caspase-1/IL-1beta axis in cigarette smoke-induced pulmonary inflammation and COPD. Eur Respir J. 2011;38:1019–28. https://doi.org/10.1183/09031936.00158110.
Kanellopoulos JM, Almeida-da-Silva CLC, Ruutel Boudinot S, Ojcius DM. Structural and functional features of the P2X4 receptor: an immunological perspective. Front Immunol. 2021;12: 645834. https://doi.org/10.3389/fimmu.2021.645834.
Karimi R, Tornling G, Grunewald J, Eklund A, Skold CM. Cell recovery in bronchoalveolar lavage fluid in smokers is dependent on cumulative smoking history. PLoS ONE. 2012;7: e34232. https://doi.org/10.1371/journal.pone.0034232.
Uhlen M, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. https://doi.org/10.1126/science.1260419.
Qureshi OS, Paramasivam A, Yu JC, Murrell-Lagnado RD. Regulation of P2X4 receptors by lysosomal targeting, glycan protection and exocytosis. J Cell Sci. 2007;120:3838–49. https://doi.org/10.1242/jcs.010348.
Cao Q, et al. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J Cell Biol. 2015;209:879–94. https://doi.org/10.1083/jcb.201409071.
Martin TP, et al. CaMKIIdelta interacts directly with IKKbeta and modulates NF-kappaB signalling in adult cardiac fibroblasts. Cell Signal. 2018;51:166–75. https://doi.org/10.1016/j.cellsig.2018.07.008.
Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017. https://doi.org/10.1038/sigtrans.2017.23.
Thompson KE, et al. Fusion-activated cation entry (FACE) via P2X(4) couples surfactant secretion and alveolar fluid transport. FASEB J. 2013;27:1772–83. https://doi.org/10.1096/fj.12-220533.
Winkelmann VE, et al. Inflammation-induced upregulation of P2X4 expression augments mucin secretion in airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2019;316:L58–70. https://doi.org/10.1152/ajplung.00157.2018.
Ferrari D, Wesselborg S, Bauer MK, Schulze-Osthoff K. Extracellular ATP activates transcription factor NF-kappaB through the P2Z purinoreceptor by selectively targeting NF-kappaB p65. J Cell Biol. 1997;139:1635–43. https://doi.org/10.1083/jcb.139.7.1635.
Korcok J, Raimundo LN, Ke HZ, Sims SM, Dixon SJ. Extracellular nucleotides act through P2X7 receptors to activate NF-kappaB in osteoclasts. J Bone Miner Res. 2004;19:642–51. https://doi.org/10.1359/JBMR.040108.
Theatre E, Bours V, Oury C. A P2X ion channel-triggered NF-kappaB pathway enhances TNF-alpha-induced IL-8 expression in airway epithelial cells. Am J Respir Cell Mol Biol. 2009;41:705–13. https://doi.org/10.1165/rcmb.2008-0452OC.
Boumechache M, Masin M, Edwardson JM, Gorecki DC, Murrell-Lagnado R. Analysis of assembly and trafficking of native P2X4 and P2X7 receptor complexes in rodent immune cells. J Biol Chem. 2009;284:13446–54. https://doi.org/10.1074/jbc.M901255200.
Sakaki H, et al. P2X4 receptor regulates P2X7 receptor-dependent IL-1beta and IL-18 release in mouse bone marrow-derived dendritic cells. Biochem Biophys Res Commun. 2013;432:406–11. https://doi.org/10.1016/j.bbrc.2013.01.135.
Chen K, et al. ATP-P2X4 signaling mediates NLRP3 inflammasome activation: a novel pathway of diabetic nephropathy. Int J Biochem Cell Biol. 2013;45:932–43. https://doi.org/10.1016/j.biocel.2013.02.009.
Amores-Iniesta J, et al. Extracellular ATP activates the NLRP3 inflammasome and is an early danger signal of skin allograft rejection. Cell Rep. 2017;21:3414–26. https://doi.org/10.1016/j.celrep.2017.11.079.
Martinez-Garcia JJ, et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat Commun. 2019;10:2711. https://doi.org/10.1038/s41467-019-10626-x.
Acknowledgements
The authors thank Madelon Hoßfeld (Freiburg, Germany) for the support in histopathologic sample preparation and analysis as well as for the assistance in qPCR data processing and murine sample preparation for morphometric emphysema assessment. The authors also want to thank Korcan Ayata (Department of Biomedicine, Gastroenterology Unit, University of Basel, Switzerland) and Barbara Hammer (Department of Pulmonology, Inner Medicine II, Medical University, Vienna, Austria) for intellectual input.
Funding
The research reported in this publication was supported by a Grant of the Boehringer-Ingelheim Foundation to Prof. Dr. M. Idzko. The content is solely the responsibility of the authors and does not necessarily represent the official views of Boehringer-Ingelheim Foundation.
Author information
Authors and Affiliations
Contributions
SS planned and conducted the murine experiments. SS provided and analysed the biologic samples, was involved in data processing, statistical analysis, data interpretation, and wrote the main manuscript. IM helped in planning, carrying out and analysing the THP-1 experiments, provided biologic samples and supervised data analysis as well as interpretation. MI was responsible for the overall study design. MI assisted in data analysis, interpretation and supported the main manuscript preparation. AZ assisted in the execution of the experiments and was involved in statistical analysis, data interpretation, and writing the main manuscript. IM was involved in Fig. 4 preparation. SS and AZ prepared Figs. 1, 2, 3, 4 and all Additional file data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The studies involving human material were reviewed and approved by the Ethics Committee of the Albert-Ludwigs University Freiburg (EC Nr. 06.2006 & 10.2006). All subjects gave written informed consent in accordance with ICH-GCP. The animal experiments were approved by the local animal ethics committee of the regional authority Freiburg (Regierungspräsidium Freiburg), which complies with EU law and guidelines (G-12/106, G-14/108).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary tables and figures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Schneider, S., Merfort, I., Idzko, M. et al. Blocking P2X purinoceptor 4 signalling alleviates cigarette smoke induced pulmonary inflammation. Respir Res 23, 148 (2022). https://doi.org/10.1186/s12931-022-02072-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-022-02072-z