
Abstract
Background
Patients with fibrotic hypersensitivity pneumonitis (HP) show variable clinical courses, and some experience rapid deterioration (RD), including acute exacerbation (AE). However, little is known about AE in fibrotic HP. Here, we retrospectively examined the incidence, risk factors, and outcomes of AE in fibrotic HP.
Methods
The incidence rates of AE were calculated in 101 patients with biopsy-proven HP. AE was defined as the worsening of dyspnoea within 30 days, with new bilateral lung infiltration and no evidence of infection or other causes of dyspnoea.
Results
During follow-up (median: 30 months), 18 (17.8%) patients experienced AE. The 1, 3, and 5 year incidence rates of AE were 6.0, 13.6, and 22.8%, respectively. Lower diffusing capacity of the lung for carbon monoxide (DLCO) and a radiologic usual interstitial pneumonia (UIP)-like pattern were risk factors for AE. In-hospital mortality after AE was 44.4%. Median survival from diagnosis was significantly shorter in patients with AE (26.0 months) than in those with no-AE RD (55.0 months; p = 0.008) or no RD (not reached; p < 0.001). AE remained a significant predictor of all-cause mortality (hazard ratio, 8.641; 95% confidence interval, 3.388–22.040; p < 0.001) after adjustment for age, body mass index, lung function, lymphocyte levels in bronchoalveolar lavage fluid, and the presence of a UIP-like pattern.
Conclusions
AE was not uncommon among patients with fibrotic HP and significantly affected prognosis. A lower DLCO value and radiologic UIP-like pattern at diagnosis were associated with the development AE in patients with fibrotic HP.
Similar content being viewed by others


Introduction
Hypersensitivity pneumonitis (HP) is an interstitial lung disease (ILD) that results from inhalation of organic antigens in susceptible individuals [1]. According to the recently published guidelines for diagnosis of hypersensitivity pneumonitis, HP is classified into fibrotic or nonfibrotic [2], since the presence of radiologic or histopathologic fibrosis is important in determining prognosis [3,4,5]. The progressive fibrosing nature of fibrotic HP is also characteristic of idiopathic pulmonary fibrosis (IPF), a prototype of fibrosing ILD [6], and the radiologic pattern of usual interstitial pneumonia (UIP) is often indistinguishable between cases of fibrotic HP and IPF [7]. Furthermore, genomic risk factors such as the MUC5B promotor polymorphism and shorter telomere lengths, known to be associated with the development and progression of IPF, are also associated with the extent of fibrosis in HP [8].
In IPF, the development of acute exacerbation (AE) is a well-known risk factor for a poor outcome, with an in-hospital mortality rate of approximately 50% [9,10,11,12]. It has been noted that AE can occur not only in IPF but also in other fibrosing ILDs, including HP [13,14,15,16,17,18]. However, only few studies have examined AE in HP. In 2008, Olson et al. reported a case series on AE in chronic HP [15]. Four patients developed AE, which resulted in respiratory failure requiring mechanical ventilation; three of them died and one had to undergo lung transplantation. In their retrospective study involving 100 patients with chronic bird fancier’s lung, Miyazaki et al. also found a high mortality rate following AE (12 of 14 patients, 85.7%) [18]. Although these results demonstrate detrimental outcomes, it remains challenging to determine how AE affects patient prognosis in fibrotic HP owing to the limited amount of data and knowledge on the incidence and risk factors of AE [15, 18]. Therefore, in this study, we aimed to determine the incidence rate and risk factors of AE and its effect on the survival of patients with fibrotic HP.
Methods
Study patients
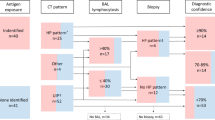
A total of 101 patients diagnosed with definite HP based on histopathological findings (surgical lung biopsy: 99, transbronchial lung biopsy: 2) were identified between January 2002 and December 2017 at Asan Medical Center, Seoul, Republic of Korea. All patients showed radiologic and/or histopathological fibrosis. Their exposure history to causative antigens is shown in Table S1 in Additional file 1. All diagnoses were made through multidisciplinary discussions. The study protocol was approved by the Institutional Review Board of Asan Medical Center (IRB No.: 2017-1081). The requirement for informed consent was waived due to the retrospective nature of the study.
Clinical data
Clinical and survival data of all patients were retrospectively collected from medical records, telephone interviews, and/or records from the National Health Insurance of Korea. Spirometry was performed, and the diffusing capacity of the lung for carbon monoxide (DLCO) and total lung capacity (TLC) were measured according to the ATS/European Respiratory Society recommendations [19,20,21]. Results are expressed as percentages of the normal predicted values. The 6-min-walk test and bronchoalveolar lavage (BAL) were performed in accordance with previously published guidelines [22, 23].
Records of follow-up visits, which usually occurred every 3–6 months, and hospitalisation data were reviewed to identify the development of complications such as pneumonia, AE, pneumothorax, and pulmonary hypertension. Rapid deterioration (RD) was defined as an acute worsening of dyspnoea, requiring hospitalisation and presenting with new radiologic abnormalities [9]. RD included AE as well as other acute respiratory events including pneumonia or pneumothorax. AE was defined, using the criteria previously used in IPF by Collard et al. (2007), as a worsening of dyspnoea within 30 days, with new bilateral lung infiltration and no evidence of infection or other alternative causes of dyspnoea (e.g. pulmonary embolism and left heart failure) [24]. AE was labelled as ‘suspected AE’ when the aetiology of acute respiratory worsening was unknown but did not fulfil all the AE criteria owing to missing data. When infection was strongly suspected clinically (symptoms such as purulent sputum and rapid improvement and response to antibiotic treatment alone) but causative organism was not identified, it was categorised as ‘suspected infection’.
Radiologic assessment
High-resolution computed tomography (HRCT) images of all patients were evaluated by two thoracic radiologists (J.C., E.J.C.) who were blinded to the clinical data. Based on HRCT findings, patients were classified as having a UIP-like pattern or not. Discrepancies were resolved by a consensus. A UIP-like pattern was diagnosed when HRCT findings were compatible with a UIP or probable UIP pattern according to the HRCT classification of Fleischner Society IPF diagnostic guidelines with modification [25]. Briefly, a UIP-like pattern was defined as a reticular pattern with traction bronchiectasis or bronchiolectasis with/without honeycombing. Given that mosaic attenuation is frequently observed in HP [26] and distribution of fibrosis in HP may differ from that in IPF [27], the presence of mosaic attenuation and distribution of fibrosis were not considered as features of alternative diagnosis in this study [28].
Statistical analysis
Data are presented as mean ± standard deviation or median [interquartile range] for continuous variables and percentages for categorical variables. The Student’s t-test or Mann–Whitney test was used for continuous variables, and the chi-squared test and Fisher’s exact test were used to compare categorical variables. Kaplan–Meier estimates and the log-rank test were used for survival analysis. Cumulative incidence rates of RD and AE were also estimated using Kaplan–Meier survival analysis. Risk factors for RD and AE, and predictors of all-cause mortality were analysed using a Cox proportional hazards analysis. Variables with p < 0.05 in the unadjusted analysis were entered into multivariable models. Logistic regression analysis was used to determine risk factors for in-hospital mortality. All p-values were two-tailed, and p-values < 0.05 were considered statistically significant. Data were analysed using the Statistical Package for the Social Sciences software version 23.0 (IBM Corp., Armonk, N.Y., USA).
Results
Incidence
The median follow-up duration was 30.0 months (interquartile range, 15.0–51.0 months). During follow-up, 33 patients (32.7%) experienced RD. The 1-, 3-, and 5-year cumulative incidence rates of RD were 14.0%, 25.4%, and 38.0%, respectively (Fig. 1). The aetiologies of RD are shown in Table 1. AE was the most common cause of RD (54.5%), followed by infection (36.4%). The 1, 3, and 5 year cumulative incidence rates of AE were 6.0, 13.6, and 22.8%, respectively (Fig. 1).

The 1, 3, and 5 year cumulative incidence rates of RD and AE in patients with fibrotic HP. AE acute exacerbation, HP hypersensitivity pneumonitis, RD rapid deterioration
Risk factors for RD and AE
The mean age of all patients was 58.9 years, and 60.4% were female. Patients who developed RD had a significantly lower forced vital capacity (FVC), DLCO, and TLC and a shorter 6-min-walk distance (6MWD) at diagnosis than did those with no RD (Table 2). A significantly lower DLCO, shorter 6MWD, and greater number of patients with a UIP-like pattern were found among patients with AE than among those with no RD. In addition, the proportion of patients with a UIP-like pattern on HRCT was significantly higher in patients with AE than in those with no-AE RD.
To identify factors significantly associated with the development of AE and RD, Cox proportional hazards analyses were performed. A lower DLCO and the presence of a UIP-like pattern were identified as significant risk factors for AE both in the unadjusted and multivariable analyses (Table 3). Regarding RD, older age and lower DLCO, were identified as significant risk factors in the multivariable analysis (Table S2 in Additional file 1).
Effect on overall survival
Patients who experienced RD, with or without AE, showed a significantly poorer survival following diagnosis than did those without RD (Fig. 2). The median survival of patients with no-AE RD and AE was 55.0 months (95% confidence interval [CI], 36.3–73.7 months) and 26.0 months (95% CI, 13.5–38.5 months), respectively. Their median overall survival was significantly shorter than that of patients with no RD (median survival not reached; p = 0.013 and p < 0.001, respectively). Among the patients with RD, those with AE had a significantly shorter survival than those with no-AE RD (p = 0.008).

Survival from the initial diagnosis in patients with AE, no-AE RD, and no RD. Patients with AE showed a significantly poorer survival than those with no-AE RD or with no RD. AE acute exacerbation, RD rapid deterioration
The predictors of all-cause mortality are shown in Table 4. In the unadjusted analysis, the factors found to be significantly associated with mortality were older age; a lower BMI, FVC, DLCO, and TLC; lower lymphocyte levels in BAL fluid; the presence of a UIP-like pattern; and the development of AE. The development of AE remained as a significant predictor even after adjustment for other variables, and it appeared to exert the greatest effect on mortality risk (hazard ratio, 8.641; 95% CI, 3.388–22.040; p < 0.001).
Survival after hospitalisation
Clinical characteristics at the time of hospitalisation were compared between patients with AE and those with bilateral infection to identify factors that could help distinguish between them (Table S3 in Additional file 1). The total white blood cell (WBC) count and percentage of neutrophils in the BAL fluid were significantly higher in patients with bilateral infection than in those with AE (WBC: 8066.0/µL vs 206.4/µL, p = 0.011; neutrophils: 50.4 vs 13.4%, p = 0.019). However, there was no significant difference in age, sex, disease duration, fever, level of C-reactive protein, and lung function.
The mortality outcomes following the hospitalisation of patients with AE and those with bilateral infection are shown in Fig. 3. In-hospital mortality after AE was 44.4% (8/18), whereas none of the patients with bilateral infection died. Patients who experienced AE showed significantly poorer outcomes; the 30 day, 90 day, and 1 year mortality were significantly higher in patients with AE (33.3, 55.6, 83.3%, respectively) than in those with bilateral infection (no death; all p-values < 0.05).

Survival following the development of AE and bilateral infection. Patients with AE showed a significantly poorer survival than did those with bilateral infection. AE acute exacerbation
Clinical characteristics of survivors and non-survivors of AE and their treatment regimens are illustrated in Tables S4 and S5, respectively, in Additional file 1. There was no significant difference in the treatment regimens between survivors and non-survivors including the use of cytotoxic agents. A significantly lower partial pressure of oxygen to fraction of inspired oxygen ratio measured at the time of hospitalisation was found to be significantly associated with in-hospital mortality following AE (odds ratio, 0.983; 95% CI, 0.967–0.999; p = 0.040), as shown in Table 5.
Discussion
In this study, we evaluated the incidence, risk factors, and outcomes of AE in patients with fibrotic HP. The 1, 3, and 5 year cumulative incidence rates of AE were 6.0, 13.6, and 22.8%, respectively. A lower DLCO and the presence of a UIP-like pattern were significant risk factors for AE. AE was associated with a poor prognosis and high in-hospital mortality rate (44.4%), and it appeared to have the greatest effect on overall survival.
As previous studies have shown, AE occurs not only in IPF but also in non-IPF fibrosing ILD [14,15,16,17,18, 29]. According to a recent retrospective study by Suzuki et al. that included 462 IPF and 557 non-IPF ILD patients, AE had a significant effect on mortality, regardless of diagnosis [30]. The 90- ay mortality rate following AE was similar between IPF and non-IPF ILD (47 and 38%, p = 0.345). However, patients with HP comprised only 5.2% of the non-IPF ILD patients in that study, thus limiting the generalisability of the result in the HP population. There is also lack of data regarding the incidence of AE in HP. In the aforementioned study by Suzuki et al., the incidence rate of AE in HP was 6.05 per 100 patient-years and that in IPF was 8.38 per 100 patient-years [30]. Considering the results of a previous study on IPF patients conducted at our institution [9], the cumulative incidence rates of AE seem to be lower in patients with fibrotic HP than in those with IPF (1 and 3 year incidence rates were 6.0 and 13.6%, respectively, in fibrotic HP; 14.2 and 20.7%, respectively, in IPF). To accurately estimate the incidence rate of AE in fibrotic HP, further prospective studies are needed.
In this study, a lower DLCO and the presence of a UIP-like pattern on HRCT at diagnosis were risk factors for AE. This result is in line with a previous report. In a retrospective study involving 100 patients with bird fancier’s lung, patients who developed AE had a lower TLC and DLCO at baseline than did those without AE [18]. In IPF, lower lung function is also known as a risk factor for AE [9, 31,32,33]; it is likely that patients with advanced disease are at a greater risk of AE, regardless of the underlying pathology. A UIP-like pattern on HRCT was another significant risk factor for AE. In the aforementioned study evaluating patients with chronic bird fancier’s lung, patients with UIP-like lesions on surgical lung biopsy experienced AE more frequently. No other studies have evaluated whether a radiologic UIP-like pattern is associated with AE in HP. However, a UIP pattern on HRCT was associated with a higher risk of AE in non-IPF ILDs such as rheumatoid arthritis-associated ILD [16].
The high in-hospital mortality rate following AE in our study, which was almost similar to that of IPF, should be noted [9, 11]. Interestingly, there was a significant difference in the outcomes of AE and bilateral infection. With regard to IPF, findings suggest that the outcome is similar in idiopathic and triggered AE [6, 9, 34]. Thus, the previous definition of AE in IPF, which is limited to idiopathic acute respiratory worsening, was revised to include all events—both idiopathic and triggered—characterised by acute lung injury [34]. On the contrary, when we classified our patients based on whether they had AE (idiopathic worsening) or bilateral infection, patients with infection showed a better outcome than did those with AE. So far, the definition of AE in IPF has been widely adopted in previous studies on non-IPF ILD [13, 16]; given that our study demonstrates a difference between the outcomes of idiopathic worsening (AE) and infection, the cause of acute respiratory worsening may be important in predicting outcomes in fibrotic HP. However, the interpretation should be cautious because our analysis included only small number of patients. Further studies are required to evaluate this issue.
This study has some limitations First, this was a retrospective study conducted at a single tertiary hospital. However, in our study, the clinical characteristics of patients were similar to those of other studies evaluating patients with HP [18, 35]. Second, the study patients were collected over 15 years during which diagnostic approach might have changed. To ensure the diagnosis, we included patients with biopsy confirmed HP. One may argue this might have resulted in including only patients with atypical features who therefore required a histological confirmation of their diagnosis. However, clinical features highly suggestive of HP were fairly frequently found among study patients (e.g., history of antigen exposure and lympho-dominance in BAL fluid in 85.1% and 41.6% of the patients, respectively). Third, BAL or endotracheal aspiration was not performed in all patients with RD; therefore, the distinction between AE and infection may not be perfect. In some patients, BAL could not be performed due to instability, but microbial tests, including respiratory virus PCR panels, were performed for all patients. Our diagnostic approach reflects the real clinical practice in which the differential diagnosis between AE and infection is often difficult. Finally, the definition of a UIP-like pattern used in this study was modified from the HRCT classification of IPF diagnostic guidelines to include a feature inconsistent with UIP (e.g. mosaic attenuation). In the absence of a consensus on the definition of the UIP-like pattern in fibrosing ILDs other than IPF, the modification may be inevitable in some subtypes of ILDs, as previously described [28, 36]. In a recent randomised controlled trial of progressive fibrosing ILD, the presence of mosaic attenuation or centrilobular nodules was also accepted. Furthermore, when categorised using the definition, patients with a UIP-like pattern tended to have a more rapid FVC decline [36], suggesting the usefulness of this definition.
In conclusion, AE was not uncommon in patients with fibrotic HP and, similar to IPF, exerted a significant effect on prognosis. A lower DLCO and the presence of a UIP-like pattern on HRCT at diagnosis were associated with AE in patients with fibrotic HP.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- 6MWD:
-
6-Minute walk distance
- AE:
-
Acute exacerbation
- ATS:
-
American Thoracic Society
- BAL:
-
Bronchoalveolar lavage
- CI:
-
Confidence interval
- DLCO:
-
Diffusing capacity of the lung for carbon monoxide
- FVC:
-
Forced vital capacity
- HP:
-
Hypersensitivity pneumonitis
- HRCT:
-
High-resolution computed tomography
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- RD:
-
Rapid deterioration
- TLC:
-
Total lung capacity
- UIP:
-
Usual interstitial pneumonia
- WBC:
-
White blood cell
References
Vasakova M, Morell F, Walsh S, Leslie K, Raghu G. Hypersensitivity pneumonitis: perspectives in diagnosis and management. Am J Respir Crit Care Med. 2017;196:680–9.
Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, Bargagli E, Chung JH, Collins BF, Bendstrup E, et al. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2020;202:e36–69.
Mooney JJ, Elicker BM, Urbania TH, Agarwal MR, Ryerson CJ, Nguyen MLT, Woodruff PG, Jones KD, Collard HR, King TE Jr, Koth LL. Radiographic fibrosis score predicts survival in hypersensitivity pneumonitis. Chest. 2013;144:586–92.
Chiba S, Tsuchiya K, Akashi T, Ishizuka M, Okamoto T, Furusawa H, Tateishi T, Kishino M, Miyazaki Y, Tateishi U, et al. Chronic hypersensitivity pneumonitis with a usual interstitial pneumonia-like pattern: correlation between histopathologic and clinical findings. Chest. 2016;149:1473–81.
Hanak V, Golbin JM, Hartman TE, Ryu JH. High-resolution CT findings of parenchymal fibrosis correlate with prognosis in hypersensitivity pneumonitis. Chest. 2008;134:133–8.
Kolb M, Bondue B, Pesci A, Miyazaki Y, Song JW, Bhatt NY, Huggins JT, Oldham JM, Padilla ML, Roman J, Shapera S. Acute exacerbations of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27:180071.
Morell F, Villar A, Montero MA, Munoz X, Colby TV, Pipvath S, Cruz MJ, Raghu G. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med. 2013;1:685–94.
Ley B, Newton CA, Arnould I, Elicker BM, Henry TS, Vittinghoff E, Golden JA, Jones KD, Batra K, Torrealba J, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. 2017;5:639–47.
Song JW, Hong SB, Lim CM, Koh Y, Kim DS. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J. 2011;37:356–63.
Al-Hameed FM, Sharma S. Outcome of patients admitted to the intensive care unit for acute exacerbation of idiopathic pulmonary fibrosis. Can Respir J. 2004;11:117–22.
Kim DS, Park JH, Park BK, Lee JS, Nicholson AG, Colby T. Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur Respir J. 2006;27:143–50.
Kishaba T, Tamaki H, Shimaoka Y, Fukuyama H, Yamashiro S. Staging of acute exacerbation in patients with idiopathic pulmonary fibrosis. Lung. 2014;192:141–9.
Suda T, Kaida Y, Nakamura Y, Enomoto N, Fujisawa T, Imokawa S, Hashizume H, Naito T, Hashimoto D, Takehara Y, et al. Acute exacerbation of interstitial pneumonia associated with collagen vascular diseases. Respir Med. 2009;103:846–53.
Park IN, Kim DS, Shim TS, Lim CM, Lee SD, Koh Y, Kim WS, Kim WD, Jang SJ, Colby TV. Acute exacerbation of interstitial pneumonia other than idiopathic pulmonary fibrosis. Chest. 2007;132:214–20.
Olson AL, Huie TJ, Groshong SD, Cosgrove GP, Janssen WJ, Schwarz MI, Brown KK, Frankel SK. Acute exacerbations of fibrotic hypersensitivity pneumonitis: a case series. Chest. 2008;134:844–50.
Hozumi H, Nakamura Y, Johkoh T, Sumikawa H, Colby TV, Kono M, Hashimoto D, Enomoto N, Fujisawa T, Inui N, et al. Acute exacerbation in rheumatoid arthritis-associated interstitial lung disease: a retrospective case control study. BMJ Open. 2013;3:e003132.
Tachikawa R, Tomii K, Ueda H, Nagata K, Nanjo S, Sakurai A, Otsuka K, Kaji R, Hayashi M, Katakami N, Imai Y. Clinical features and outcome of acute exacerbation of interstitial pneumonia: collagen vascular diseases-related versus idiopathic. Respiration. 2012;83:20–7.
Miyazaki Y, Tateishi T, Akashi T, Ohtani Y, Inase N, Yoshizawa Y. Clinical predictors and histologic appearance of acute exacerbations in chronic hypersensitivity pneumonitis. Chest. 2008;134:1265–70.
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP, Gustafsson P, et al. Standardisation of spirometry. Eur Respir J. 2005;26:319–38.
Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, Burgos F, Casaburi R, Coates A, Enright P, et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J. 2005;26:720–35.
Wanger J, Clausen JL, Coates A, Pedersen OF, Brusasco V, Burgos F, Casaburi R, Crapo R, Enright P, van der Grinten CP, et al. Standardisation of the measurement of lung volumes. Eur Respir J. 2005;26:511–22.
Holland AE, Spruit MA, Troosters T, Puhan MA, Pepin V, Saey D, McCormack MC, Carlin BW, Sciurba FC, Pitta F, et al. An official European Respiratory Society/American Thoracic Society technical standard: field walking tests in chronic respiratory disease. Eur Respir J. 2014;44:1428–46.
Meyer KC, Raghu G, Baughman RP, Brown KK, Costabel U, du Bois RM, Drent M, Haslam PL, Kim DS, Nagai S, et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185:1004–14.
Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE Jr, Lasky JA, Loyd JE, Noth I, Olman MA, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176:636–43.
Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, Goldin JG, Hansell DM, Inoue Y, Johkoh T, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6:138–53.
Lynch DA. CT phenotypes in hypersensitivity pneumonitis . Chest. 2019;155:655–6.
Silva CI, Muller NL, Lynch DA, Curran-Everett D, Brown KK, Lee KS, Chung MP, Churg A. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology. 2008;246:288–97.
Jacob J, Hirani N, van Moorsel CHM, Rajagopalan S, Murchison JT, van Es HW, Bartholmai BJ, van Beek FT, Struik MHL, Stewart GA, et al. Predicting outcomes in rheumatoid arthritis related interstitial lung disease. Eur Respir J. 2019. https://doi.org/10.1183/13993003.00869-2018.
Churg A, Wright JL, Tazelaar HD. Acute exacerbations of fibrotic interstitial lung disease. Histopathology. 2011;58:525–30.
Suzuki A, Kondoh Y, Brown KK, Johkoh T, Kataoka K, Fukuoka J, Kimura T, Matsuda T, Yokoyama T, Fukihara J, et al. Acute exacerbations of fibrotic interstitial lung diseases. Respirology. 2019. https://doi.org/10.1111/resp.13682.
Collard HR, Yow E, Richeldi L, Anstrom KJ, Glazer C. Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir Res. 2013;14:73.
Kondoh Y, Taniguchi H, Katsuta T, Kataoka K, Kimura T, Nishiyama O, Sakamoto K, Johkoh T, Nishimura M, Ono K, Kitaichi M. Risk factors of acute exacerbation of idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2010;27:103–10.
Ohshimo S, Ishikawa N, Horimasu Y, Hattori N, Hirohashi N, Tanigawa K, Kohno N, Bonella F, Guzman J, Costabel U. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir Med. 2014;108:1031–9.
Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, Lee JS, Maher TM, Wells AU, Antoniou KM, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2016;194:265–75.
Fernandez Perez ER, Swigris JJ, Forssen AV, Tourin O, Solomon JJ, Huie TJ, Olson AL, Brown KK. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest. 2013;144:1644–51.
Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381:1718–27.
Acknowledgements
None.
Funding
This study was supported by a grant from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and Technology (NRF-2019R1A2C2008541).
Author information
Authors and Affiliations
Contributions
JWS was responsible for the conception and design of the study. JWS also served as a guarantor and takes full responsibility for the content of the manuscript, including the data analysis. JK, YJK, JC, EJC, and JWS were responsible for the creation, revision, and final approval of the manuscript. JWS was responsible for data acquisition. JK and YJK performed the statistical analysis and data interpretation. JC and EJC performed the radiologic analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the amended Declaration of Helsinki and the protocol was approved by the Institutional Review Board of Asan Medical Center (2017–1081). Informed consent was waived due to the retrospective nature of the study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Additional tables.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kang, J., Kim, Y.J., Choe, J. et al. Acute exacerbation of fibrotic hypersensitivity pneumonitis: incidence and outcomes. Respir Res 22, 152 (2021). https://doi.org/10.1186/s12931-021-01748-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-021-01748-2