
Abstract
Background
CHF6001 is a novel inhaled phosphodiesterase-4 inhibitor. This Phase IIa study assessed the effects of CHF6001 on markers of inflammation in induced sputum and blood in patients with chronic obstructive pulmonary disease (COPD).
Methods
This was a multicentre, three-period (each 32 days), three-way, placebo-controlled, double-blind, complete-block crossover study. Eligible patients had COPD, chronic bronchitis, and were receiving inhaled triple therapy for ≥2 months. Patients received CHF6001 800 or 1600 μg, or matching placebo twice daily via multi-dose dry-powder inhaler (NEXThaler). Induced sputum was collected pre-dose on Day 1, and post-dose on Days 20, 26 and 32. Blood was sampled pre-dose on Day 1, and pre- and post-dose on Day 32.
Results
Of 61 randomised patients, 54 (88.5%) completed the study. There were no significant differences between groups for overall sputum cell count, or absolute numbers of neutrophils, eosinophils or lymphocytes. CHF6001 800 μg significantly decreased the absolute number and percentage of macrophages vs placebo.
In sputum, compared with placebo both CHF6001 doses significantly decreased leukotriene B4, C-X-C motif chemokine ligand 8, macrophage inflammatory protein 1β, matrix metalloproteinase 9, and tumour necrosis factor α (TNFα). In blood, both CHF6001 doses significantly decreased serum surfactant protein D vs placebo. CHF6001 1600 μg significantly decreased TNFα ex-vivo (after incubation with lipopolysaccharide).
Conclusion
The data from this study show that CHF6001 inhaled twice daily has anti-inflammatory effects in the lungs of patients with COPD already treated with triple inhaled therapy.
Trial registration
The study is registered on ClinicalTrials.gov (NCT03004417).
Similar content being viewed by others


Background
The pathogenesis and progression of chronic obstructive pulmonary disease (COPD) is, in part, due to chronic inflammation [1]. However the nature and severity of inflammation in COPD varies, and pharmacological anti-inflammatory treatments are unlikely to be effective in all patients; a precision medicine approach is needed to selectively target patients to increase the chance of therapeutic success [2].
Phosphodiesterase-4 (PDE4) is an enzyme that mediates the breakdown of cyclic adenosine monophosphate (cAMP), with PDE4 inhibition having anti-inflammatory effects in a broad range of cell types. The orally administered PDE4 inhibitor roflumilast prevents exacerbations in patients with COPD, although is effective only in a specific subgroup: individuals with chronic bronchitis and a history of exacerbations [3,4,5,6]. However, systemic exposure after oral administration can cause side effects such as nausea, weight loss and gastrointestinal disturbance, which may limit its use in clinical practice.
CHF6001 is a novel PDE4 inhibitor [7, 8], currently in clinical development that has been specifically designed and formulated as an extrafine formulation (i.e., with mass median aerodynamic diameter ≤ 2 μm) to be delivered via inhalation and to have a low systemic exposure. This allows CHF6001 to reach therapeutic concentration in the target organ, the lung, yet reduces exposure in the systemic circulation thus limiting systemic adverse effects. The current manuscript describes the results of a Phase IIa study that aimed to assess the effects of CHF6001 on markers of inflammation in induced sputum and blood in patients with COPD who have a chronic bronchitis phenotype. Since in clinical practice a PDE4 inhibitor is often administered in addition to inhaled triple therapy (i.e., inhaled corticosteroid [ICS] plus long-acting muscarinic antagonist [LAMA] plus long-acting β2-agonist [LABA]) [9], the study recruited patients who were receiving inhaled triple therapy prior to study start, and who continued this therapy for the duration of the study.
Methods
Study design
This was a multicentre, three-period, three-way, placebo-controlled, double-blind, complete block crossover study. No later than 21 days after a screening visit, eligible patients attended a randomisation visit, followed by three, 32-day treatment periods, each separated by a 28 to 42-day washout, with a follow-up visit 12 days after completion of the third treatment period (Fig. 1). During each treatment period, patients attended visits on Days 1, 20, 26 and 32.

Study design. Abbreviation: BID, twice daily
Induced sputum was collected pre-dose on Day 1 of each treatment period (for the first treatment period, the sample could be collected up to 10 days prior to Day 1), and 2 h post-dose on Days 20, 26 and 32 (see the Additional file 1 for further detail on sputum and blood analysis methods). Blood samples were taken pre-dose on Day 1, and pre-dose and at 30 min, and 1, 1.5, 2, 3, 4, 6, 8 and 12 h post-dose on Day 32. Other pre-dose assessments on Days 1 and 32 were spirometry (forced expiratory volume in 1 s [FEV1] and forced vital capacity [FVC]), forced oscillometry, and COPD Assessment Test (CAT), with Baseline Dyspnea Index recorded on Day 1 and Transition Dyspnea Index (TDI) on Day 32. Central and peripheral airways mechanics were assessed using forced oscillometry, with the following values determined: inspiratory and expiratory resistance (at 5 and 19 Hz), inspiratory and expiratory reactance, tidal expiratory flow limitation and the percentage of flow limited breaths.
Patients were instructed not to inhale salbutamol (rescue medication) for 6 h prior to each visit, and not to inhale concomitant COPD maintenance therapy for 12 h prior to the visit for twice-daily (BID) medication or 24 h for once-daily medication. Patients fasted for at least 10 h prior to the Day 32 visits, with alcohol, xanthine-containing beverages or food, and grapefruit not permitted from 48 h prior to the visit.
The study was approved by independent ethics committees for each institution (see the Additional file 1 for details), and was performed in accordance with the principles of the declaration of Helsinki, and the International Conference on Harmonization notes for guidance on Good Clinical Practice (ICH/CPMP/135/95). The study is registered on ClinicalTrials.gov (NCT03004417). There were no protocol amendments.
Patients
Eligible patients were male or female, ≥40 years of age, current or ex-smokers with smoking history ≥10 pack-years, a diagnosis of COPD, post-bronchodilator FEV1/FVC ratio < 0.70 and FEV1 ≥ 30% and ≤ 70% predicted, CAT score ≥ 10, and a history of chronic bronchitis (defined as chronic cough and sputum production for more than three months per year for at least two years). All eligible patients were to have been receiving inhaled triple therapy daily for at least two months. In addition, patients were to be able to produce an adequate induced sputum sample, defined as a load ≥300 mg, with viability factor ≥ 70%, < 30% epithelial cells and a neutrophil differential count of ≥60%. All patients provided written informed consent prior to study start.
The key exclusion criteria were a moderate or severe COPD exacerbation within six weeks prior to entry or between screening and randomisation, and the use of a PDE4 inhibitor within two months prior to entry. Full inclusion and exclusion criteria are listed in Additional file 1, as are non-permitted COPD concomitant medication.
Interventions
Treatments administered were CHF6001 800 or 1600 μg BID (total daily doses of 1600 or 3200 μg) or matching placebo, all via multi-dose dry-powder inhaler (NEXThaler®). On Day 1 of the first treatment period, patients were randomised to one of six treatment sequences, according to a balanced block randomisation scheme prepared by the sponsor, such that each patient received all three treatments. All patients, investigators, site staff and employees of the sponsor were blinded to treatment for the duration of the study.
Outcomes
Given the nature of the study, all of the objectives were exploratory:
-
To evaluate the effect of CHF6001 on biomarkers of inflammation in induced sputum and in blood, and on pulmonary function and symptoms in comparison with placebo;
-
To evaluate the safety and tolerability of CHF6001;
-
To assess the pharmacokinetic profile of CHF6001 at steady state.
Statistical analysis
There were no formal hypotheses or sample size calculations due to the exploratory nature of this study. A sample size of 42 patients was expected to be appropriate for the study purpose; with an estimated non-evaluable rate of 30%, approximately 60 patients were required to be randomised.
A patient had to have a minimum of one acceptable sputum sample on Day 20, 26 or 32 to be included in the sputum biomarker evaluation for that treatment period, with the available cell count and biomarker data averaged for each patient. The mean change from baseline (pre-dose on Day 1 of each treatment period) was then compared between treatments using an analysis of covariance (ANCOVA) that included treatment, patient and period as fixed effects, and baseline values as covariate. The mean change from baseline to Day 32 for biomarkers in blood were analysed using the same model as for sputum data. All data were log-transformed before analysis, and the results are presented as ratio of geometric means.
The mean changes from baseline to Day 32 for lung function, CAT and TDI were analysed using similar methodology to sputum biomarkers, but were not log-transformed. Plasma pharmacokinetic endpoints calculated at steady state (Day 32) included maximum concentration (Cmax,ss), time to maximum concentration (Tmax,ss), concentration area under the curve from 0 to 12 h (AUC0–12,ss), and clearance adjusted for absolute bioavailability (CL/Fss). The sputum concentration of CHF6001 was also evaluated at 2 h post-dose, using the mean of all values measured on Days 20, 26 and 32. Pharmacokinetic variables were summarised using geometric means and coefficients of variation, except for Tmax,ss, which is presented as median (range).
The Safety population is all randomised patients who received at least one dose of study medication. The Pharmacodynamic population is all patients in the Safety population with available evaluations in at least two treatment periods, excluding patients with major protocol deviations affecting the pharmacodynamic evaluations. The Pharmacokinetic population consists of all patients from the Safety population excluding patients without any valid pharmacokinetic assessment and with major protocol deviations affecting the pharmacokinetic evaluations.
Results
Patients
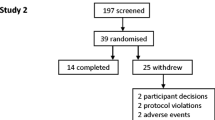
The study ran from October 2016 to December 2017 at six sites in the UK and Germany. Patient disposition throughout the study is shown in Fig. 2. All randomised patients were included in the Safety population. Three patients were excluded from the Pharmacodynamic population as they did not have data available in at least two treatment periods. One patient was excluded from the Pharmacokinetic population as no pharmacokinetic data were available. The mean post-bronchodilator FEV1 of the 61 randomised patients was 50.2% predicted, while the mean CAT score was 20.7 (Table 1).

Screening, randomisation and study completion. Abbreviation: BID, twice daily
Sputum cell counts and inflammatory biomarkers
There were no significant differences between treatments for overall sputum cell count, or the absolute numbers of neutrophils, eosinophils or lymphocytes (Fig. 3 shows the ratio of CHF6001 vs placebo). For macrophages, CHF6001 800 μg significantly decreased the absolute number vs placebo; there was no difference vs placebo for 1600 μg. Similarly, there was no consistent effect of CHF6001 on the percentage of neutrophils, eosinophils, lymphocytes or macrophages, although 800 μg significantly increased the percentage of neutrophils and decreased the percentage of macrophages vs placebo (Fig. 3).

Ratio of geometric means for CHF6001 to placebo for overall cell count, and absolute and relative differential cell counts in sputum (Pharmacodynamic population). Data are the ratios of geometric means and 95% CI. *p < 0.05. Abbreviation: BID, twice daily. A total of 56 patients were included in the CHF6001 800 μg Pharmacodynamic population, 57 in the CHF6001 1600 μg population and 57 in the placebo population
Compared with placebo, both CHF6001 doses significantly decreased the levels of leukotriene B4 (LTB4), C-X-C motif chemokine ligand 8 (CXCL8), macrophage inflammatory protein 1β (MIP-1β; also known as C-C motif chemokine ligand [CCL] 4), matrix metalloproteinase 9 (MMP9), and tumour necrosis factor α (TNFα) (Fig. 4, with absolute values in Additional file 1: Table S1). There was no clear CHF6001 dose-related trend for any of these inflammatory biomarkers. CHF6001 1600 μg significantly increased interleukin 6 (IL-6) levels compared with placebo, whereas CHF6001 800 μg significantly decreased monocyte chemotactic protein-1 (MCP1; also known as CCL2) levels compared with placebo with the higher dose failing to reach significance.

Ratio of geometric means for CHF6001 to placebo for markers of inflammation in sputum (Pharmacodynamic population). Data are the ratios of geometric means and 95% CI. *p < 0.05. Abbreviation: BID, twice daily. A total of 56 patients were included in the CHF6001 800 μg Pharmacodynamic population, 57 in the CHF6001 1600 μg population and 57 in the placebo population
Blood inflammatory biomarkers
Overall, there were few changes in any blood inflammatory biomarker (Fig. 5, with absolute values in Additional file 1: Table S2). Both CHF6001 doses significantly decreased serum surfactant protein D (SP-D) levels compared with placebo. CHF6001 1600 μg significantly decreased TNFα ex-vivo (i.e., assessed after incubation of whole blood with lipopolysaccharide); the decrease with CHF6001 800 μg was almost significant (p = 0.057). By contrast, non-stimulated TNFα in serum was not affected by CHF6001 (Fig. 5).

Ratio of geometric means for CHF6001 to placebo for markers of inflammation in blood (Pharmacodynamic population). Data are the ratios of geometric means and 95% CI. *p < 0.05. Abbreviation: BID, twice daily. A total of 56 patients were included in the CHF6001 800 μg Pharmacodynamic population, 57 in the CHF6001 1600 μg population and 57 in the placebo population
Lung function and symptoms
Neither CHF6001 dose differed from placebo in terms of lung function (FEV1 and FVC) or symptoms (Additional file 1: Table S3), and there were no consistent treatment–placebo differences in any of the oscillometry parameters (Additional file 1: Table S4).
Pharmacokinetics
On Day 32, CHF6001 systemic exposure was broadly proportional to dose; Tmax and clearance was similar for the two doses (Table 2). Although there was high variability in the sputum concentration of CHF6001, dose-proportionality was observed at 2 h post-dose in steady state conditions (after Day 20), with the concentration in sputum approximately 2000-fold higher than in plasma (Table 2).
Safety
Overall, a similar proportion of adverse events were reported with each treatment (Table 3). The most commonly reported adverse event in all three groups was nasopharyngitis, which was reported by more patients with placebo than either CHF6001 dose. More importantly, only two events were considered related to treatment, neither of which was serious, and no events had a fatal outcome. Gastrointestinal adverse events were reported by seven (12.1%), six (10.2%) and six (10.3%) patients during treatment with CHF6001 800 μg, CHF6001 1600 μg and placebo respectively (see the Additional file 1 for a list of the preferred terms). Only one of these events was considered related to study drug (mild dry mouth during treatment with CHF6001 1600 μg), which had resolved by the study end and required no change in study drug. Two patients had severe adverse events, both leading to study drug withdrawal. One of these occurred during treatment with CHF6001 1600 μg – staphylococcal wound infection, not considered related to study drug. None of the other adverse events that led to study drug withdrawal were considered related to study drug. There were no major differences between treatments in any of the haematology or biochemistry data. There were also no differences between either dose of CHF6001 and placebo in systolic or diastolic blood pressure, heart rate or Fridericia’s corrected QT interval.
Discussion
The PDE4 inhibitor CHF6001 given twice daily by inhalation significantly decreased the levels of a variety of inflammatory biomarkers in sputum, such as LTB4, CXCL8, MIP-1β, MMP9, and TNFα, but had no consistent effect on sputum inflammatory cell numbers. CHF6001 also significantly decreased SP-D levels in the blood. Notably, these anti-inflammatory effects were achieved with both doses tested in addition to background ICS-containing triple therapy.
The sputum cytokines and chemokines downregulated by CHF6001 are known to be relevant to the pathophysiology of COPD. LTB4, CXCL8, MMP9, and TNFα play important roles in COPD inflammation, with CXCL8 and LTB4 acting as neutrophil chemoattractants, TNFα causing amplification of inflammation, and MMP-9 being a protease that can target lung elastin [10]. Studies using isolated human alveolar macrophage have shown that PDE4 inhibition reduces TNFα and chemokine secretion [11, 12], compatible with our observations of TNFα and MIP-1β suppression in sputum. MIP-1β levels are elevated in the lungs of patients with COPD [10], resulting in CCR5 activation, and subsequent recruitment of T cells, eosinophils and macrophages during COPD exacerbations [10].
Among the inflammatory cells, macrophage levels were numerically reduced in sputum by CHF6001, with a significant effect for the 800 μg dose. There are multiple studies showing the role and importance of macrophages to the pathophysiology of COPD. The number of lung macrophages is increased in patients with COPD compared to controls, with greater numbers associated with more severe disease [13]. Further, macrophages become dysfunctional in patients with COPD [14, 15], with a reduced ability to perform phagocytosis and efferocytosis [15, 16], and can release a wide range of inflammatory mediators, including during COPD exacerbations [15]. The inflammatory mediators suppressed by CHF6001 are known to be secreted by macrophages [17], and this could be a key target cell for PDE4 inhibitors in COPD. Given CHF6001 had no consistent statistically significant effects on the numbers or percentages of macrophages in sputum (with statistical significance reached for the lower CHF6001 dose but not the higher dose), our results suggest that CHF6001 may act mainly to reduce secretion of inflammatory mediators rather than the trafficking of cells through tissues. The increased level of IL-6 with the higher CHF6001 dose (and not with the lower dose) is an unexpected finding, but is likely to be a chance false positive result, since during treatment with placebo there was a significant reduction from baseline for this biomarker. Importantly, corticosteroids have a limited effect on the secretion of many of these mediators [17]. There was a trend to decreased sputum eosinophil levels with both CHF6001 doses, a result that is consistent with that of other compounds that target PDE4 [12, 18, 19]. Since in the current study all patients were also receiving ICS, the small effect on sputum eosinophil levels with CHF6001 might have been blunted by the background therapy.
In blood, ex-vivo TNFα production by LPS (a model that mimics the typical systemic inflammation occurring during an exacerbation) was significantly decreased with the higher CHF6001 dose. These data suggest that CHF6001 could have some systemic anti-inflammatory activity upon interaction with pathogenic material (despite the low systemic exposure). This anti-inflammatory activity may translate into clinical efficacy during exacerbations; this requires further investigation. The only in-vivo blood inflammatory biomarker that was consistently decreased was SP-D, which exerts antimicrobial effects and dampens inflammation in a range of tissues, including the lung [20]. Importantly, circulating SP-D is a biomarker of lung injury, with decreases associated with improvements in health status in patients with COPD [21, 22]. SP-D is a secretory product of non-ciliated bronchiolar cells [23], suggesting an active involvement in surfactant metabolism and/or host defence within small airways. This is particularly important in view of the extrafine formulation of CHF6001, which might have the potential to decrease SP-D leakage from the small airways to the systemic circulation and ameliorate small airways integrity. As expected, given the current study was not powered or designed to evaluate effects on lung function or symptoms (with all patients receiving inhaled triple therapy), CHF6001 did not show any effect on these endpoints. It should be noted that only modest improvements of lung function are typically observed even in larger studies using the oral PDE4 inhibitor roflumilast [3,4,5,6], with a lack of effect on symptoms also common [5, 6], while two other inhaled PDE4 inhibitors did not show consistent effects on lung function [24, 25].
The pharmacokinetic data clearly demonstrate dose-proportionality for CHF6001, both in terms of systemic exposure and sputum concentration. Importantly, the concentration of CHF6001 in the sputum was approximately 2000-fold that in the systemic circulation. These data are compatible with the positive biological effects of CHF6001 on inflammatory mediators in the sputum, and explain the lack of typical systemic PDE4-inhibitor side effects. These pharmacokinetic results validate the inhaled route of delivery as a way of both overcoming tolerability issues and avoiding systemic exposure.
Overall, CHF6001 demonstrated a good safety profile, with few adverse events considered related to study drug, and no major differences between treatments (including placebo) in vital signs or laboratory data. Notably, there was no dose-relationship with the adverse events typically experienced with roflumilast, especially those associated with the gastrointestinal tract.
This study has some limitations. First, despite clear dose-proportionality for systemic exposure of CHF6001 we were not able to show a dose-response for the majority of sputum and blood inflammatory markers. Second, we did not show an effect on cell numbers apart from macrophages for one of the tested doses – surprising given the anti-inflammatory effect of CHF6001 on cytokines and chemokines. Finally, the study was too short to fully evaluate the safety and tolerability profile of CHF6001. Key strengths of the study are the detailed sputum and blood sampling, and that all patients were receiving inhaled triple therapy throughout the study, consistent with how a PDE4 inhibitor is often used (and as per the Global Initiative for Chronic Obstructive Lung Disease recommendations [1]).
In conclusion, this study demonstrated that inhaled CHF6001 has lung anti-inflammatory activity when administered twice daily in addition to triple therapy in patients with COPD and chronic bronchitis. The high lung relative to systemic exposure provided by inhaled delivery improves the therapeutic index of CHF6001 by delivering anti-inflammatory effects while minimising the possibility of typical PDE4 inhibitor side effects.
Availability of data and materials
Chiesi commits to sharing with qualified scientific and medical Researchers, conducting legitimate research, patient-level data, study-level data, the clinical protocol and the full clinical study report of Chiesi Farmaceutici S.p.A.-sponsored interventional clinical trials in patients for medicines and indications approved by the European Medicines Agency and/or the US Food and Drug Administration after 1st January 2015, following the approval of any received research proposal and the signature of a Data Sharing Agreement. Chiesi provides access to clinical trial information consistently with the principle of safeguarding commercially confidential information and patient privacy. To date, the current study is out of scope of the Chiesi policy on Clinical Data Sharing.
Other information on Chiesi’s data sharing commitment, access and research request’s approval process are available in the Clinical Trial Transparency section of http://www.chiesi.com/en/research-and-development/.
References
Singh D, Agusti A, Anzueto A, Barnes PJ, Bourbeau J, Celli BR, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease: the GOLD science committee report 2019. Eur Respir J. 2019:1900164. https://doi.org/10.1183/13993003.00164-2019.
Singh D, Roche N, Halpin D, Agusti A, Wedzicha JA, Martinez FJ. Current controversies in the pharmacological treatment of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2016;194:541–9. https://doi.org/10.1164/rccm.201606-1179PP.
Calverley PMA, Rabe KF, Goehring U-M, Kristiansen S, Fabbri LM, Martinez FJ, et al. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009;374:685–94. https://doi.org/10.1016/S0140-6736(09)61255-1.
Fabbri LM, Calverley PMA, Izquierdo-Alonso JL, Bundschuh DS, Brose M, Martinez FJ, et al. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet. 2009;374:695–703. https://doi.org/10.1016/S0140-6736(09)61252-6.
Martinez FJ, Rabe KF, Sethi S, Pizzichini E, McIvor A, Anzueto A, et al. Effect of roflumilast and inhaled corticosteroid/long-acting β2-agonist on chronic obstructive pulmonary disease exacerbations (RE2SPOND). A randomized clinical trial. Am J Respir Crit Care Med. 2016;194:559–67. https://doi.org/10.1164/rccm.201607-1349OC.
Martinez FJ, Calverley PMA, Goehring U-M, Brose M, Fabbri LM, Rabe KF. Effect of roflumilast on exacerbations in patients with severe chronic obstructive pulmonary disease uncontrolled by combination therapy (REACT): a multicentre randomised controlled trial. Lancet Elsevier. 2015;385:857–66. https://doi.org/10.1016/S0140-6736(14)62410-7.
Villetti G, Carnini C, Battipaglia L, Preynat L, Bolzoni PT, Bassani F, et al. CHF6001 II: a novel phosphodiesterase 4 inhibitor, suitable for topical pulmonary administration - in vivo preclinical pharmacology profile defines a potent anti-inflammatory compound with a wide therapeutic window. J Pharmacol Exp Ther. 2015;352:568–78. https://doi.org/10.1124/jpet.114.220558.
Moretto N, Caruso P, Bosco R, Marchini G, Pastore F, Armani E, et al. CHF6001 I: a novel highly potent and selective phosphodiesterase 4 inhibitor with robust anti-inflammatory activity and suitable for topical pulmonary administration. J Pharmacol Exp Ther. 2015;352:559–67. https://doi.org/10.1124/jpet.114.220541.
Kardos P, Mokros I, Sauer R, Vogelmeier CF. Health status in patients with COPD treated with roflumilast: two large noninterventional real-life studies: DINO and DACOTA. Int J Chron Obstruct Pulmon Dis. 2018;13:1455–68. https://doi.org/10.2147/COPD.S159827.
Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118:3546–56. https://doi.org/10.1172/JCI36130.
Buenestado A, Grassin-Delyle S, Guitard F, Naline E, Faisy C, Israël-Biet D, et al. Roflumilast inhibits the release of chemokines and TNF-α from human lung macrophages stimulated with lipopolysaccharide. Br J Pharmacol. 2012;165:1877–90. https://doi.org/10.1111/j.1476-5381.2011.01667.x.
Grootendorst DC, Gauw SA, Verhoosel RM, Sterk PJ, Hospers JJ, Bredenbroker D, et al. Reduction in sputum neutrophil and eosinophil numbers by the PDE4 inhibitor roflumilast in patients with COPD. Thorax. 2007;62:1081–7. https://doi.org/10.1136/thx.2006.075937.
Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–53. https://doi.org/10.1056/NEJMoa032158.
Dewhurst JA, Lea S, Hardaker E, Dungwa JV, Ravi AK, Singh D. Characterisation of lung macrophage subpopulations in COPD patients and controls. Sci Rep. 2017;7:7143. https://doi.org/10.1038/s41598-017-07101-2.
Bewley MA, Budd RC, Ryan E, Cole J, Collini P, Marshall J, et al. Opsonic phagocytosis in chronic obstructive pulmonary disease is enhanced by Nrf2 agonists. Am J Respir Crit Care Med. 2018;198:739–50. https://doi.org/10.1164/rccm.201705-0903OC.
Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81:289–96. https://doi.org/10.1046/j.1440-1711.2003.t01-1-01170.x.
Higham A, Booth G, Lea S, Southworth T, Plumb J, Singh D. The effects of corticosteroids on COPD lung macrophages: a pooled analysis. Respir Res. 2015;16:98. https://doi.org/10.1186/s12931-015-0260-0.
Rabe KF, Watz H, Baraldo S, Pedersen F, Biondini D, Bagul N, et al. Anti-inflammatory effects of roflumilast in chronic obstructive pulmonary disease (ROBERT): a 16-week, randomised, placebo-controlled trial. Lancet Respir Med Elsevier. 2018;6:827–36. https://doi.org/10.1016/S2213-2600(18)30331-X.
Martinez FJ, Rabe KF, Calverley PMA, Fabbri LM, Sethi S, Pizzichini E, et al. Determinants of response to roflumilast in severe COPD: pooled analysis of two randomized trials. Am J Respir Crit Care Med. 2018;198:1268–78. https://doi.org/10.1164/rccm.201712-2493OC.
Sorensen GL. Surfactant protein D in respiratory and non-respiratory diseases. Front Med. 2018;5:18. https://doi.org/10.3389/fmed.2018.00018.
Sin DD, Leung R, Gan WQ, Man SP. Circulating surfactant protein D as a potential lung-specific biomarker of health outcomes in COPD: a pilot study. BMC Pulm Med. 2007;7:13. https://doi.org/10.1186/1471-2466-7-13.
Sin DD, Man SFP, Marciniuk DD, Ford G, FitzGerald M, Wong E, et al. The effects of fluticasone with or without salmeterol on systemic biomarkers of inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:1207–14. https://doi.org/10.1164/rccm.200709-1356OC.
Crouch E, Parghi D, Kuan SF, Persson A. Surfactant protein D: subcellular localization in nonciliated bronchiolar epithelial cells. Am J Physiol Cell Mol Physiol. 1992;263:L60–6. https://doi.org/10.1152/ajplung.1992.263.1.L60.
Vestbo J, Tan L, Atkinson G, Ward J, UK-500 001 Global Study Team. A controlled trial of 6-weeks’ treatment with a novel inhaled phosphodiesterase type-4 inhibitor in COPD. Eur Respir J. 2009;33:1039–44. https://doi.org/10.1183/09031936.00068908.
Watz H, Mistry SJ, Lazaar AL, IPC101939 investigators. Safety and tolerability of the inhaled phosphodiesterase 4 inhibitor GSK256066 in moderate COPD. Pulm Pharmacol Ther. 2013;26:588–95. https://doi.org/10.1016/j.pupt.2013.05.004.
Acknowledgements
Results of this study have been presented at the American Thoracic Society 2019 annual conference.
The authors would like to thank the investigators and patients at the investigative sites for their support of this study. Dave Singh is supported by the National Institute for Health Research (NIHR) Manchester Biomedical Research Centre (BRC).
Funding
This study was funded by Chiesi Farmaceutici SpA. Chiesi Farmaceutici SpA, was responsible for the design and analysis of the study, oversaw its conduct and was responsible for study report preparation. Writing support was provided by David Young of Young Medical Communications and Consulting Ltd. This support was funded by Chiesi Farmaceutici SpA.
Author information
Authors and Affiliations
Contributions
The study was conceived and designed by DS, HW, GL, MG and MAN. DS, KMB, BC, OK, BL and HW contributed to data acquisition, with GL overseeing study conduct, and AE and MAN overseeing medical data integrity. The data were analysed by MG and SG, and all authors revised the manuscript for intellectual content and approved its publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by independent ethics committees for each institution (see the Additional file 1 for details). All patients provided written informed consent prior to study start.
Consent for publication
Not applicable.
Competing interests
Dave Singh received personal fees from Chiesi during the conduct of this study. Outside the submitted work, he reports grants and personal fees from AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Glenmark, Menarini, Mundipharma, Novartis, Pfizer, Pulmatrix, Therevance, and Verona, and personal fees from Cipla, Genentech and Peptinnovate.
Kai Michael Beeh declares that no personal payments were received from any pharmaceutical entity in the past five years. He is a full time employee of insaf Respiratory Research Institute. The institution has received compensation for services on advisory boards or consulting for Ablynx, Almirall, AstraZeneca, Berlin Chemie, Boehringer, Chiesi, Cytos, Mundipharma, Novartis, Pohl Boskamp, Zentiva. The institution has received compensation for speaker activities in scientific meetings supported by Almirall, AstraZeneca, Berlin Chemie, Boehringer, Cytos, ERT, GSK, Novartis, Pfizer, Pohl Boskamp, Takeda. The institution has received compensation for design and performance of clinical trials from Almirall, Altana/Nycomed, AstraZeneca, Boehringer, Cytos, GSK, Infinity, Medapharma, MSD, Mundipharma, Novartis, Parexel, Pearl Therapeutics, Pfizer, Revotar, Teva, Sterna, and Zentiva.
Brendan Colgan has nothing to disclose.
Oliver Kornmann’s institution received fees from Chiesi for conducting this study as a participating site. Dr. Kornmann reports personal fees as speaker or Advisory Board member from AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Sanofi and Novartis.
Brian Leaker has nothing to disclose.
Henrik Watz reports personal fees from Chiesi during the conduct of the study. Outside the submitted work, Dr. Watz reports personal fees from Bayer, personal fees from GSK, personal fees from Boehringer Ingelheim, personal fees from Novartis, personal fees from AstraZeneca, personal fees from BerlinChemie, and personal fees from Roche.
Germano Lucci, Silvia Geraci, Mirco Govoni, Aida Emirova, and Marie Anna Nandeuil are all employees of Chiesi, the sponsor of this trial.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Supplementary methods and results. Methods and results supporting main body of the manuscript. (PDF 216 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Singh, D., Beeh, K.M., Colgan, B. et al. Effect of the inhaled PDE4 inhibitor CHF6001 on biomarkers of inflammation in COPD. Respir Res 20, 180 (2019). https://doi.org/10.1186/s12931-019-1142-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-019-1142-7