
Abstract
Background
Pulmonary hypertension (PH) is a progressive and fatal disease. While cigarette smoke can change DNA methylation status, the role of such molecular alterations in smoke-associated PH is unclear.
Methods
A PH rat model was developed by exposing animals to cigarette smoke for 3 months. Right ventricular systolic pressure was measured with a right heart catheter. Histological changes (right ventricular hypertrophy index, medial wall thickness in pulmonary arteries (PAs)) and DNMT1 protein levels in rat PAs or primary human PA smooth muscle cells (HPASMCs) exposed to cigarette smoke extract were assessed. Methylation sequencing and MassArray® were used to detect genomic and RASEF promoter methylation status, respectively. After DNMT1 knockdown and cigarette smoke extract exposure, HPASMCs behavior (proliferation, migration) and RASEF methylation status were examined; RASEF mRNA expression was evaluated by real-time-polymerase chain reaction. RASEF overexpression viral vectors were used to assess the impact of RASEF on rat PH and HPASMCs remodeling.
Results
Higher right ventricular systolic pressure, medial wall thickness, and right ventricular hypertrophy index values were observed in the smoking group rats. Smoke exposure increased DNMT1 expression and RASEF methylation levels in rat PAs and HPASMCs. Cigarette smoke extract induced HPASMCs behavioral changes and RASEF hypermethylation followed by silencing, while DNMT1 knockdown markedly inhibited these changes. RASEF overexpression distinctly inhibited PH and HPASMCs remodeling, possibly through phospho-AKT (Ser473), PCNA, and MMP9 downregulation.
Conclusions
Cigarette smoke caused PA remodeling in PH rats related to RASEF hypermethylation. These results expand our understanding of key epigenetic mechanisms in cigarette smoke-associated PH and potentially provide a novel therapeutic target for PH.
Similar content being viewed by others


Introduction
Pulmonary hypertension (PH) is defined as an increase in mean pulmonary arterial pressure above 25 mmHg at rest. PH is a complex, deadly, and refractory disease associated with various respiratory and cardiovascular diseases [1]. An estimated 30–50% of patients with advanced chronic obstructive pulmonary disease (COPD) suffer from PH [2, 3], while 1–3% patients with mild COPD have severe PH [4]. Few studies have investigated the molecular mechanisms of PH associated with COPD (PH-COPD).
Pulmonary arterial smooth muscle remodeling (PASMR) can lead to media thickening, a main pathological characteristic of PH [5, 6], due to increased proliferation, apoptotic resistance, and migration of PA smooth muscle cells (SMCs) [7]. Except for hypoxia [8], cigarette smoking (the major risk factor for COPD) can directly facilitate the remodeling of PA smooth muscle (PASM) [9, 10] and could partly explain the pathogenesis of PH-COPD.
DNA methylation, the most common genome epigenetic modification that can regulate chromosomal stability and gene expression under the catalytic action of DNA methyltransferases (DNMTs) [11]; eukaryotic DNMTs consist of a maintenance methyltransferase (DNMT1) and two de novo methyltransferases (DNMT3a and DNMT3b) [12]. Genomic methylation status can be easily altered by environmental factors [13]. Cigarette and tobacco smoke have been shown to strongly modify genome methylation [14, 15], which is related to many diseases, such as COPD [16], lung cancer [17], and atherosclerosis [18]. Altered DNA methylation (hypermethylation) has also been shown to play an important role in group I PH [19, 20]. DNMT1 overexpression has been found in PH tissues and involved in hypoxia-induced HPASMCs proliferation and apoptotic resistance [21]. 5-Azacytidine (5-Aza), a selective DNMT1 inhibitor, has been shown to inhibit platelet-derived growth factor-induced aortic and airway SMC proliferation and migration, and has a protective role in atherosclerosis and asthma [22, 23]. However, the roles of DNMT1 in cigarette smoke (CS)-associated PH have not been investigated.
Rab GTPases are vital regulators of intracellular membrane traffic [24], and Rab proteins influence the transport and/or function of signal transducers and growth factors. RASEF (also known as Rab45) is an atypical GTPase, as a tumor suppressor gene in cutaneous malignant melanoma [25]. Maat et al. found RASEF promotor hypermethylation was inversely related with the survival of uveal melanoma patients [26]. In addition, RASEF was shown to distinctly promote apoptosis of chronic myeloid leukemia progenitor cells via activation of p38 signal [27].
As a proliferative disease similar to tumors, PH may also be improved by many tumor suppressor genes, such as P53, P21 and PPAR-γ [28, 29]. However, the relationship between RASEF and PH is unknown. Therefore, this study investigated the role of RASEF on CS-induced remodeling of PASM and rat PH.
Materials and methods
Animals models
Adult male Sprague–Dawley rats (180–220 g) were acquired from the Experimental Animal Center of Tongji Medical College (Wuhan, China). All animal experiments were carried out according to the Animal Care and Use guidelines of the Chinese Council on Animal Care. Twenty rats were randomly and equally divided into two groups (n = 10 each) and exposed to either fresh air (the air group) or cigarette smoke (the smoking group). The smoking group rats were exposed to the smoke from 10 cigarettes (Hong Jin Long, 1.2 mg nicotine, 15 mg tar per cigarette, Wuhan, China) in a ventilated whole body exposure chamber for 1 h each time, two times per day for a total of 3 months according to previous protocols [30, 31]. All rats were euthanized with sodium pentobarbital.
RASEF overexpression in PH rats
Thirty rats were randomly divided into two groups and exposed to fresh air (n = 10) or cigarette smoke (n = 20) as described above. Then, rats exposed to cigarette smoke were equally subdivided (n = 10 each) and infected with either adeno-associated virus type-1 (Hanheng Biotechnology, China) expressing green fluorescent protein (AAV1.GFP) or AAV1 containing rat RASEF cDNA (AAV1.RASEF) by tracheal injection (1 × 1011 viral genomes/rat) as described previously [32]; the air group rats received AAV1.GFP. All rats were sacrificed by sodium pentobarbital 6 weeks after infection.
Hemodynamic measurements and histological analysis
A 3F polyethylene catheter and the PowerLab system (AD Instruments, Australia) were used to test right ventricular (RV) systolic pressure (RVSP) in vivo as described previously [33]. After hemodynamic measurements were completed, rats were sacrificed as described above and hearts divided into the RV and left ventricle plus septum (LV + S). RV and LV + S tissues weighed and used to calculate the RV hypertrophy index (RVHI), which is the mass ratio of the RV to the LV + S. Left lung tissue was fixed, and 4-μm paraffin sections were made and stained with hematoxylin and eosin. The wall thickness of pulmonary arterioles (outside diameter: 50–150 μm) was then measured using an optical microscope (Olympus BX61, Tokyo, Japan) [34].
HPASMCs culture and transfection
HPASMCs were purchased from American Type Culture Collection (MD, USA) and grown in Dulbecco’s Modified Eagle’s Medium-F12 containing 10% fetal bovine serum. Cigarette smoke extract (CSE) obtained from Research Cigarettes (Code 3R4F, University of Kentucky, USA) was acquired as described elsewhere [35]. DNMT1 small interfering RNA (siRNA; 50 nM) was transfected into HPASMCs using Lipofectamine 2000 (Invitrogen, USA) for silencing DNMT1. DNMT1 siRNA target sequences were as follows: the first 5′-GCACCUCAUUUGCCGAAUATT-3′; the second 5′- GGGACUGUGUCUCUGUUAUTT-3′. DNMT1 overexpression pcDNA3.1 plasmid vector was also transfected into HPASMCs using Lipofectamine 2000 (Vigene Biosciences, China). The RASEF overexpression adenovirus vector (Ad.RASEF; Vigene Biosciences, China) was also transfected into HPASMCs (MOI 250).
Western blot
Total proteins were extracted from rat PAs or HPASMCs, and their concentrations were measured with a BCA kit (Servicebio, China). Primary antibodies against β-actin (Sungene, China), DNMT1 (ABclonal, USA), RASEF, matrix metalloproteinase 9 (MMP9) (Abcam, UK), phospho-AKT (ser-473), AKT (Cell Signaling Technologies, USA), and proliferating cell nuclear antigen (PCNA; Proteintech, China) were used. Bands were detected by a ChemiDoc MP System (Bio-Rad Laboratories, USA), and the intensity was analyzed by ImageJ software.
Real-time polymerase chain reaction (RT-PCR)
Total RNA was extracted from rat PAs and HPASMCs, reverse transcribed, and then subjected to RT-PCR using corresponding kits according to the manufacturer’s instructions (Takara, Japan). The primers used were as follows: human β-actin, 5′-AGAAAATCTGGCACCACACCT-3′ (forward) and 5′-GATAGCACAGCCTGGATAGCA-3′ (reverse); rat β-actin, 5′-CGTAAAGACCTCTATGCCAACA-3′ (forward) and 5′-CGGACTCATCGTACTCCTGCT-3′ (reverse); human DNMT1, 5′-AGGCGGCTCAAAGATTTGGAA-3′ (forward) and 5′-GCAGAAATTCGTGCAAGAGATTC-3′ (reverse); human RASEF, 5′-AGATTGTACTTGCTGGGGACG-3′ (forward) and 5′-GAGCTGCAGAACTGTTCGTT-3′ (reverse); rat RASEF, 5′-ACGGGATCTGGAACTAATCCG-3′ (forward) and 5′-GGCACTTCTAAGGCCGTCAT-3′ (reverse). The ratio for the mRNA of interest was normalized by β-actin.
Immunofluorescence and immunohistochemistry
Rat lung sections were stained with mouse anti-α-smooth muscle actin (Abcam, UK) and rabbit anti-RASEF (Abcam, UK) primary antibodies followed by fluorescein isothiocyanate-conjugated anti-mouse immunoglobulin G and phycoerythrin-conjugated anti-rabbit immunoglobulin G (Jackson ImmunoResearch, USA). HPASMCs were incubated with a primary anti-DNMT1 (ABclonal, USA) antibody followed by phycoerythrin-conjugated anti-rabbit immunoglobulin G. And 4′,6-diamidino-2-phenylindole was used to stain nuclei. Immunofluorescence was observed by fluorescence microscope. Immunohistochemical staining was performed according to the manufacturer’s instructions (Boster Biological Technology, China). Primary antibodies against RASEF, MMP9 (Abcam, UK), and phospho-AKT (ser-473) (Cell Signaling Technology, USA) were used.
DNA methylation analysis
Genomic DNA of rat PAs was extracted using a QIAamp® DNA Mini kit (Qiagen, Germany) according to the manufacturer’s instructions, and genome-wide methylation sequencing using MethylRAD technology was carried out by the Shanghai OE Biotech Company (China). The CpG methylation status of RASEF promotor (from − 224 to + 196 bp) in rat PAs was determined by a MassARRAY® system, which was carried out by the OE Biotech Company. The methylated primers used were as follows: forward 5′- aggaagagag GTATTTGGGAATGAGTTGGGTT-3′, and reverse 5′- cagtaatacgactcactatagggagaaggct CAACAACAATTAAAAACAAAAACCTC-3′. HPASMCs were pretreated with 2 μM 5-Aza or DNMT1 siRNA for 24 h and then incubated with 2% cigarette smoke extract (CSE) for 24 h. Cellular DNA was extracted as described above, and the CpG methylation status of the RASEF promotor (from − 164 to + 215 bp) was detected by Compass Biotechnology (Beijing, China) using a MassARRAY® system. The methylated primers used were 5′-aggaagagagGTAGGGTTTTTTTTGGAAGGA-3′ (forward) and 5′-cagtaatacgactcactatagggagaaggctCAACCAAATCTCCCCCACCTAC-3′ (reverse).
Cellular viability and cell cycle assay
In order to study the role of DNMT1 on HPASMCs proliferation, HPASMCs were treated with 2 μM 5-Aza or DNMT1 siRNA for 24 h followed by incubation with 2% CSE for 48 h. Cell proliferation/viability was then assessed with a Cell Counting Kit-8 (CCK8; Promotor, China) and cell cycle detection kit (KeyGEN BioTECH, China) according to the manufacturer’s instructions. CCK8 and cell cycle analyses were completed using an automated spectrophotometric plate reader (PerkinElmer, USA) and flow cytometer (BD Bioscience, USA), respectively. In addition, the proliferation of HPASMCs infected with Ad.NC or Ad.RASEF for 24 h then 2% CSE for another 48 h was measured by cell cycle analysis and 5-ethynyl-2′-deoxyuridine (EdU) staining (RiboBio, China). EdU was labeled with Apollo 567, and cells were observed via fluorescence microscope.
Cellular apoptosis detection
HPASMCs were transfected with Ad.RASEF for 24 h followed by 2% CSE for another 48 h. Then, cells were harvested, and annexin V-fluorescein isothiocyanate and propidium iodide were added to the cell suspension. Flow cytometry was used to measure apoptosis. Experiments were repeated at least three times.
Migration assay
Membranes with 8-μm pores in 24-well Transwell® plates (Coring, USA) were employed for migration assays. HPASMCs were transfected with DNMT1 siRNA or Ad.RASEF for 24 h before being digested and counted. About 50,000 cells were added to the upper chamber for 24 h to adhere. Next, 600 μL of standard culture medium with or without 2% CSE was added to the lower compartment, and the upper chamber liquid was replaced with 200 μL of medium including 5% fetal bovine serum. After 24 h, cells within the membranes of bottom chambers were fixed, stained with 0.1% crystal violet, and imaged. Experiments were repeated at least three times.
Statistical analysis
Data were analyzed using GraphPad Prism software (version 5.0) and expressed as the mean ± standard error of mean. Student’s t-test was used for comparisons between two groups. One-way analysis of variance followed by a Newman–Keuls post-test was utilized for multiple group comparisons. A P < 0.05 was considered statistically significant.
Results
Cigarette smoke induced PH in rats
The RVSP was significantly higher in the smoking group (37.2 ± 3.5 mmHg) than in the air group (22.2 ± 1.0 mmHg, P < 0.01, Fig. 1a, b). The RVHI and mass ratio of the RV to body weight were increased in the smoking group (Fig. 1c, d). The wall thickness of pulmonary arterioles was also obviously increased in the smoking group (Fig. 1e, f).

Influence of cigarette smoke on rat hemodynamics and histopathology. Rat RVSP (a, b), RVHI [RV to LV + S mass ratio] (c), RV to body weight mass ratio (d), and pulmonary arteriole wall thickness (e, f). Scale bar = 50 μm. Data are displayed as the mean ± standard error of the mean (n = 10 per group); **P < 0.01, ***P < 0.001 versus control
Cigarette smoke increased DNMT1 expression in pulmonary arterial smooth muscle
DNMT1 protein were higher in rats PA smooth muscle of the smoking group than controls (Fig. 2a, b). In addition, exposure of HPASMCs to 2%CSE increased cell viability (Fig. 2c) and DNMT1 protein levels (Fig. 2d-f).

Effect of cigarette smoke on DNMT1 levels in rat PAs and HPASMCs. a, b Western blot showed that cigarette smoke increased DNMT1 protein levels in rat PAs. c CCK8 assay revealed an increase in HPASMCs cell viability after treatment with different concentrations of CSE for 48 h. d, e DNMT1 protein expression in HPASMCs treated with CSE for 48 h. f Immunofluorescence results indicate an increase in DNMT1 protein levels (increased fluorescence) in CSE-treated HPASMCs. Data are displayed as the mean ± standard error of the mean (n = 3); (b) *P < 0.05 vs. the air group; (c, e) *P < 0.05, **P < 0.01, ***P < 0.001 vs. Con
5-Aza and DNMT1 siRNA inhibited CSE-induced HPASMCs viability, cell cycle, migration
5-Aza (0.5–10 μM), a selective inhibitor of DNMT1, and DNMT1 siRNA both reduced DNMT1 protein levels in HPASMCs (Figs. 3a, b and 4a-c, respectively). Pretreatment with 5-Aza (2 μM) inhibited CSE-induced the increase in cell viability (Fig. 3d) but did not influence apoptosis (Fig. 3c) in HPASMCs. DNMT1 siRNA also suppressed CSE-induced proliferation and cell cycle transition from G0/G1 to S or G2/M phase (Fig. 4d-f). DNMT1 siRNA also inhibited CSE-induced cell migration (Fig. 4g, h).

Effect of 5-Aza on CSE-induced HPASMCs proliferation and apoptosis changes. HPASMCs pretreatment with various concentration of 5-Aza (0, 0.5, 1, 2, 5, and 10 μM) for 48 h decreased DNMT1 protein levels (a, b) but did not affect the number of apoptotic cells (c). d CCK8 assay revealed pretreatment with 5-Aza (2 μM) inhibited CSE-induced increases in HPASMCs viability. Data are displayed as the mean ± standard error of the mean (n = 3); *P < 0.05, **P < 0.01

Effect of DNMT1 siRNA on CSE-induced HPASMCs proliferation and migration changes. a-c DNMT1 siRNA reduced DNMT1 mRNA and protein expression in HPASMCs. Pretreatment of HPASMCs with DNMT1 siRNA suppressed 48-h CSE-induced increases in cell viability (d) and cell cycle transition from G0/G1 to S or G2/M phase (e, f). g, h DNMT1 siRNA transfection prevented 24-h CSE-induced migration of HPASMCs. Scale bar = 100 μm. Data are displayed as the mean ± standard error of the mean (n = 3); *P < 0.05, **P < 0.01, ***P < 0.001
Cigarette smoke reduced RASEF expression in rat PAs
DNA methylation sequencing based on MethylRAD technology revealed many differences in genes and/or promotors (TSS200 and TSS1500) in rat PAs of the air group and the smoking group (Fig. 5a-b). And the detailed data was in Additional file 1. Among the different loci, one CpG of the RASEF promotor (Rnor_6.0: chromosome 5, NC_005104.4–90,418,747) was found to be hypermethylated in PAs of the smoking group (Fig. 5c). RASEF promoter in rat PAs of the smoking group was hypermethylated (Fig. 5d, e). Moreover, RASEF mRNA level in rat PAs in the smoking group was lower (Fig. 5f). RASEF-stained smooth muscle cells in PAs in the smoking group were also decreased (Fig. 5g).

Cigarette smoke influenced RASEF promoter methylation and transcription in rat PAs. Genome-wide DNA methylation sequencing and heat map of rat PAs revealed a number of changes in gene distribution associated with different methylation sites (a, b). c Cigarette smoke caused hypermethylation of a CpG within the RASEF promoter (located at NC_005104.4–90,418,763) in rat PAs. d, e The methylation status of CpGs in RASEF promotor in rat PAs was measured by MassARRAY®. f RASEF mRNA levels were lower in the smoking PH versus control rat PAs. g RASEF immunofluorescence was much lower in the pulmonary arterioles of PH rats versus controls. Data are displayed as the mean ± standard error of the mean (n = 4); *P < 0.05, ***P < 0.001 vs. Air group
DNMT1 knockdown inhibited CSE-induced RASEF downregulation in HPASMCs
RASEF protein level in HPASMCs was reduced after stimulation with 2% CSE for 48 h or 72 h (Fig. 6a). Similarly, DNMT1 overexpression via transfection of DNMT1-vector significantly reduced RASEF mRNA expression (Fig. 6e, f). Treatment with 5-aza (2 μM) for 24–72 h increased RASEF protein expression (Fig. 6b). 2% CSE also inhibited RASEF mRNA expression in HPASMCs; however, 5-Aza or DNMT1siRNA could partly reverse this effect (Fig. 6c, d). More importantly, the CpG island in the RASEF promotor (from − 164 to + 215 bp) was hypermethylated under CSE treatment, while pretreatment of HPASMCs with either DNMT1 siRNA or 5-Aza suppressed the methylation caused by CSE (Fig. 6g-i).

Influence of DNMT1 siRNA and 5-Aza on CSE-induced RASEF hypermethylation and transcription in HPASMCs. CSE reduced RASEF protein expression in HPASMCs (a), while 5-Aza (2 μM) increased them (b). c, d Pretreatment with either 5-Aza (2 μM) or DNMT1 siRNA inhibited CSE-induced downregulation of RASEF mRNA. e, f HPASMCs transfected with a DNMT1 overexpression vector reduced RASEF mRNA expression. g, i CSE-induced RASEF promoter CpG hypermethylation was prevented by pretreatment with either DNMT1 siRNA or 5-Aza (2 μM). Data are displayed as the mean ± standard error of the mean (n = 3); *P < 0.05, **P < 0.01
AAV1.RASEF inhibited cigarette smoke-induced PH pathology
Tracheal injection of AAV1 enabled successful infection of rat PA smooth muscle (Fig. 7a). Infection with AAV1.RASEF was found to reverse cigarette smoke (CS)-induced downregulation of RASEF (Fig. 7c-d). Moreover, both RVSP changes and RV hypertrophy caused by CS were significantly improved in AAV1.RASEF-treated rats (Fig. 8a-d). AAV1.RASEF infection also obviously reduced the thickness of pulmonary arteriole walls (Fig. 8e, f).

Effect of AAV1.RASEF on cigarette smoke-induced RASEF downregulation. a One month after tracheal injection of AAV1, the rat PAs were successfully infected. b Intervention plan for experimental rats. c AAV1.RASEF inhibited cigarette smoke-induced RASEF gene silencing in rat PAs. d The protein expression of RASEF in rat PAs. Data are displayed as the mean ± standard error of the mean(n = 6); ***P < 0.001

AAV1.RASEF effect on cigarette smoke-triggered rat PH. AAV1.RASEF infection significantly improved cigarette smoke-induced changes in the RVSP (a, b), RVHI [RV to LV + S mass ratio] (c), RV to body weight mass ratio (d), and wall thickness of rat pulmonary arterioles (e, f). Scale bar = 50 μm. Data are displayed as the mean ± standard error of the mean (n = 8); **P < 0.01, ***P < 0.001
Ad.RASEF inhibited CSE-induced HPASMCs cell cycle and migration changes
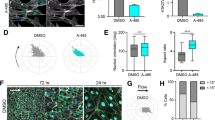
Ad.RASEF transfection successfully induced overexpression of RASEF protein in HPASMCs (Fig. 9a). Increase in EdU-positive staining cells caused by CSE treatment was significantly inhibited by Ad.RASEF transfection (Fig. 9b). CSE-induced cell cycle changes, including increased S phase and reduced G0/G1 phase populations, were also reversed by Ad.RASEF; and Ad.RASEF also reduced G2/M phase populations (Fig. 9c, f). Moreover, Ad.RASEF promoted HPASMCs apoptosis (Fig. 7d, g), and CSE-induced HPASMCs migration was reversed by Ad.RASEF (Fig. 9e, h).

Influence of Ad.RASEF on CSE-induced HPASMCs proliferation and migration changes. a Ad.RASEF transfection increased RASEF protein levels in HPASMCs. Transfection of HPASMCs with Ad.RASEF inhibited CSE-induced increases in cell proliferation [EdU positivity] (b), reduced S and G2/M phase and increased G0/G1 phase populations (c, f), promoted apoptosis (d, g), and prevented HPASMCs migration (e, h). Scale bar = 100 μm. Data are displayed as the mean ± standard error of the mean (n = 3); *P < 0.05, **P < .001, ***P < 0.001
RASEF overexpression reduced cigarette smoke -induced AKT activity
2% CSE increased PCNA and MMP9 protein levels in HPASMCs accompanied by upregulation of phospho-AKT (Ser473). However, these effects were inhibited by RASEF overexpression (Fig. 10a). And similar effects were also seen in vivo (Fig. 10b).

Effect of RASEF overexpression on cigarette smoke -induced AKT activation. a Transfection of HPASMCs with Ad.RASEF prevented 48-h CSE-induced increases in phospho-AKT (Ser473), PCNA, and MMP9 protein levels. b Protein expressions of RASEF, MMP9, and P-AKT (ser-473) in rat PAs were examined by immunochemistry. Scale bar = 50 μm
Discussion
In this study, cigarette smoking (CS) exposure led to rat PH and induced proliferation and migration of HPASMCs, followed by increased DNMT1 mRNA and protein expression, RASEF silencing, and RASEF promotor hypermethylation in vivo and in vitro. However, pretreatment with either DNMT1 siRNA or 5-Aza inhibited CSE-induced HPASMCs proliferation, migration, and RASEF expression and methylation changes. Furthermore, RASEF overexpression improved CS-induced rat PH pathophysiology and inhibited CSE-induced HPASMCs proliferation and migration. Taken together, these results emphasize the important roles of RASEF methylation in CS-associated PH development.
The most commonly used rodent models of PH include those induced by monocrotaline or chronic hypoxia, which model Group 3 PH (PH due to lung diseases and/or hypoxia) [36, 37]. However, Group 3 PH also includes COPD-associated PH without hypoxemia, which requires greater consideration of the pathogenic roles of cigarette smoke [4]. Long-term (≥6 months) cigarette smoke exposure has been shown to cause emphysema in rodents [38], and short-term (3–4 months) exposure can induce rat PH [39, 40]. In the present study, rats exposed to CS for 3 months had increased RVSP, obvious PA remodeling, and RV hypertrophy.
As with other epigenetic changes (e.g., microRNA, histone modification, etc.), DNA methylation has also been implicated in the pathogenesis of PH, particularly in PA remodeling [19, 20]. Recent studies have shown that DNMT1 or its inhibitor, 5-Aza, influenced vascular smooth muscle phenotype and behaviors (e.g., proliferation and migration) resulting from hypoxia or platelet-derived growth factor treatment [21, 22]. The current results showed CS exposure increased DNMT1 mRNA and protein levels in rat PAs and HPASMCs, while DNMT1 knockdown inhibited CSE-induced proliferation and migration of HPASMCs. These results suggest that DNMT1 expression is closely related to CS-associated PA remodeling. However, the detailed mechanism of how DNA methylation influences CS-induced PA remodeling via regulation of DNMT1 expression remains unclear.
Although there are no reports regarding RASEF involvement in PA remodeling or PH pathology, other Rab family members have been found to be connected with vascular SMC function. For example, Rab5a knockdown has been shown to inhibit the proliferation and migration of human aorta SMCs, as well AKT activation [41], and Rab25 reportedly influences cerebral artery SMC vasoconstriction through regulating the abundance of Cav1.2 [42]. Here, CS was found to decrease RASEF mRNA and protein expression in rat PAs and HPASMCs, while RASEF overexpression alleviated CS-induced PH in vivo and inhibited CSE-induced HPASMCs proliferation and migration in vitro. Hence, RASEF plays an important role in CS-associated PA remodeling.
Of note, RASEF silencing in melanoma resulted from its promotor hypermethylation, and was associated with the prognosis of melanoma patients [26, 43]. Interestingly, we found the methylation level of CpGs in RASEF promotor was significantly elevated in rat PAs in the smoking group. In order to illuminate the relationship between DNA methylation, DNMT1, and RASEF in CS-associated PA remodeling, we examined RASEF promoter methylation status and mRNA expression in HPASMCs under the combined stimuli including CSE and DNMT1siRNA or 5-aza. While CSE increased RASEF promotor methylation and RASEF expression in HPASMCs, pretreatment with either DNMT1 siRNA or 5-Aza inhibited these effects. Overall, these results suggest that CS induced PA remodeling likely via DNMT1-mediated RASEF hypermethylation in PASMCs.
AKT signaling plays an important role in vessel maturation [44] and vascular remodeling in PH models [45, 46]. Garat et al. demonstrated that tricirbine (an AKT inhibitor) attenuated hypoxia-induced PA media thickening and RV hypertrophy [47]. Although AKT has a vital role in vascular SMC proliferation [48] and migration [49], few studies have shown the link between AKT and CS-associated vascular remodeling [35]. In our study, CS was found to upregulate phospho-AKT (Ser 473), PCNA (proliferation marker), and MMP9 protein levels in HPASMCs, while RASEF overexpression inhibited these changes. Our previous study clarified the important role of MMP9 in PASMC migration in a monocrotaline-induced PH model [33], and some studies have reported AKT signaling could regulate MMP9 expression [50, 51]. The current result that CS upregulated MMP9 protein expression in HPASMCs is similar to that of Ghosh et al. [52]. More importantly, RASEF overexpression notably reversed CS-induced upregulation of P-AKT and MMP9 not only in vitro, but also in vivo. So we believe that RASEF plays a protective role in CS-induced PA remodeling possibly via regulation of P-AKT/MMP9 (Fig. 11).

Schematic illustration of the potential role of DNA methylation of RASEF in the pathogenesis of pulmonary hypertension
There are also some limitations in our study. Though CSE treatment was found to enhance HPASMCs proliferation and migration as well as increase phospho-AKT (Ser 473), PCNA, and MMP9 levels, whether AKT directly or indirectly causes upregulation of MMP9 under PH conditions in vitro and/or in vivo to be seen. Nonetheless, Ghosh et al. previously reported AKT was the upstream of MMP9 in elastase-treated rat aortic SMCs [53]. In addition, whether RASEF, a member of Ras family, regulated AKT activation via Raf or other signal is uncertain and need to be studied in the future [54].
Conclusions
DNMT1 mediated RASEF promotor hypermethylation plays an important role in cigarette smoke-induced PA remodeling. Therefore, RASEF may be a novel therapeutic target for treatment of cigarette smoke-associated PH.
Abbreviations
- COPD:
-
Chronic obstructive pulmonary disease
- COPD-PH:
-
pulmonary hypertension associated with COPD
- CS:
-
Cigarette smoking
- CSE:
-
Cigarette smoke extract
- DNMT1:
-
DNA methyltransferase 1
- HPASMCs:
-
Primary human pulmonary arterial smooth muscle cells
- MCT:
-
Monocrotaline
- PAR:
-
Pulmonary arterial remodeling
- PAs:
-
Pulmonary arteries
- PASMR:
-
Pulmonary arterial smooth muscle remodeling
- PDGF:
-
Platelet-derived growth factor
- RASEF:
-
RAS And EF-hand domain containing
- RVHI:
-
Right ventricle hypertrophy index
- RVSP:
-
Right ventricle systolic pressure
- WT:
-
Wall thickness
References
Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67–119.
Barnes PJ, Celli BR. Systemic manifestations and comorbidities of COPD. Eur Respir J. 2009;33:1165–85.
Andersen KH, Iversen M, Kjaergaard J, Mortensen J, Nielsen-Kudsk JE, Bendstrup E, et al. Prevalence, predictors, and survival in pulmonary hypertension related to end-stage chronic obstructive pulmonary disease. J Heart Lung Transplant. 2012;31:373–80.
Minai OA, Chaouat A, Adnot S. Pulmonary hypertension in COPD: epidemiology, significance, and management: pulmonary vascular disease: the global perspective. Chest. 2010;137:39S–51S.
Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43:25S–32S.
Sakao S, Tatsumi K. Vascular remodeling in pulmonary arterial hypertension: multiple cancer-like pathways and possible treatment modalities. Int J Cardiol. 2011;147:4–12.
Goncharova EA, Ammit AJ, Irani C, Carroll RG, Eszterhas AJ, Panettieri RA, et al. PI3K is required for proliferation and migration of human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L354–63.
Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, et al. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1alpha. Am J Respir Crit Care Med. 2014;189:314–24.
Ferrer E, Peinado VI, Castaneda J, Prieto-Lloret J, Olea E, Gonzalez-Martin MC, et al. Effects of cigarette smoke and hypoxia on pulmonary circulation in the Guinea pig. Eur Respir J. 2011;38:617–27.
Wang XD, Li F, Ma DB, Deng X, Zhang H, Gao J, et al. Periostin mediates cigarette smoke extract-induced proliferation and migration in pulmonary arterial smooth muscle cells. Biomed Pharmacother. 2016;83:514–20.
Ehrlich M, Lacey M. DNA methylation and differentiation: silencing, upregulation and modulation of gene expression. Epigenomics. 2013;5:553–68.
Bird A. DNA methylation de novo. Science. 1999;286:2287–8.
Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8:e63812.
Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4:132.
Tsaprouni LG, Yang TP, Bell J, Dick KJ, Kanoni S, Nisbet J, et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics. 2014;9:1382–96.
Qiu W, Wan E, Morrow J, Cho MH, Crapo JD, Silverman EK, et al. The impact of genetic variation and cigarette smoke on DNA methylation in current and former smokers from the COPDGene study. Epigenetics. 2015;10:1064–73.
Duruisseaux M, Esteller M. Lung cancer epigenetics: from knowledge to applications. Semin Cancer Biol. 2018;51:116-28.
Reynolds LM, Wan M, Ding J, Taylor JR, Lohman K, Su D, et al. DNA methylation of the aryl hydrocarbon receptor repressor associations with cigarette smoking and subclinical atherosclerosis. Circ Cardiovasc Genet. 2015;8:707–16.
Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121:2661–71.
Liu D, Yan Y, Chen JW, Yuan P, Wang XJ, Jiang R, et al. Hypermethylation of BMPR2 promoter occurs in patients with heritable pulmonary arterial hypertension and inhibits BMPR2 expression. Am J Respir Crit Care Med. 2017;196:925–8.
Zhang Y, Xu J. MiR-140-5p regulates hypoxia-mediated human pulmonary artery smooth muscle cell proliferation, apoptosis and differentiation by targeting Dnmt1 and promoting SOD2 expression. Biochem Biophys Res Commun. 2016;473:342–8.
Zhuang J, Luan P, Li H, Wang K, Zhang P, Xu Y, et al. The yin-Yang dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Arterioscler Thromb Vasc Biol. 2017;37:84–97.
Ning Y, Huang H, Dong Y, Sun Q, Zhang W, Xu W, et al. 5-Aza-2′-deoxycytidine inhibited PDGF-induced rat airway smooth muscle cell phenotypic switching. Arch Toxicol. 2013;87:871–81.
Li G, Marlin MC. Rab family of GTPases. Methods Mol Biol. 2015;1298:1–15.
Jonsson G, Bendahl PO, Sandberg T, Kurbasic A, Staaf J, Sunde L, et al. Mapping of a novel ocular and cutaneous malignant melanoma susceptibility locus to chromosome 9q21.32. J Natl Cancer Inst. 2005;97:1377–82.
Maat W, Beiboer SH, Jager MJ, Luyten GP, Gruis NA, van der Velden PA. Epigenetic regulation identifies RASEF as a tumor-suppressor gene in uveal melanoma. Invest Ophthalmol Vis Sci. 2008;49:1291–8.
Nakamura S, Takemura T, Tan L, Nagata Y, Yokota D, Hirano I, et al. Small GTPase RAB45-mediated p38 activation in apoptosis of chronic myeloid leukemia progenitor cells. Carcinogenesis. 2011;32:1758–72.
Mizuno S, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez-Arroyo J, Voelkel NF, et al. p53 gene deficiency promotes hypoxia-induced pulmonary hypertension and vascular remodeling in mice. Am J Physiol Lung Cell Mol Physiol. 2011;300:L753–61.
Hansmann G, Zamanian RT. PPARgamma activation: a potential treatment for pulmonary hypertension. Sci Transl Med. 2009;1:12ps4.
Wang R, Xu YJ, Liu XS, Zeng DX, Xiang M. Knockdown of connective tissue growth factor by plasmid-based short hairpin RNA prevented pulmonary vascular remodeling in cigarette smoke-exposed rats. Arch Biochem Biophys. 2011;508:93–100.
Wang T, Han SX, Zhang SF, Ning YY, Chen L, Chen YJ, et al. Role of chymase in cigarette smoke-induced pulmonary artery remodeling and pulmonary hypertension in hamsters. Respir Res. 2010;11:36.
Hadri L, Kratlian RG, Benard L, Maron BA, Dorfmuller P, Ladage D, et al. Therapeutic efficacy of AAV1.SERCA2a in monocrotaline-induced pulmonary arterial hypertension. Circulation. 2013;128:512–23.
Li Q, Wang J, Zhu X, Zeng Z, Wu X, Xu Y, et al. Dihydromyricetin prevents monocrotaline-induced pulmonary arterial hypertension in rats. Biomed Pharmacother. 2017;96:825–33.
Everett AD, Le Cras TD, Xue C, Johns RA. eNOS expression is not altered in pulmonary vascular remodeling due to increased pulmonary blood flow. Am J Phys. 1998;274:L1058–65.
Sun D, Li Q, Ding D, Li X, Xie M, Xu Y, et al. Role of Kruppel-like factor 4 in cigarette smoke-induced pulmonary vascular remodeling. Am J Transl Res. 2018;10:581–91.
Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–32.
Ryan J, Bloch K, Archer SL. Rodent models of pulmonary hypertension: harmonisation with the world health organisation's categorisation of human PH. Int J Clin Pract Suppl. 2011;65:15–34.
Churg A, Cosio M, Wright JL. Mechanisms of cigarette smoke-induced COPD: insights from animal models. Am J Physiol Lung Cell Mol Physiol. 2008;294:L612–31.
Zhao L, Wang J, Wang L, Liang YT, Chen YQ, Lu WJ, et al. Remodeling of rat pulmonary artery induced by chronic smoking exposure. J Thorac Dis. 2014;6:818–28.
Jiang X, Yuan L, Li P, Wang J, Wang P, Zhang L, et al. Effect of simvastatin on 5-HT and 5-HTT in a rat model of pulmonary artery hypertension. Cell Physiol Biochem. 2015;37:1712–24.
Tan JY, Jia LQ, Shi WH, He Q, Zhu L, Yu B. Rab5amediated autophagy regulates the phenotype and behavior of vascular smooth muscle cells. Mol Med Rep. 2016;14:4445–53.
Bannister JP, Bulley S, Leo MD, Kidd MW, Jaggar JH. Rab25 influences functional Cav1.2 channel surface expression in arterial smooth muscle cells. Am J Physiol Cell Physiol. 2016;310:C885–93.
Kaplon J, Homig-Holzel C, Gao L, Meissl K, Verdegaal EM, van der Burg SH, et al. Near-genomewide RNAi screening for regulators of BRAF(V600E) -induced senescence identifies RASEF, a gene epigenetically silenced in melanoma. Pigment Cell Melanoma Res. 2014;27:640–52.
Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, et al. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat Med. 2005;11:1188–96.
Gary-Bobo G, Houssaini A, Amsellem V, Rideau D, Pacaud P, Perrin A, et al. Effects of HIV protease inhibitors on progression of monocrotaline- and hypoxia-induced pulmonary hypertension in rats. Circulation. 2010;122:1937–47.
Chen B, Xue J, Meng X, Slutzky JL, Calvert AE, Chicoine LG. Resveratrol prevents hypoxia-induced arginase II expression and proliferation of human pulmonary artery smooth muscle cells via Akt-dependent signaling. Am J Physiol Lung Cell Mol Physiol. 2014;307:L317–25.
Garat CV, Crossno JT Jr, Sullivan TM, Reusch JE, Klemm DJ. Inhibition of phosphatidylinositol 3-kinase/Akt signaling attenuates hypoxia-induced pulmonary artery remodeling and suppresses CREB depletion in arterial smooth muscle cells. J Cardiovasc Pharmacol. 2013;62:539–48.
Stabile E, Zhou YF, Saji M, Castagna M, Shou M, Kinnaird TD, et al. Akt controls vascular smooth muscle cell proliferation in vitro and in vivo by delaying G1/S exit. Circ Res. 2003;93:1059–65.
Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circ Res. 2007;100:607–21.
Cheng JC, Chou CH, Kuo ML, Hsieh CY. Radiation-enhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-kappaB signal transduction pathway. Oncogene. 2006;25:7009–18.
Zhang BG, Du T, Zang MD, Chang Q, Fan ZY, Li JF, et al. Androgen receptor promotes gastric cancer cell migration and invasion via AKT-phosphorylation dependent upregulation of matrix metalloproteinase 9. Oncotarget. 2014;5:10584–95.
Ghosh A, Pechota A, Coleman D, Upchurch GR Jr, Eliason JL. Cigarette smoke-induced MMP2 and MMP9 secretion from aortic vascular smooth cells is mediated via the Jak/stat pathway. Hum Pathol. 2015;46:284–94.
Ghosh A, Lu G, Su G, McEvoy B, Sadiq O, DiMusto PD, et al. Phosphorylation of AKT and abdominal aortic aneurysm formation. Am J Pathol. 2014;184:148–58.
Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt cross-talk. J Biol Chem. 2002;277:31099–106.
Author contributions
JX conceived the study and the hypotheses; LL and QL designed the experiments; QL and JW performed the experiments; LL and QL analyzed the data; QL and JX wrote the paper. All authors read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81570033,81570047, 81370145, 81370156,), National key basic research and development program (973 Program, No. 20l5CB553403), Chinese medical association research project (No. 2013BAI09B00), National Key Technologies R&D Program (2016YFC1303900 and 2016YFC1304700), National Key Research and Development Program in China (2016YFC0903600) .
Availability of data and materials
Any data generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Methylation differences in CpG in PAs of the rats with different treatment (xls). The number 164, 165, 187 and 188 were from the air group, and the last (171,174, 189 and 197) were the smoking group rats. (XLS 1785 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, Q., Wu, J., Xu, Y. et al. Role of RASEF hypermethylation in cigarette smoke-induced pulmonary arterial smooth muscle remodeling. Respir Res 20, 52 (2019). https://doi.org/10.1186/s12931-019-1014-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-019-1014-1