
Abstract
Mitochondrial mass and quality are tightly regulated by two essential and opposing mechanisms, mitochondrial biogenesis (mitobiogenesis) and mitophagy, in response to cellular energy needs and other cellular and environmental cues. Great strides have been made to uncover key regulators of these complex processes. Emerging evidence has shown that there exists a tight coordination between mitophagy and mitobiogenesis, and their defects may cause many human diseases. In this review, we will first summarize the recent advances made in the discovery of molecular regulations of mitobiogenesis and mitophagy and then focus on the mechanism and signaling pathways involved in the simultaneous regulation of mitobiogenesis and mitophagy in the response of tissue or cultured cells to energy needs, stress, or pathophysiological conditions. Further studies of the crosstalk of these two opposing processes at the molecular level will provide a better understanding of how the cell maintains optimal cellular fitness and function under physiological and pathophysiological conditions, which holds promise for fighting aging and aging-related diseases.
Similar content being viewed by others

Introduction
Mitochondria serve as power plants that generate adenosine 5’-triphosphate (ATP) through oxidative phosphorylation (OXPHOS) for the cell. They also contribute to the regulation of calcium homeostasis, intracellular signaling transduction, cellular proteostasis, heme and lipid biosynthesis, reactive oxygen species (ROS) production, and programmed cell death [1,2,3,4,5,6]. To fulfill such diverse and critical roles in the cell, mitochondria undergo constant fission and fusion cycles to maintain their shape, network, and inheritance. This constant turnover helps maintain their fitness and normal cellular functions. Dysregulation of these critical processes has been causally linked to a myriad of diseases, including metabolic disorders, neurodegenerative diseases, heart and vascular diseases, inflammatory diseases, hematological diseases, and cancers [7,8,9,10,11,12].
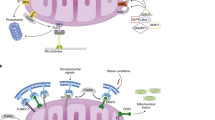
Mitochondria are not generated de novo in eukaryotic cells. The preexisting mitochondria are distributed between the two daughter cells following cell division [13]. Mitochondria have circular DNA (mitochondrial DNA, mtDNA), and mitochondrial biogenesis (mitobiogenesis) involves the replication, transcription, and translation of mtDNA-encoded genes, the interorganelle transport of phospholipids, and the import of nuclear-encoded proteins into mitochondria through the protein translocation machinery of the outer and inner membranes [14]. Mitobiogenesis is a balanced process that also occurs in parallel to the process of removing mitochondria, ensuring that an optimal number of mitochondria persist within the cell. During evolution, cells have gained several strategies to monitor and remove damaged or superfluous mitochondria. One of the major mechanisms of removal is mitophagy, a selective form of autophagy that promotes mitochondrial degradation via the mitolysosomal pathway. In the past decade, both ubiquitin- and receptor-mediated mitophagy pathways have been described [15]. It is less clear how the two opposing processes of mitobiogenesis and mitophagy are coordinated at the molecular level and how they maintain a healthy population of mitochondria (Fig. 1). In this review, we discuss recent advances toward elucidating the molecular mechanisms underlying the coordination of mitobiogenesis and mitophagy. We review the molecular regulation of both mitophagy and mitobiogenesis processes and give special attention to the crosstalk between them that fine tunes the balance of mitochondrial mass and quality.

Coordination between mitophagy and mitobiogenesis. Mitophagy and mitobiogenesis are two opposing processes that work together to maintain mitochondrial quality and quantity in response to various physiological and environmental signals
Mitobiogenesis
Mitobiogenesis is the process of increasing the number and size of mitochondria in response to higher energy demands and other cellular cues [16]. Given that the majority of mitochondrial proteins are nuclear-encoded, the transcription and translation of nuclear and mitochondrial genes must be tightly coordinated to ensure the creation of new mitochondria [17]. For instance, the assembly of the respiratory chain necessitates the synchronization of the gene expression of the nuclear and mitochondrial genomes [18]. Most mitochondrial precursor proteins generated in the cytoplasm are imported and sorted into mitochondrial subcompartments by the TOM/TIM complex, and the assembly of mitochondrial-encoded and nuclear-encoded subunits faces potential difficulty [18, 19]. Unassembled respiratory chain subunit buildup is linked to elevated ROS generation and mitochondrial proteotoxic stress, both of which have negative effects on the cell [20, 21]. As a result, diverse signaling cascades and transcriptional complexes are needed to ensure appropriate mitobiogenesis, which is regulated by several types of coactivators and transcription factors (Fig. 2) [22]. In addition to transcription factors, mitobiogenesis is also regulated by mitochondrial retrograde signals, such as ATP, ROS and Ca2+ [23]. The roles of ATP and ROS in mitobiogenesis are discussed below. Many studies have shown that disruption of mitochondrial membrane potential (Δψm) affects mitochondrial uptake of Ca2+, and increased cytosolic Ca2+in turn activates various Ca2+-dependent kinases and initiates signaling cascades that regulate mitobiogenesis [24].
Transcriptional regulators of mitobiogenesis
The nuclear transcription factors that were first discovered to play a role in the regulation of mitobiogenesis were nuclear respiratory factors 1 (NRF1) and 2 (NRF2) (Fig. 2) [25, 26]. NRF1 acts as a positive transcriptional regulator because it has a C-terminal transcriptional activation domain (TAD) that binds the promoters of several mitochondrial-related genes [26]. It has now been well documented that NRF1 serves as the master regulator of mitobiogenesis [27]. NRF1 controls the expression of genes encoding respiratory complexes, heme biosynthesis pathway enzymes, components of the mtDNA transcription and replication machinery, and the mitochondrial transport and assembly apparatus [28]. The first mitochondrial gene that was revealed to be upregulated by NRF1 was cytochrome C, and a specific NRF1 binding site was discovered in the region upstream of the cytochrome C promoter that was necessary for maximal promoter activity [29]. Shortly after, NRF1 was discovered to activate multiple genes involved in respiratory chain complexes [26, 30]. NRFs may also play an integrative role in nuclear-mitochondrial interactions since they increase the expression of key mitochondrial transport machinery components such as TOMM70, TOMM34, and TOMM20 [31]. Additionally, NRF1 activation triggers the transcription of the mitochondrial transcription factor A (TFAM), mitochondrial transcription factor B1 (TFB1M) and B2 (TFB2M) genes, whose products are important regulators involved in the direct control of mtDNA transcription, stimulating mtDNA transcription and its maintenance [22, 32].
NRF2 was initially recognized for its function in the transactivation of the adenovirus E4 gene, also known as GA-binding protein transcription factor (GABP) [33]. It is composed of two separate proteins, NRF2α and NRF2β, which come together to form a tetrameric α2β2 complex. The DNA-binding domain (DBD) or transcriptional activation domain (TAD) is present in NRF2α and NRF2β separately, but both components are required for the generation of a functional complex [34]. NRF2 plays a significant role in the regulation of the transcription of genes involved in the respiratory chain and mitochondrial transcription. Many cytochrome c oxidase (COX) component genes as well as numerous other genes associated with the construction and operation of the respiratory chain have now been unveiled to have functional NRF2 binding sites [28]. Although it has a weaker influence on the activity of the TFAM promoter than NRF1, NRF2 can also bind to the TFAM promoter [32]. In most cases, promoters have both NRF1 and NRF2 binding sites, indicating a mechanism for their coordinated regulation during mitobiogenesis [28].
The orphan nuclear receptor known as estrogen-related receptor α (ERRα) was first identified to control fatty acid oxidation and estrogen signaling. Further studies have demonstrated the crucial function of ERRα in the induction of peroxisome proliferator-activated receptor coactivator 1α (PGC-1α) and 1β (PGC-1β) in mitobiogenesis (Fig. 2) [35, 36]. Many mitochondrial genes include ERRα binding sites, and when ERRα was knocked down, mitobiogenesis driven by PGC-1α or PGC-1β was also reduced [35, 36]. It is noteworthy to state that NRF1 and NRF2 are thought to activate the gene expression of the mitochondrial respiratory chain downstream from the PGC-1α/ERRα axis [37, 38]. Along with controlling mitobiogenesis and function, ERRα can bind to p53, independent of the p53 mutational status. This was found to be essential in preventing ROS generation in cancer cells [39]. In addition, brown adipose tissue (BAT) levels of mitobiogenesis and the expression of genes related to mitochondrial OXPHOS were dependent on ERRα to meet the energy requirements necessary for thermogenesis [40]. Hence, when ERRα is lacking, mice are unable to regulate their body temperatures in response to exposure to cold [40].
By regulating the expression of numerous endogenous antioxidants, nuclear factor erythroid 2-like 2 (NFE2L2) has become a key regulator of antioxidant response element (ARE)-dependent transcription and serves as a master regulator of intracellular redox homeostasis (Fig. 2) [41]. NFE2L2 is typically destined to be ubiquitinated by Kelch-like ECH-associated protein 1 (Keap1), a component of the Cullin 3 (CUL3)-based E3 ubiquitin ligase complex, and is degraded by the ubiquitin‒proteasome pathway within the cytoplasm [42]. When the binding of Keap1 to NFE2L2 is reduced by electrophilic substances or ROS, NFE2L2 is stabilized and translocates to the nucleus, and NFE2L2-dependent cytoprotective genes are activated [43, 44]. It is interesting to note that the NRF1 promoter contains 4 AREs, and overexpression of heme oxygenase-1 (HO-1) activates the NRF1-dependent mitobiogenesis pathway by enhancing the nuclear translocation of NFE2L2 [45]. Moreover, the NFE2L2 activator dimethyl fumarate stimulates mitobiogenesis both in vitro and in vivo [46]. Using NFE2L2 knockout mice, it has been demonstrated that NFE2L2 is essential for mitobiogenesis. In NFE2L2-deficient animals, the esophageal epithelium showed downregulation of 11 mitobiogenesis-related genes, which led to a reduction in mitochondrial mass [47]. In addition, following exercise training, mitochondrial mass in skeletal muscle was also decreased in NFE2L2-defective animals [48].
Nuclear coactivators
The master regulator of mitobiogenesis, PGC-1α, is a transcriptional coregulator that integrates the activity of many transcription factors, including NRF1, NRF2, and ERRα (Fig. 2) [28]. PGC-1α was originally cloned following yeast two-hybrid screening that utilized peroxisome proliferator-activated receptor γ (PPARγ) as bait. Furthermore, PGC-1α mRNA is dramatically upregulated in mouse BAT and skeletal muscle in response to cold exposure as well as exercise in a cAMP-PKA-CREB-dependent manner, leading to mitobiogenesis and increased respiration [49, 50]. The expression of UCP1, a crucial gene involved in thermogenesis, is also enhanced by ectopic expression of PGC-1α, which is critical for BAT thermogenesis [49, 51]. Simultaneously, the expression of PGC-1α is also significantly induced upon oxidative stress, in turn enhancing the expression of some antioxidant proteins, which can prevent excessive mitochondrial ROS production following mitobiogenesis [52, 53]. Taken together, these findings support the involvement of PGC-1α in mitobiogenesis [49]. Further research has shown that PGC-1α increases the transcriptional activity of NRF1, stimulates the production of NRF1 and NRF2, and increases the expression of TFAM and numerous mitochondrial respiratory chain genes, leading to mitobiogenesis [54]. Additionally, PGC-1α is ubiquitinated and degraded in cancer cells under conditions of protracted glucose shortage that depend on RNF2, and the inhibition of the E3 ligase RNF34 is what causes PGC-1α to stabilize in response to cold exposure in BAT [55, 56]. Many studies have established the dominant involvement of PGC-1α in mitobiogenesis, and the influence of PGC-1α-related mitobiogenesis has also been connected to a number of illnesses, including metabolic disease, cardiomyopathy, neurodegenerative disease, cancer, and kidney disease [57,58,59,60,61,62,63].
PGC-1β is a homolog of PGC-1α, sharing a similar tissue distribution and amino acid protein sequence (Fig. 2) [64]. The expression of PGC-1β mRNA, however, is increased during brown fat cell development rather than being triggered by exposure to cold, suggesting that these two proteins have different roles throughout various physiological processes [64]. The fact that PGC-1β promotes transcription by interacting with several transcription factors, including NRF1, ERRα, and ERRβ, suggests that it is also crucial for mitobiogenesis. However, PGC-1α or PGC-1β deficiency had no effect on mitobiogenesis during brown fat differentiation, indicating a complimentary role for these two coactivators in differentiation-induced mitobiogenesis [65]. Indeed, silencing of PGC-1β in brown preadipocytes with PGC-1α deficiency entirely eliminated the increase in mitochondrial mass and respiration during brown fat formation [65]. Other evidence suggests that PGC-1 coactivators serve an important but complementary role in mitobiogenesis, and this comes from PGC-1α and PGC-1β double deletion animals that died shortly after birth with tiny hearts and a lack of mitobiogenesis in both the heart and BAT [66].
PPARγ-related coactivator 1 (PRC; PPRC1) was first discovered as a transcriptional coactivator. It shares structural similarities and a similar mechanism of action with PGC-1, is widely expressed in mouse tissues, and does not increase in levels in the BAT of mice exposed to cold temperatures (Fig. 2) [67]. PRC interacts with NRF1 and CREB directly and promote NRF1/CREB-dependent gene transcription [67, 68]. Moreover, PRC controls the transcription of NRF2-dependent genes by interacting with host cell factor-1 (HCF-1), and when PRC expression is reduced, TFB2M, COX-II, and cytochrome b are downregulated [69]. PRC expression was rapidly increased by serum stimulation of quiescent cells, which was in contrast to PGC-1α. This suggested that PRC serves complementary roles in controlling mitobiogenesis [67]. The critical role of PRC has also been demonstrated using PRC loss-of-function or knockdown in cultured cells. The expression of respiratory chain proteins and mitobiogenesis were impaired in both cases [67, 70]. Moreover, gene arrays have shown that silencing PRC reduced the expression of almost 50 genes associated with mitochondria, such as respiratory chain subunits, mitochondrial protein import, and assembly factors [70]. In addition, double knockouts of PGC-1α and PGC-1β in skeletal muscle or the adult heart did not result in a loss in mitochondrial mass, which indicated that PRC may play a role in mitobiogenesis in the absence of PGC-1α and PGC-1β [71, 72], a finding that warrants further investigation.
Signaling pathways that activate mitobiogenesis
The spatiotemporal modulation of mitobiogenesis in response to a variety of stimuli, such as energy need, hormone cues, and environmental stimuli, involves a number of signaling pathways. The coordination of transcription factor networks for mitobiogenesis depends on the integration of different signaling pathways (Fig. 2). Through phosphorylation of PGC-1α and increased PGC-1α stability, AMP-activated protein kinase (AMPK), the main molecular sensor activated by cellular stressors that causes ATP depletion, was found to be a key regulator of mitobiogenesis [73]. While AMPK is active, NAD+ levels also rise, which promotes sirtuin 1 (SIRT1) and PGC-1α activity [74]. The AMPK/SIRT1/PGC1α signaling axis is essential for the deacetylation of PGC-1α by SIRT1, which is necessary for adaptive metabolic programming to occur during energy restriction and exercise [74].
As a secondary messenger molecule, calcium and its signaling pathways also control mitobiogenesis in a calcium/calmodulin-dependent protein kinase (CAMK)-dependent manner in skeletal muscle [75]. PGC-1α and mitobiogenesis are specifically promoted in mouse skeletal muscle by constitutively active CAMK expression, and an increase in mitochondria in L6 myotubes is driven by increasing cytosolic Ca2+, which could be prevented by using a CAMK inhibitor [75, 76]. PGC-1α expression and activity are controlled by p38 MAPK, and a CAMK inhibitor prevented p38 activation in response to increased cytosolic Ca2+, demonstrating that p38 MAPK is a downstream target of CAMK [77,78,79,80]. Consequently, exercise-induced mitobiogenesis in skeletal muscle was shown to be mediated through the calcium/CAMK/p38 MAPK/PGC-1 pathway, which is crucial for skeletal muscle adaptation to exercise training.
Another clearly established mechanism, the cAMP-PKA pathway, is crucial for mitobiogenesis in BAT in response to cold exposure. cAMP-PKA phosphorylates CREB at serine (Ser)-133, which activates transcription factors, and directly activates PGC-1 expression via CREB, which is a necessary pathway for mediating glucose homeostasis during fasting [81, 82]. The cAMP-stimulating drugs forskolin and CAMK were unable to induce PGC-1α expression when the cAMP response element (CRE) site was mutated, demonstrating a critical function for CREB in this process. Activating transcription factor 2 (ATF-2), a different CRE-binding protein, is activated by p38 MAPK and is thought to be the primary regulator of PGC-1 gene transcription in brown adipocytes in response to cAMP rather than CREB [77]. Moreover, the expression of PGC-1 is regulated by members of the myocyte enhancer factor 2 (MEF2) family of transcription factors, and PGC-1, CAMK, and p38MAPK all work together to activate MEF2 functions [77, 83].

Molecular mechanisms and regulation of mitobiogenesis. The coactivators PGC-1α, PGC-1β, and PRC bind to the transcription factors NFR1/2, ERRα, and NEF2L2 to speed up the transcription of genes involved in mitobiogenesis. MEF2, ATF-2, CREB, and other transcription factors all work together to promote PGC-1α expression, and PGC-1α also improves the transcriptional activity of MEF2 and ATF-2. To trigger mitobiogenesis in response to various physiological and environmental cues, such as cold exposure, exercise, nutritional restriction, and cytokines, these factors are integrated and controlled by several signaling pathways, including the AMPK/SIRT1/PGC1α system, the calcium/CAMK/p38 MAPK/PGC-1α pathway, and the cAMP-PKA pathway
Mitophagy
Macroautophagy, also referred to as autophagy, is a cellular catabolic mechanism that destroys protein aggregates, damaged organelles, and invasive microorganisms through double-membrane vesicles called autophagosomes, which are then degraded in lysosomes. Autophagy is tightly regulated by autophagy-related (Atg) proteins [84,85,86,87]. Autophagy is a crucial system for metabolite recycling, minimizing the toxicity caused by damaged organelles or aberrant proteins, and removing pathogenic microbes, which is critical for maintaining cellular fitness [88, 89]. Dysregulation of autophagy has been linked to a variety of illnesses, such as cancer, heart disease, neurological disorders, and metabolic diseases [90,91,92]. Although autophagy was formerly thought to be a nonselective process, compelling evidence has shown that organelle-specific autophagy is primarily a selective process that is responsible for maintaining the quality and homeostasis of almost all organelles [93, 94]. The term “mitophagy,” which was first used by Lemasters [95], refers to a conserved cellular process that enables the selective destruction of unhealthy or unnecessary mitochondria by autophagy. In most cases, mitophagy is triggered upon oxidative stress and serves as a cellular defense mechanism to remove damaged mitochondria that produce high levels of ROS [96]. As a key cellular quality control mechanism, mitophagy is essential for maintaining the optimal amounts and normality of the functions of mitochondria [97,98,99]. Mitophagy is also able to promote cell survival by eliminating damaged mitochondria because the release of cytochrome c into the cytosol during mitochondrial damage is associated with the induction of programmed cell death through apoptosis [100, 101]. Furthermore, upregulation of mitophagy by rapamycin, an mTORC1 inhibitor, resulted in a decrease in mtDNA mutant load and rescued cellular bioenergetic function in MELAS or Leber’s hereditary optic neuropathy cell lines, indicating the essential role of mitophagy in reducing mtDNA mutations [102, 103]. As discussed below, mitophagy is primarily regulated by ubiquitin- or mitophagy receptor-dependent mechanisms (Fig. 3) [97, 104].
Receptor-mediated mitophagy
Mitophagy receptors are proteins that contain the LC3 interaction region (LIR) domain and are located in the mitochondrial outer or inner membrane [15]. In response to various mitochondrial stressors, such as hypoxia, loss of mitochondrial membrane potential, and oxidative stress, mitophagy receptor proteins are engaged in the selective destruction of damaged mitochondria through their interactions with the essential autophagy protein LC3 [15, 105]. In recent years, a number of mitophagy receptors have been discovered, and the function of these proteins in physiological and pathophysiology processes has also been demonstrated [99].
FUNDC1
FUNDC1 is a conserved outer mitochondrial membrane (OMM) protein with 3 transmembrane domains, containing an LIR at the N-terminus that is exposed to the cytosol [106] (Fig. 3). FUNDC1 is normally phosphorylated by Src kinase at tyrosine (Tyr)-18 and CK2 at Ser13. Upon mitochondrial stresses, FUNDC1 becomes dephosphorylated by phosphatase PGAM5 or other phosphatases, leading to the enhanced interaction between FUNDC1 and LC3 and subsequent selective mitophagy [106, 107]. Additionally, hypoxia causes Unc-51-like autophagy activating kinase 1 (ULK1) to translocate to the mitochondria and engage with FUNDC1, phosphorylating Ser17 and enhancing the interaction between FUNDC1 and LC3 [108]. Through interaction with and suppression of PGAM5 and phosphorylation of Ser13 in FUNDC1, the essential anti-apoptotic protein BCL-xL negatively regulates FUNDC1-mediated mitophagy [109]. Multimeric PGAM5 dephosphorylates FUNDC1 to trigger mitochondrial fission and the clearance of damaged mitochondria in response to mild cellular stress, and it can also dephosphorylate BCL-xL at Ser62, which was shown to be necessary for anti-apoptotic actions following severe cellular stress [110]. Moreover, when mitophagy is triggered by membrane potential dissipation, PGAM5 is also cleaved by the rhomboid protease PARL, which facilitates the interaction between PGAM5 and FUNDC1 in a Syntaxin-17 (SYN17)-dependent manner [111]. In addition, FUNDC1 directly interacts with dynamic mitochondrial proteins such as the mitochondrial fission protein DRP1 and the inner membrane fusion protein OPA1 [112]. Hypoxia reduces the capacity of FUNDC1 to interact with OPA1 and instead increases its association with DRP1, which causes mitochondrial fragmentation and selective mitochondrial elimination [112, 113]. The ubiquitin-protein ligase MARCH5 and deubiquitinase USP19 also regulate the protein stability of FUNDC1 and its function, respectively [114]. In the early stages of hypoxia, MARCH5 ubiquitinates FUNDC1 at lysine (Lys)-119, which causes FUNDC1 to degrade and prevents the improper elimination of healthy mitochondria [114]. Meanwhile, during hypoxia, a small percentage of FUNDC1 protein is deubiquitinated by USP19 at Lys119, allowing FUNDC1 translocation to MAMs and DRP1-mediated mitochondrial division [115]. In addition, FUNDC1 is acetylated and deacetylated at K104 by Tip60 and HDAC3, which regulates the dimerization of FUNDC1 and thus its ubiquitination by MARCH5 [116].
Given that mitophagy plays a crucial role in maintaining cellular homeostasis, it is not surprising that dysregulation of FUNDC1-mediated mitophagy has emerged as a crucial factor in many physiological processes and pathophysiological conditions, such as cardiac progenitor cell differentiation [117], angiogenesis and neoangiogenesis [118], liver cancer [119], breast cancer [120], metabolic disorders [121,122,123], cardiovascular diseases [124,125,126,127,128,129,130], renal anemia [131], liver injury [132, 133], cerebral ischemia‒reperfusion injury [134], intestinal ischemia/reperfusion injury [135], kidney ischemia/reperfusion injury [136], and chronic obstructive pulmonary disease [137, 138]. Additionally, FUNDC1-mediated mitophagy may inhibit mtDNA release and suppress inflammasome activation and the inflammatory response, which are advantageous mechanisms for cellular homeostasis [119, 123, 131].
BNIP3 and NIX
BCL2 and adenovirus E1B 19 kDa interacting protein 3 (BNIP3) was first discovered by using yeast two-hybrid assays that utilized adenovirus E1B 19 kDa protein as bait. BNIP3 was categorized as a dimeric mitochondrion-localized pro-apoptotic protein with a potential BH3 (Bcl-2 homology 3) domain [139, 140]. The homolog of BNIP3 is NIX, which shares considerable sequence homology and pro-apoptotic activity with BNIP3 (Fig. 3) [141]. The promoters of BNIP3 and NIX both include functional HIF-1-responsive elements (HREs) and are elevated by hypoxia, indicating their possible roles in hypoxia-induced apoptosis [142,143,144]. Mechanistically, expression of either BNIP3 or NIX may result in the opening of the mitochondrial permeability transition pore (mPTP), loss of ΔΨm, and an increase in ROS generation, which leads to both apoptotic and necrotic cell death [145,146,147,148].
Subsequent research has demonstrated that autophagy rather than cell death is induced in hypoxic microenvironments in a BNIP3- and NIX-dependent manner [149]. Furthermore, the impaired sequestration of mitochondria into autophagosomes in Nix mutant erythroid cells prevents mitochondrial clearance, revealing a crucial function for NIX-mediated mitophagy in the maturation of erythrocytes [150, 151]. Moreover, NIX was shown to be a mitophagy receptor protein with an LIR that specifically interacts with LC3/GABARAP proteins. This association was shown to be required for the clearance of mitochondria during erythrocyte development [152]. Similarly, the BNIP3 homodimer also interacts with LC3 directly through its amino-terminal LIR, acting as an autophagy receptor for the careful degradation of mitochondria and endoplasmic reticulum [153].
The function and stability of BNIP3 and NIX are also regulated by phosphorylation. At the BNIP3 LIR motif, Ser17 and Ser24 phosphorylation promotes its interaction with LC3B and GATE16 [154]. ULK1 was shown to be the kinase that phosphorylates BNIP3 at Ser17, and this association increases the stability of the BNIP3 protein [155]. Moreover, JNK1/2-mediated phosphorylation of BNIP3 at Ser60 and threonine (Thr)-66 regulates protein stability and prevents proteasomal degradation of BNIP3 during hypoxia [156]. Furthermore, phosphorylation of NIX at Ser34 and Ser35 improves its ability to bind with LC3B and recruit autophagosomes to mitochondria [157]. Additionally, cells treated with cadmium to promote mitophagy exhibited increased phosphorylation of Ser81, which was indicated to be essential for NIX-mediated mitophagy [158, 159]. In contrast, PRKA phosphorylates NIX at Ser212, causing NIX translocation to the cytosol and the inhibition of NIX-induced mitophagy and insulin signaling [160].
Several additional novel mitophagy receptors have been discovered recently (Fig. 3). The mammalian homologue of the yeast mitophagy receptor ATG32, BCL-2-like protein 13 (BCL2L13), directly binds to LC3 through the LIR domain to trigger mitophagy [161]. In mammalian cells, FK506 binding protein 8 (FKBP8) serves as an additional outer mitochondrial membrane (OMM) mitophagy receptor that, in an LIR-dependent manner, attracts LC3A to injured mitochondria [162]. Moreover, upon OMM disruption, the inner mitochondrial membrane (IMM) protein PHB2 binds LC3 via LIR, which is essential for Parkin-dependent mitophagy [163].
Ubiquitin-mediated mitophagy
Parkin is a cytosolic E3 ubiquitin ligase, and mutations in Parkin genes are responsible for the pathogenesis of autosomal recessive parkinsonism (ARP) [164, 165]. A serine/threonine kinase known as PTEN-induced kinase 1 (PINK1), which is located in mitochondria, was also discovered to be connected to ARP in later studies (Fig. 3) [166]. Early research has indicated that the Parkin/PINK1 pathway contributes to mitochondrial dynamic regulation [167,168,169], as well as maintaining mitochondrial function [170, 171]. The function of Parkin in mitophagy was not fully understood until a key study, carried out in 2008 [172], showed that Parkin facilitates the autophagic clearance of damaged mitochondria by being specifically attracted to mitochondria with diminished ΔΨm after treatment with a mitochondrial uncoupler [172].
A number of subsequent Investigations provided a clearer picture of Parkin/PINK1-driven mitophagy at the molecular level (Fig. 3). Under steady-state conditions, PINK1 is transported to the mitochondrial intermembrane space for proteolytic cleavage and the cleavaged PINK1 is transported to cytosol and degraded by proteaosome [173, 174]. Nevertheless, PINK1 import is prevented by depolarization of the ΔΨm, which stabilizes PINK1 on the OMM and causes Parkin to be recruited to damaged mitochondria [175]. Upon translocation, the E3 activity of Parkin is also increased by the direct phosphorylation of the ubiquitin-like domain at Ser65 by PINK1 [176, 177]. Furthermore, Parkin E3 activity must be fully activated for PINK1 to phosphorylate ubiquitin at Ser65 and release its autoinhibition of catalytic cysteine [178,179,180]. Autophosphorylation of Ser228 and Ser402 controls the activity of PINK1 and facilitates Parkin recruitment to the mitochondria [181]. After localization on mitochondria, Parkin ubiquitylates and degrades several mitochondrial dynamic-related proteins, such as Miro and mitofusins, causing a pause in mitochondrial trafficking and segregation of injured mitochondria from the healthy mitochondrial network [182,183,184,185]. Meanwhile, the autophagy receptor proteins optineurin (OPTN), NDP52 and NBR1 are also recruited to ubiquitinated mitochondria via their ubiquitin binding domain [186, 187]. Later, LC3 is recruited to damaged mitochondria by the omegasome protein double FYVE-containing protein 1 (DFCP1), enabling OPTN-mediated initiation of the autophagosome via its LIR [186]. Further research has shown that the phosphoro-ubiquitin produced by PINK1 attracts NDP52 and OTPN to injured mitochondria in a Parkin-independent manner, acting as an autophagy signal that may be increased by Parkin on mitochondria [188]. Interestingly, TBK1, a key kinase in the innate immune antiviral response, controls the translocation of OPTN to mitochondria, and OPTN but not NDP52 is essential for autophagosome formation after mitochondrial depolarization [189, 190]. The formation of autophagosome membranes around ubiquitinated mitochondria is dependent on the binding of NDP52 and OPTN to the core autophagy proteins FIP200 and ATG9A, respectively [191, 192].
Paternal mitochondria are thought to be eliminated through Parkin-mediated mitophagy in tandem with mitochondrial E3 ubiquitin protein ligase-1 (MUL1), which was demonstrated during the development of the mouse embryo [193]. Parkin and MUL1 appear to have a redundant function in the autophagic clearance of mitochondria upon membrane depolarization, as evidenced by the greatly reduced paternal mitochondrial degradation that is observed upon the depletion of both proteins rather than either one alone [193]. Similar to FUNDC1, Parkin/PINK1-mediated mitophagy reduces inflammation driven by the release of mtDNA, indicating that both receptor- and ubiquitin-mediated mitophagy play important roles in maintaining mitochondrial quality in vivo [194]. However, one study that utilized mito-QC reporter mice revealed that basal levels of mitophagy occurred independently of PINK1 [195].
Signaling pathways involved in the activation of mitophagy
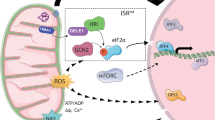
The signaling mechanisms that initiate mitophagy, in contrast to those associated with mitobiogenesis, have not yet been thoroughly studied. Several pathways have been shown to be involved in the regulation of mitophagy, but the precise underlying molecular mechanisms remain elusive (Fig. 3). According to one study, blockade of p38 or ERK2 effectively inhibited mitophagy but not autophagy triggered by starvation or hypoxia, which indicated that these two kinases are necessary for mitophagy [196]. Additionally, inhibiting the activity of kinases upstream of p38 and ERK2 could also prevent mitophagy, further demonstrating the importance of these signaling pathways in mitophagy [196]. However, Parkin phosphorylation at Ser131 by p38 MAPK reduced its activity and the mitophagy that resulted from the overexpression of A53T-synuclein [197]. Similarly, ERK1/2 activation of ULK1 caused its ubiquitination and degradation, thereby impairing mitophagy [198]. Hence, further studies are required to reach a consensus on precisely how p38 and ERK activated by various stimuli function in the regulation of mitophagy.
Hypoxia-inducible factor (HIF)-1α controls the expression of the mitophagy receptors BNIP3 and NIX, which are necessary in hypoxia-induced mitophagy. When HIF-1α is defective, the reduction in mtDNA and mass caused by extended exposure of MEFs to hypoxia is entirely blocked but could be recovered when BNIP3 was reintroduced [199]. When HIF-1α was knocked out in renal tubules, ischemia/reperfusion (I/R)-induced mitophagy in the kidney was also suppressed, and this mitophagy defect could be corrected by BNIP3 overexpression [200]. HIF-1α also has essential roles in mitophagy induced by the iron chelator DFP, a chemical hypoxia-mimicking agent, wherein mitophagy was abolished by the combined depletion of BNIP3 and NIX [201]. Thus, in both in vivo and in vitro studies, the HIF-1α-BNIP3/NIX signaling pathway was shown to be crucial for the induction of mitophagy by hypoxia.
Direct interaction and phosphorylation of ULK1, an essential regulator of mitobiogenesis, by AMPK was also shown to control mitophagy [202]. AMPK phosphorylates ULK1 at Ser317 and Ser777 under nutrient deprivation conditions, which promotes ULK1 activity and autophagy [203]. Mitophagy deficiency and accumulation of mitochondria occur in the absence of AMPK or ULK1, demonstrating that AMPK-dependent phosphorylation of ULK1 is necessary for mitophagy and mitochondrial homeostasis [204]. Moreover, exercise-induced mitophagy in skeletal muscle is dependent on ULK1 phosphorylation at Ser555, highlighting the significance of AMPK-ULK1 signaling in the induction of mitophagy in vivo [205]. In addition, in response to mitochondrial stress, Parkin is phosphorylated at Ser108 by ULK1 in an AMPK-dependent manner, which is necessary for PINK1-mediated phosphorylation of Parkin at Ser65 and activation of Parkin-mediated mitochondrial clearance [206]. Moreover, ULK1 directly phosphorylates mitophagy receptors such as FUNDC1, BNIP3, NIX, and BCL2L13 to facilitate mitophagy receptor-mediated mitophagy [207].

Mechanisms of mitophagy. Mitophagy receptors such as FUNDC1, BNIP3, NIX, BCL2L13, and FKBP8 localize to the OMM and interact with LC3 directly through LIR to induce mitochondrial clearance. The phosphorylation status of these mitophagy receptors and their association with LC3 are influenced by multiple kinases (CK2, SRC, UKL1, JNK1/2, PRKA), phosphatases such as PGAM5, and a variety of stimuli. Together, these influencing factors control the initiation and progression of mitophagy. Dephosphorylation of FUNDC1 promotes its separation from OPA1 and interaction with DRP1, which in turn facilitates mitochondrial fission and the targeted elimination of damaged mitochondria. The E3 ligase MARCH5 and deubiquitinase USP19 also control the protein level of FUNDC1 and mitochondrial fission activity. HIF-1α controls the levels of the proteins BNIP3 and NIX, which are essential for hypoxia-induced mitophagy. Parkin recruitment to damaged mitochondria depends on the stability of PINK1 on the OMM, which occurs as a result of ΔΨm dissipation. Parkin ubiquitinates several OMM proteins, while PINK1 phosphorylates and activates Parkin. Autophagy initiation proteins (ATG9, FIP200) are attracted to damaged mitochondria by several autophagy adaptor proteins, including NDP52 and OPTN, which are brought to the OMM by phosphorylated poly-Ub chains. OPTN is phosphorylated by TBK1, enhancing its interaction with poly-Ub chains and serving as a feed-forward mechanism promoting mitophagy
The interplay between mitobiogenesis and mitophagy
Maintaining optimal mitochondrial content and function requires tightly controlled orchestration of the mitophagy and mitobiogenesis processes. Strong evidence supports the claim that a variety of stimuli can simultaneously elicit mitophagy and mitobiogenesis. In lipopolysaccharide (LPS)-exposed primary mouse lung endothelial cells, MAPK kinase 3 (MKK3) deficiency activated PGC-α-regulated mitobiogenesis and Parkin/PINK1-regulated mitophagy concurrently, resulting in a healthy mitochondrial network and protection against sepsis [208]. Similarly, loss of the putative nutrient-sensing regulator GCN5-like 1 (GCN5L1) resulted in the simultaneous activation of transcription factor EB (TFEB)-mediated mitophagy and PGC-1α-mediated mitobiogenesis, thereby increasing mitochondrial turnover [209]. Moreover, melatonin therapy increased the expression of Parkin/PINK1 and PGC-1α/NRF1 and was beneficial in the prevention of liver fibrosis by enhancing mitobiogenesis and mitophagy [210]. Additionally, thyroid hormone increased the quality of mitochondria by inducing PGC-1α and ULK1 in HepG2 cells and mouse liver tissues, which suggested the potential use of ERRα agonists in metabolic illnesses through enhancing mitochondrial function [211]. Moreover, thyroid hormone increases fatty acid oxidation and mitochondrial respiration in mouse BAT by promoting both mitobiogenesis and MTOR-mediated mitophagy [212]. In mouse renal tissue, the secreted glycoprotein progranulin (PGRN) induces mitobiogenesis and mitophagy through the PGRN-Sirt1-PGC-1α/FoxO1 signaling axis, reducing the effects of excessive hyperglycemia on mitochondrial dysfunction [213]. Myogenic differentiation also requires the coordinated regulation of mitophagy and mitobiogenesis, and the upregulation of p62-mediated mitophagy in the early stages of myogenic differentiation is necessary for subsequent PGC-1α-mediated mitobiogenesis and myogenic differentiation [214]. In C. elegans, the processes linking mitobiogenesis and mitophagy have been deciphered (Fig. 4) [215]. The nematode homolog of NFE2L2, SKN-1, is a key regulator of mitobiogenesis and has a role in the control of the expression of the mitophagy receptor DCT-1/NIX in worms [215]. The accumulation of damaged mitochondria caused by DCT-1 or PINK1 knockdown results in the upregulated expression of SKN-1, which is a necessary mechanism involved in counteracting lifetime reduction by mitophagy impairment [215]. In accordance, the cardiac tissues of Bnip3 and Nix double-knockout mice had elevated quantities of mtDNA and mitochondrial proteins [216]. Further work is required to determine whether this axis is evolutionarily conserved and whether NIX is a transcriptional target gene of NFE2L2.
Parkin/PINK1 in mitobiogenesis
In contrast to BNIP3 and NIX deficiency, Parkin/PINK1 deficiency in neuronal cells suppresses mitobiogenesis, demonstrating that mitophagy coordinates the regulation of mitobiogenesis (Fig. 4). PARIS (ZNF746) is a transcriptional repressor of PGC-1 and NRF1, and the stability of PARIS is controlled by Parkin. As a result, when Parkin is absent, PARIS accumulates, and PGC-1 and NRF1 are downregulated [217]. Conditional depletion of Parkin causes a progressive loss of dopamine neurons in adult animals, which contributes to the pathogenesis of Parkinson’s disease (PD) [217]. As anticipated, Parkin deficiency in the adult mouse ventral midbrain resulted in decreased mitobiogenesis and mitochondrial mass, which could be reversed by PARIS knockdown [218]. In Drosophila, the loss of dopamine neurons, mitobiogenesis-related defects, and motor deficits were causally linked to PINK1 and Parkin deficiencies, and these effects were identified to be PARIS dependent. Additionally, phosphorylation of Parkin and PARIS by PINK1 promotes PARIS ubiquitination and proteasomal degradation by Parkin [219]. Moreover, the pathogenesis of PD is significantly influenced by a loss of Parkin-induced mitobiogenesis deficiency rather than mitophagy impairment [220]. Indeed, neurons created from induced pluripotent stem cells (iPSCs) derived from PD patients with Parkin mutations have compromised mitobiogenesis pathways [221]. Moreover, Parkin knockdown suppresses mtDNA transcription and replication in proliferating cells, indicating that Parkin is directly involved in the control of mitobiogenesis [222]. In SH-SY5Y cells and hepatocellular carcinoma cells, mitochondrial electron transport chain (ETC) function, mitochondrial cristae, and mtDNA levels were decreased when PINK1 was knocked down, indicating a critically important role of PINK1 in mitobiogenesis and in maintaining the activity of the mitochondrial ETC [223, 224]. However, in white adipose tissues (WAT), PARIS protein levels remain unchanged, and PGC-1 is increased when Parkin is absent, indicating that the Parkin-PARIS axis regulates PGC1 in a tissue-specific manner [225]. By increasing PGC-1α protein stability in a mitochondrial superoxide-activated NAD(P)H quinone dehydrogenase 1 (Nqo1)-dependent manner in WAT, deletion of Parkin stimulates mitobiogenesis and protects mice from obesity-causing high-fat diets [225]. When Parkin is absent, the expression of PGC-1 and mitobiogenesis driven by acute exercise in skeletal muscle remain unaffected, indicating that other signaling pathways control mitobiogenesis in this circumstance [226]. It is interesting to note that Parkin overexpression increases PGC-1α expression and mitobiogenesis in the skeletal muscles of aged mice, thereby attenuating fibrosis and oxidative stress caused by aging [227]. Additionally, overexpressing Parkin in Drosophila skeletal muscle resulted in an increase in PGC-1α expression and mitochondrial activity [228]. Since Parkin-induced mitophagy decreases mitochondrial mass, it is conceivable that mitobiogenesis is induced to maintain mitochondrial number through upregulation of PGC-1α levels to counterbalance mitophagy when Parkin is overexpressed. Indeed, mitobiogenesis is induced in a TFEB- and PGC1-1α-dependent manner following FCCP-induced mitophagy in SH-SY5Y cells [229]. These findings collectively show that Parkin/PINK1 regulate both mitophagy and mitobiogenesis, and their involvement has significant ramifications for regulating mitochondrial homeostasis and cellular energy metabolism.
PGC-1α-NRF1-FUNDC1 axis in mitochondrial biogenesis and mitophagy
Investigation of Fundc1 transcriptional regulation revealed the direct connection between mitobiogenesis and mitophagy (Fig. 4) [230]. Binding of NRF1 to the classic consensus site in the promoter region of Fundc1 was identified [230, 231]. By using electrophoretic mobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP) analysis, it was determined that NRF1 directly binds to the Fundc1 promoter. When NRF1 was knocked down, the expression of FUNDC1 was lowered, but this effect was reversed when NRF1 was reintroduced [230]. As mentioned above, the transcriptional activity of NRF1 is stimulated by cofactor PGC-1α, so it is conceived that the expression of Fundc1 gene should be regulated by PGC-1α. Indeed, we discovered that PGC-1α and FUNDC1 expression were positively associated in both the BAT tissues of cold-exposed mice and in vitro cultivated adipocytes [230]. The PGC-1α/NRF1 axis is critical for the regulation of Fundc1 expression, as evidenced by both the direct binding of NRF1 and PGC-1α to the Fundc1 promoter and the stimulation of FUNDC1 protein production by PGC-1α overexpression [230]. It has been shown that enhanced mitophagy that occurs in skeletal muscles of mice accompanied with acute exercise was attenuated when PGC-1α was deficient and it would be interesting to determine whether FUNDC1 is implicated in this process [232]. Notably, the Eukaryotic Promoter Database indicates a putative NRF1 binding site in the promoter of Bnip3 and that both NFE2L2 and NRF1 are necessary for the insulin-like growth factor I (IGF-1)-induced activation of the BNIP3 protein [233]. However, it is presently unclear whether NRF1 or other transcription factors involved in mitobiogenesis directly affect the expression of Bnip3.
Mouse BAT produces more mitochondria after exposure to cold temperatures, and given that FUNDC1 is a mitophagy receptor, it is conceivable that the mitophagy pathway may also be activated during this process. According to a recent study, the crucial thermogenesis gene uncoupling protein 1 (UCP1) was responsible for elevated mitophagy in BAT from mice following cold exposure [234]. Indeed, mitophagy is activated in cold-exposed BAT, as shown by electron microscopy (EM) visualization of mitochondria inside autophagosomes and the upregulation of autophagy-related proteins [230, 234]. The interaction between FUNDC1 and LC3 was also shown to be amplified in cold-challenged BAT [230]. Brown adipocyte-specific Fundc1-deficient mice were used to further demonstrate the significance of FUNDC1 in cold-challenge induction of BAT mitophagy. The mitophagy produced by cold exposure in mouse BAT was dramatically suppressed when FUNDC1 was absent, and this suppression was accompanied by an accumulation of damaged mitochondria, demonstrating that FUNDC1-mediated mitophagy serves as an essential mediator of mitochondrial quality control during mitobiogenesis [230]. Accordingly, Fundc1-depleted BAT had decreased mtDNA and mitochondrial function in response to cold exposure, causing poor thermogenesis and cold sensitivity in the animals [230]. Intriguingly, UCP1 induction in BAT by cold exposure was hampered when FUNDC1 was defective due to the lower levels of PGC-1 protein compared to wild-type controls [230]. Given that PGC-1α is a predominant regulator of the expression of UCP1 during cold exposure and that UCP1 plays a crucial role in BAT thermogenesis, it is possible that Fundc1 loss impairs PGC-1α induction and results in the observed thermogenic abnormalities [230, 235]. The feedback regulation of PGC-1α by Fundc1 deficiency was also observed in primary cardiomyocytes, and the increased mRNA levels of PGC-1α, NRF1, and TFAM in CK2α-deleted cells were diminished when FUNDC1 was knocked down, suggesting that mitobiogenesis and mitochondrial turnover are accelerated by FUNDC1-mediated mitophagy [236]. PGC-1α and NRF1 directly regulate the expression of Fundc1, which in turn provides a direct mechanism for the coordinated regulation of both mitophagy and mitobiogenesis. The rapid clearance of defective mitochondria during mitobiogenesis, which is crucial for maintaining mitochondrial quality and quantity as well as tissue functions, may be caused by activation of PGC-1α/NRF1 control of FUNDC1-dependent mitophagy. It has been shown that inducing mitophagy in a way that is dependent on the small GTPases Rheb and NIX promotes mitochondrial OXPHOS, which in turn stimulates mitochondrial renewal, indicating that mitophagy is committed to the successful renewal of mitochondria [237]. Therefore, by simultaneously activating the two opposing processes of mitophagy and mitobiogenesis, it is possible to speed up mitochondrial turnover and facilitate cells to acquire more healthy mitochondria in response to an increase in energy demand.
Furthermore, when FUNDC1-regulated mitophagy is insufficient, cold-induced mitobiogenesis in BAT is similarly repressed, offering a feedback mechanism to monitor mitobiogenesis and to maintain mitochondrial homeostasis and the ideal number of mitochondria [235]. Our unpublished research has shown that the lack of FUNDC1 increases the interaction between PGC-1α and its E3 ligase while decreasing the protein stability of PGC-1α. However, the details of this retrograde regulation need to be further clarified. Similarly, rosiglitazone-induced mitobiogenesis and upregulation of PGC-1α protein were diminished in differentiated 3T3-L1 adipocytes when Bnip3 was knocked down, suggesting that efficient mitophagy is required for proper mitobiogenesis [238]. Further evidence supports that the coordination of mitophagy and mitobiogenesis is necessary for supplying energy during differentiation, as indicated by the increased expression of PGC-1α with another mitophagy receptor, BCL2L13, during adipocyte differentiation. Both mitobiogenesis and adipogenic differentiation were suppressed when BCL2L13 was knocked down [239]. The tight coordination of mitobiogenesis and mitophagy allows cells to adjust their mitochondrial content in response to physiological demands, stress, and other intracellular or environmental stimuli, such as cold, hypoxia, and nutrient supply. When mitophagy is impaired, compromised mitochondrial function results in the suppression of mitobiogenesis by decreasing the activity of PGC-1α either by regulating its stability or by repressing its expression to avoid the further accumulation of damaged mitochondria, mitochondrial hyperactivity, and increased production of ROS [217, 236]. In many neurodegenerative diseases, both mitobiogenesis and mitophagy are impaired in neuronal cells, and an imbalance in this regulatory circuit may be responsible for the mitochondria-mediated vicious cycle of oxidative stress [240,241,242,243].

Coordination of mitochondrial biogenesis and mitophagy. PGC-1α is the primary regulator of mitobiogenesis, and in an NRF1-dependent manner, PGC-1α also regulates the expression of the mitophagy receptor Fundc1. The NFE2L2 homolog SKN-1 regulates the expression of the BNIP3/NIX homolog DCT-1 in C. elegans. In addition, PGC-1α activity can be decreased by mitophagy defects by either controlling its stability (Fundc1 deficiency) or suppressing its expression in a Parkin-PARIS-CREB pathway-dependent manner
The interplay of mitobiogenesis and mitophagy involved in the metabolism and fitness of organisms during health and disease
To regulate mitochondrial content and quality within cells, there must be a balance between mitobiogenesis and mitophagy. When this balance is disrupted, the functions of organs are hampered, and a variety of disorders are more likely to develop. When Fundc1 is specifically knocked out in mouse BAT, the mice are incapable of maintaining body temperature in response to short-term or long-term cold exposure [230]. By compromising mitochondrial turnover and quality in response to cold stress, defects in both mitophagy and mitobiogenesis brought on by the loss of FUNDC1 raise the possibility that maintaining the proper balance between these processes is essential in meeting the increased demand for mitochondrial function that arises in some tissues under diverse environmental pressures [230]. The role of autophagy or mitophagy in thermogenesis has also been investigated by using Atg5 BAT conditional knockout (cKO) mice [212]. Thyroid treatment promoted mitobiogenesis and mitophagy in mice BAT, which raised the body temperature and oxygen consumption rate (OCR) compared to those in euthyroid animals [212]. However, the body temperature of hyperthyroid Atg5 cKO mice was lower than that of control euthyroid mice, indicating that under healthy conditions, a balance between mitobiogenesis and mitophagy is required to maintain mitochondrial function and thermogenesis [212].
The interaction between mitophagy and mitobiogenesis is crucial for the early postnatal development of the heart and the switch to an exclusively aerobic metabolism [244]. This shift was characterized in cardiac myocytes by elevated mitobiogenesis and Parkin-mediated mitophagy [245]. The genes for mitobiogenesis and mitophagy were downregulated when both Mfn1 and Mfn2 were removed in midgestational and postnatal cardiac myocytes, and the mice went on to develop cardiomyopathy and died within 16 days of birth [244]. Similarly, cardiac Parkin deletion or expression of Mfn2 AA (a mutant that hinders Parkin mitochondrial translocation) caused cardiomyopathy and mortality in mice by impairing the replacement of fetal cardiomyocyte mitochondria by mature adult mitochondria [245]. The results of this study showed that the synchronization of mitobiogenesis and mitophagy can promote developmentally programmed mitochondrial turnover, which is essential for maturational metabolic switching to fatty acids in perinatal mouse hearts.
The activity of AMPK, a crucial regulator of mitobiogenesis and mitophagy, is inhibited by poly (ADP ribosyl)ation (PARylation) mediated by PARP1 [246]. The expansion of the lifespan of Drosophila occurs when PARP is inhibited in aged flies, which causes AMPK to be activated, increasing mitobiogenesis and PINK1-mediated mitophagy [246]. These findings suggest that increasing mitochondrial turnover may boost mitochondrial health and be a strategy for extending lifespan. [246]. AMPK also plays an important role in the balance between mitophagy and mitobiogenesis in MELAS syndrome, a mitochondrial disorder that is caused mainly by the m.3243 A > G mutation in mitochondrial DNA [247]. Mitobiogenesis works as an AMPK-mediated compensatory mechanism in response to increased mitophagy, and most of the pathophysiological alterations were restored by AMPK activators in MELAS fibroblast lines [247].
On the other hand, a decrease in mitochondrial abundance and function as well as a buildup of defective mitochondria caused by a deficiency in mitobiogenesis and mitophagy have been linked to the regulation of the aging process [248, 249]. Meanwhile, it is widely accepted that the interplay between mitobiogenesis and mitophagy is necessary for the function of neurons and that a failure in this connection is related to a variety of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and others [248, 250,251,252,253].
Concluding remarks
It is generally accepted that maintaining cellular and tissue homeostasis requires adequate mitochondrial number and function and that their dysregulation is intimately linked to the onset of many diseases. The regulation of mitochondrial mass and function is governed by two conserved processes, mitobiogenesis and mitophagy. If either one or both processes are disrupted, it results in an accumulation of dysfunctional mitochondria, oxidative stress, cellular aging, and ultimately cell death. Although initially considered two distinct processes, mitophagy and mitobiogenesis are now known to work in concert to maintain the health of mitochondria. More studies are warranted to investigate how cells maintain ideal quantities of mitochondria in response to multiple environmental and developmental signals. It will be interesting to see whether the factors involved in mitobiogenesis also affect the expression of other mitophagy-related proteins, in addition to the previously identified FUNDC1 and BNIP3. Future work will help to determine the mechanisms governing the feedback control of mitophagy dysfunction that leads to a deficiency in mitobiogenesis. As mitochondrial dysfunction is a key characteristic of a plethora of diseases, attempts to elucidate the crosstalk between mitobiogenesis and mitophagy will advance our understanding of the etiology and the development of novel therapeutic approaches for a variety of diseases.
Availability of data and materials
Not applicable.
References
McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16(14):R551–560.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13.
Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–12.
Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258(5 Pt 1):C755–786.
Paul BT, Manz DH, Torti FM, Torti SV. Mitochondria and Iron: current questions. Expert Rev Hematol. 2017;10(1):65–79.
Dong JYF, Lin J, He H, Song Z. The metabolism and function of phospholipids in mitochondria. Mitochondrial Commun. 2023;1:2–12.
Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders: a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. 2017;1863(5):1066–77.
Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95.
Siasos G, Tsigkou V, Kosmopoulos M, Theodosiadis D, Simantiris S, Tagkou NM, Tsimpiktsioglou A, Stampouloglou PK, Oikonomou E, Mourouzis K, et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann Transl Med. 2018;6(12):256.
Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. 2013;6:19.
Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–98.
Fontenay M, Cathelin S, Amiot M, Gyan E, Solary E. Mitochondria in hematopoiesis and hematological diseases. Oncogene. 2006;25(34):4757–67.
Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8(11):870–9.
Ploumi C, Daskalaki I, Tavernarakis N. Mitochondrial biogenesis and clearance: a balancing act. FEBS J. 2017;284(2):183–95.
Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. 2018;20(9):1013–22.
Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84.
Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23(9):459–66.
Rich PR, Marechal A. The mitochondrial respiratory chain. Essays Biochem. 2010;47:1–23.
Vercellino I, Sazanov LA. The assembly, regulation and function of the mitochondrial respiratory chain. Nat Rev Mol Cell Biol. 2022;23(2):141–61.
Larosa V, Remacle C. Insights into the respiratory chain and oxidative stress. Biosci Rep. 2018. https://doi.org/10.1042/BSR20171492.
Drose S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol. 2012;748:145–69.
Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88(2):611–38.
Granat L, Hunt RJ, Bateman JM. Mitochondrial retrograde signalling in neurological disease. Philos Trans R Soc Lond B Biol Sci. 2020;375(1801):20190415.
Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell. 2004;14(1):1–15.
Virbasius JV, Virbasius CA, Scarpulla RC. Identity of GABP with NRF-2, a multisubunit activator of cytochrome oxidase expression, reveals a cellular role for an ETS domain activator of viral promoters. Genes Dev. 1993;7(3):380–92.
Evans MJ, Scarpulla RC. NRF-1: a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 1990;4(6):1023–34.
Popov LD. Mitochondrial biogenesis: an update. J Cell Mol Med. 2020;24(9):4892–9.
Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta. 2002;1576(1–2):1–14.
Evans MJ, Scarpulla RC. Interaction of nuclear factors with multiple sites in the somatic cytochrome c promoter. Characterization of upstream NRF-1, ATF, and intron Sp1 recognition sequences. J Biol Chem. 1989;264(24):14361–8.
Chau CM, Evans MJ, Scarpulla RC. Nuclear respiratory factor 1 activation sites in genes encoding the gamma-subunit of ATP synthase, eukaryotic initiation factor 2 alpha, and tyrosine aminotransferase. Specific interaction of purified NRF-1 with multiple target genes. J Biol Chem. 1992;267(10):6999–7006.
Blesa JR, Prieto-Ruiz JA, Abraham BA, Harrison BL, Hegde AA, Hernandez-Yago J. NRF-1 is the major transcription factor regulating the expression of the human TOMM34 gene. Biochem Cell Biol. 2008;86(1):46–56.
Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci U S A. 1994;91(4):1309–13.
Watanabe H, Imai T, Sharp PA, Handa H. Identification of two transcription factors that bind to specific elements in the promoter of the adenovirus early-region 4. Mol Cell Biol. 1988;8(3):1290–300.
Rosmarin AG, Resendes KK, Yang Z, McMillan JN, Fleming SL. GA-binding protein transcription factor: a review of GABP as an integrator of intracellular signaling and protein-protein interactions. Blood Cells Mol Dis. 2004;32(1):143–54.
Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci U S A. 2004;101(17):6472–7.
Shao D, Liu Y, Liu X, Zhu L, Cui Y, Cui A, Qiao A, Kong X, Liu Y, Chen Q, et al. PGC-1 beta-regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERR alpha. Mitochondrion. 2010;10(5):516–27.
Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, et al. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A. 2004;101(17):6570–5.
Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguere V. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRalpha and gamma. Cell Metab. 2007;5(5):345–56.
De Vitto H, Ryu J, Calderon-Aparicio A, Monts J, Dey R, Chakraborty A, Lee MH, Bode AM, Dong Z. Estrogen-related receptor alpha directly binds to p53 and cooperatively controls colon cancer growth through the regulation of mitochondrial biogenesis and function. Cancer Metab. 2020;8(1):28.
Villena JA, Hock MB, Chang WY, Barcas JE, Giguere V, Kralli A. Orphan nuclear receptor estrogen-related receptor alpha is essential for adaptive thermogenesis. Proc Natl Acad Sci U S A. 2007;104(4):1418–23.
Motohashi H, O’Connor T, Katsuoka F, Engel JD, Yamamoto M. Integration and diversity of the regulatory network composed of maf and CNC families of transcription factors. Gene. 2002;294(1–2):1–12.
Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8(4):379–91.
Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23(22):8137–51.
Taguchi K, Yamamoto M. The KEAP1-NRF2 system in Cancer. Front Oncol. 2017;7:85.
Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103(11):1232–40.
Hayashi G, Jasoliya M, Sahdeo S, Sacca F, Pane C, Filla A, Marsili A, Puorro G, Lanzillo R, Brescia Morra V, et al. Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum Mol Genet. 2017;26(15):2864–73.
Chen H, Hu Y, Fang Y, Djukic Z, Yamamoto M, Shaheen NJ, Orlando RC, Chen X. Nrf2 deficiency impairs the barrier function of mouse oesophageal epithelium. Gut. 2014;63(5):711–9.
Merry TL, Ristow M. Nuclear factor erythroid-derived 2-like 2 (NFE2L2, Nrf2) mediates exercise-induced mitochondrial biogenesis and the anti-oxidant response in mice. J Physiol. 2016;594(18):5195–207.
Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92(6):829–39.
Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27(7):728–35.
Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta. 2011;1813(7):1269–78.
St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408.
Bouchez C, Devin A. Mitochondrial biogenesis and mitochondrial reactive oxygen species (ROS): a complex relationship regulated by the cAMP/PKA signaling pathway. Cells. 2019. https://doi.org/10.3390/cells8040287.
Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98(1):115–24.
Sen N, Satija YK, Das S. PGC-1alpha, a key modulator of p53, promotes cell survival upon metabolic stress. Mol Cell. 2011;44(4):621–34.
Wei P, Pan D, Mao C, Wang YX. RNF34 is a cold-regulated E3 ubiquitin ligase for PGC-1alpha and modulates brown fat cell metabolism. Mol Cell Biol. 2012;32(2):266–75.
LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16(10):992–1003.
Oberkofler H, Linnemayr V, Weitgasser R, Klein K, Xie M, Iglseder B, Krempler F, Paulweber B, Patsch W. Complex haplotypes of the PGC-1alpha gene are associated with carbohydrate metabolism and type 2 diabetes. Diabetes. 2004;53(5):1385–93.
Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94(4):525–33.
Taherzadeh-Fard E, Saft C, Akkad DA, Wieczorek S, Haghikia A, Chan A, Epplen JT, Arning L. PGC-1alpha downstream transcription factors NRF-1 and TFAM are genetic modifiers of Huntington disease. Mol Neurodegener. 2011;6(1):32.
Katsouri L, Lim YM, Blondrath K, Eleftheriadou I, Lombardero L, Birch AM, Mirzaei N, Irvine EE, Mazarakis ND, Sastre M. PPARgamma-coactivator-1alpha gene transfer reduces neuronal loss and amyloid-beta generation by reducing beta-secretase in an Alzheimer’s disease model. Proc Natl Acad Sci U S A. 2016;113(43):12292–7.
Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103(26):10086–91.
Chambers JM, Wingert RA. PGC-1alpha in disease: recent renal insights into a versatile metabolic regulator. Cells. 2020. https://doi.org/10.3390/cells9102234.
Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277(3):1645–8.
Uldry M, Yang W, St-Pierre J, Lin J, Seale P, Spiegelman BM. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metab. 2006;3(5):333–41.
Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22(14):1948–61.
Andersson U, Scarpulla RC. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol Cell Biol. 2001;21(11):3738–49.
Vercauteren K, Pasko RA, Gleyzer N, Marino VM, Scarpulla RC. PGC-1-related coactivator: immediate early expression and characterization of a CREB/NRF-1 binding domain associated with cytochrome c promoter occupancy and respiratory growth. Mol Cell Biol. 2006;26(20):7409–19.
Vercauteren K, Gleyzer N, Scarpulla RC. PGC-1-related coactivator complexes with HCF-1 and NRF-2beta in mediating NRF-2(GABP)-dependent respiratory gene expression. J Biol Chem. 2008;283(18):12102–11.
Vercauteren K, Gleyzer N, Scarpulla RC. Short hairpin RNA-mediated silencing of PRC (PGC-1-related coactivator) results in a severe respiratory chain deficiency associated with the proliferation of aberrant mitochondria. J Biol Chem. 2009;284(4):2307–19.
Rowe GC, Patten IS, Zsengeller ZK, El-Khoury R, Okutsu M, Bampoh S, Koulisis N, Farrell C, Hirshman MF, Yan Z, et al. Disconnecting mitochondrial content from respiratory chain capacity in PGC-1-deficient skeletal muscle. Cell Rep. 2013;3(5):1449–56.
Lai L, Wang M, Martin OJ, Leone TC, Vega RB, Han X, Kelly DP. A role for peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1) in the regulation of cardiac mitochondrial phospholipid biosynthesis. J Biol Chem. 2014;289(4):2250–9.
Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104(29):12017–22.
Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD + metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–60.
Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, Williams RS. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296(5566):349–52.
Ojuka EO, Jones TE, Han DH, Chen M, Holloszy JO. Raising Ca2 + in L6 myotubes mimics effects of exercise on mitochondrial biogenesis in muscle. FASEB J. 2003;17(6):675–81.
Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, Floering LM, Spiegelman BM, Collins S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol. 2004;24(7):3057–67.
Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol Cell. 2001;8(5):971–82.
Knutti D, Kressler D, Kralli A. Regulation of the transcriptional coactivator PGC-1 via MAPK-sensitive interaction with a repressor. Proc Natl Acad Sci U S A. 2001;98(17):9713–8.
Wright DC, Geiger PC, Han DH, Jones TE, Holloszy JO. Calcium induces increases in peroxisome proliferator-activated receptor gamma coactivator-1alpha and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282(26):18793–9.
Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59(4):675–80.
Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413(6852):179–83.
Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci U S A. 2003;100(12):7111–6.
Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20(3):460–73.
He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93.
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36(13):1811–36.
Mizushima N, Yoshimori T, Ohsumi Y. The role of atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32.
Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2022. https://doi.org/10.1038/s41580-022-00542-2.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41.
Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cadwell K, Cecconi F, Choi AMK, et al. Autophagy in major human diseases. EMBO J. 2021;40(19):e108863.
Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1–2):11–42.
Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–75.
Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24(1):24–41.
Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20(3):233–42.
Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8(1):3–5.
Schofield JH, Schafer ZT. Mitochondrial reactive oxygen species and mitophagy: a complex and nuanced relationship. Antioxid Redox Signal. 2021;34(7):517–30.
Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40(3):e104705.
Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20(1):31–42.
Liu L, Liao X, Wu H, Li Y, Zhu Y, Chen Q. Mitophagy and its contribution to metabolic and aging-associated disorders. Antioxid Redox Signal. 2020;32(12):906–27.
Wanderoy S, Hees JT, Klesse R, Edlich F, Harbauer AB. Kill one or kill the many: interplay between mitophagy and apoptosis. Biol Chem. 2020;402(1):73–88.
Cai J, Yang J, Jones DP. Mitochondrial control of apoptosis: the role of cytochrome c. Biochim Biophys Acta. 1998;1366(1–2):139–49.
Chung CY, Singh K, Kotiadis VN, Valdebenito GE, Ahn JH, Topley E, Tan J, Andrews WD, Bilanges B, Pitceathly RDS, et al. Constitutive activation of the PI3K-Akt-mTORC1 pathway sustains the m.3243 A > G mtDNA mutation. Nat Commun. 2021;12(1):6409.
Dai Y, Zheng K, Clark J, Swerdlow RH, Pulst SM, Sutton JP, Shinobu LA, Simon DK. Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum Mol Genet. 2014;23(3):637–47.
Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014;24(7):787–95.
Onishi M, Okamoto K. Mitochondrial clearance: mechanisms and roles in cellular fitness. FEBS Lett. 2021;595(8):1239–63.
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–85.
Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54(3):362–77.
Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15(5):566–75.
Wu H, Xue D, Chen G, Han Z, Huang L, Zhu C, Wang X, Jin H, Wang J, Zhu Y, et al. The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy. 2014;10(10):1712–25.
Ma K, Zhang Z, Chang R, Cheng H, Mu C, Zhao T, Chen L, Zhang C, Luo Q, Lin J, et al. Dynamic PGAM5 multimers dephosphorylate BCL-xL or FUNDC1 to regulate mitochondrial and cellular fate. Cell Death Differ. 2020;27(3):1036–51.
Sugo M, Kimura H, Arasaki K, Amemiya T, Hirota N, Dohmae N, Imai Y, Inoshita T, Shiba-Fukushima K, Hattori N et al. Syntaxin 17 regulates the localization and function of PGAM5 in mitochondrial division and mitophagy. EMBO J. 2018. 10.15252/embj.201798899
Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, Han Z, Chen L, Gao R, Liu L, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy. 2016;12(4):689–702.
Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, Zhuang H, Zhang X, Chen H, Li S, et al. FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J. 2016;35(13):1368–84.
Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, Wang Y, Sehgal SA, Siraj S, Wang X, et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017;18(3):495–509.
Chai P, Cheng Y, Hou C, Yin L, Zhang D, Hu Y, Chen Q, Zheng P, Teng J, Chen J. USP19 promotes hypoxia-induced mitochondrial division via FUNDC1 at ER-mitochondria contact sites. J Cell Biol. 2021. https://doi.org/10.1083/jcb.202010006.
Chen L, Zhang Q, Meng Y, Zhao T, Mu C, Fu C, Deng C, Feng J, Du S, Liu W, et al. Saturated fatty acids increase LPI to reduce FUNDC1 dimerization and stability and mitochondrial function. EMBO Rep. 2023;24(4):e54731.
Lampert MA, Orogo AM, Najor RH, Hammerling BC, Leon LJ, Wang BJ, Kim T, Sussman MA, Gustafsson AB. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy. 2019;15(7):1182–98.
Wang C, Dai X, Wu S, Xu W, Song P, Huang K. FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nat Commun. 2021;12(1):2616.
Li W, Li Y, Siraj S, Jin H, Fan Y, Yang X, Huang X, Wang X, Wang J, Liu L, et al. FUN14 domain-containing 1-mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatology. 2019;69(2):604–21.
Yang A, Peng F, Zhu L, Li X, Ou S, Huang Z, Wu S, Peng C, Liu P, Kong Y. Melatonin inhibits triple-negative breast cancer progression through the Lnc049808-FUNDC1 pathway. Cell Death Dis. 2021;12(8):712.
Ren J, Sun M, Zhou H, Ajoolabady A, Zhou Y, Tao J, Sowers JR, Zhang Y. FUNDC1 interacts with FBXL2 to govern mitochondrial integrity and cardiac function through an IP3R3-dependent manner in obesity. Sci Adv. 2020. https://doi.org/10.1126/sciadv.abc8561.
Fu T, Xu Z, Liu L, Guo Q, Wu H, Liang X, Zhou D, Xiao L, Liu L, Liu Y, et al. Mitophagy directs muscle-adipose crosstalk to alleviate dietary obesity. Cell Rep. 2018;23(5):1357–72.
Wu H, Wang Y, Li W, Chen H, Du L, Liu D, Wang X, Xu T, Liu L, Chen Q. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy. 2019;15(11):1882–98.
Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, Huang K, Xie Z, Zou MH. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation. 2017;136(23):2248–66.
Bi Y, Xu H, Wang X, Zhu H, Ge J, Ren J, Zhang Y. FUNDC1 protects against doxorubicin-induced cardiomyocyte PANoptosis through stabilizing mtDNA via interaction with TUFM. Cell Death Dis. 2022;13(12):1020.
Xu C, Cao Y, Liu R, Liu L, Zhang W, Fang X, Jia S, Ye J, Liu Y, Weng L, et al. Mitophagy-regulated mitochondrial health strongly protects the heart against cardiac dysfunction after acute myocardial infarction. J Cell Mol Med. 2022;26(4):1315–26.
Gong Y, Luo Y, Liu S, Ma J, Liu F, Fang Y, Cao F, Wang L, Pei Z, Ren J. Pentacyclic triterpene oleanolic acid protects against cardiac aging through regulation of mitophagy and mitochondrial integrity. Biochim Biophys Acta Mol Basis Dis. 2022;1868(7):166402.
Li Q, Liu Y, Huang Q, Yi X, Qin F, Zhong Z, Lin L, Yang H, Gong G, Wu W. Hypoxia acclimation protects against heart failure postacute myocardial infarction via Fundc1-mediated mitophagy. Oxid Med Cell Longev. 2022;2022:8192552.
Mao S, Tian S, Luo X, Zhou M, Cao Z, Li J. Overexpression of PLK1 relieved the myocardial ischemia-reperfusion injury of rats through inducing the mitophagy and regulating the p-AMPK/FUNDC1 axis. Bioengineered. 2021;12(1):2676–87.
Zhang W, Ren H, Xu C, Zhu C, Wu H, Liu D, Wang J, Liu L, Li W, Ma Q, et al. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife. 2016. https://doi.org/10.7554/eLife.21407.
Geng G, Liu J, Xu C, Pei Y, Chen L, Mu C, Wang D, Gao J, Li Y, Liang J, et al. Receptor-mediated mitophagy regulates EPO production and protects against renal anemia. Elife. 2021. https://doi.org/10.7554/eLife.64480.
Yan M, Yu Y, Mao X, Feng J, Wang Y, Chen H, Xie K, Yu Y. Hydrogen gas inhalation attenuates sepsis-induced liver injury in a FUNDC1-dependent manner. Int Immunopharmacol. 2019;71:61–7.
Zhou H, Zhu P, Wang J, Toan S, Ren J. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduct Target Ther. 2019;4:56.
Cai Y, Yang E, Yao X, Zhang X, Wang Q, Wang Y, Liu J, Fan W, Yi K, Kang C, et al. FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol. 2021;38:101792.
Li S, Zhou Y, Gu X, Zhang X, Jia Z. NLRX1/FUNDC1/NIPSNAP1-2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 2021;54(3):e12986.
Wang J, Zhu P, Li R, Ren J, Zhou H. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020;30:101415.
Wen W, Yu G, Liu W, Gu L, Chu J, Zhou X, Liu Y, Lai G. Silencing FUNDC1 alleviates chronic obstructive pulmonary disease by inhibiting mitochondrial autophagy and bronchial epithelium cell apoptosis under hypoxic environment. J Cell Biochem. 2019;120(10):17602–15.
Leermakers PA, Schols A, Kneppers AEM, Kelders M, de Theije CC, Lainscak M, Gosker HR. Molecular signalling towards mitochondrial breakdown is enhanced in skeletal muscle of patients with chronic obstructive pulmonary disease (COPD). Sci Rep. 2018;8(1):15007.
Chen G, Ray R, Dubik D, Shi L, Cizeau J, Bleackley RC, Saxena S, Gietz RD, Greenberg AH. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med. 1997;186(12):1975–83.
Yasuda M, Theodorakis P, Subramanian T, Chinnadurai G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J Biol Chem. 1998;273(20):12415–21.
Matsushima M, Fujiwara T, Takahashi E, Minaguchi T, Eguchi Y, Tsujimoto Y, Suzumori K, Nakamura Y. Isolation, mapping, and functional analysis of a novel human cDNA (BNIP3L) encoding a protein homologous to human NIP3. Genes Chromosomes Cancer. 1998;21(3):230–5.
Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61(18):6669–73.
Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000;97(16):9082–7.
Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K, Yu KT, Jaye M, Ivashchenko Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001;8(4):367–76.
Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20(15):5454–68.
Imazu T, Shimizu S, Tagami S, Matsushima M, Nakamura Y, Miki T, Okuyama A, Tsujimoto Y. Bcl-2/E1B 19 kDa-interacting protein 3-like protein (Bnip3L) interacts with bcl-2/Bcl-xL and induces apoptosis by altering mitochondrial membrane permeability. Oncogene. 1999;18(32):4523–9.
Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, Shi L, Dubik D, Greenberg A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem. 1999;274(1):7–10.
Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW. 2nd: dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107(20):9035–42.
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–81.
Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. 2007;104(49):19500–5.
Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454(7201):232–5.
Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51.
Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287(23):19094–104.
Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, Novak I, Dikic I, Hamacher-Brady A, Brady NR. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288(2):1099–113.
Poole LP, Bock-Hughes A, Berardi DE, Macleod KF. ULK1 promotes mitophagy via phosphorylation and stabilization of BNIP3. Sci Rep. 2021;11(1):20526.
He YL, Li J, Gong SH, Cheng X, Zhao M, Cao Y, Zhao T, Zhao YQ, Fan M, Wu HT, et al. BNIP3 phosphorylation by JNK1/2 promotes mitophagy via enhancing its stability under hypoxia. Cell Death Dis. 2022;13(11):966.
Rogov VV, Suzuki H, Marinkovic M, Lang V, Kato R, Kawasaki M, Buljubasic M, Sprung M, Rogova N, Wakatsuki S, et al. Phosphorylation of the mitochondrial autophagy receptor nix enhances its interaction with LC3 proteins. Sci Rep. 2017;7(1):1131.
Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu J, Shen Z, Jiang L, Wang L, Yang W, et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy. 2017;13(10):1754–66.
Naeem S, Qi Y, Tian Y, Zhang Y. NIX compensates lost role of parkin in cd-induced mitophagy in HeLa cells through phosphorylation. Toxicol Lett. 2020;326:1–10.
da Silva Rosa SC, Martens MD, Field JT, Nguyen L, Kereliuk SM, Hai Y, Chapman D, Diehl-Jones W, Aliani M, West AR, et al. BNIP3L/Nix-induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy. 2021;17(9):2257–72.
Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. 2015;6:7527.
Bhujabal Z, Birgisdottir AB, Sjottem E, Brenne HB, Overvatn A, Habisov S, Kirkin V, Lamark T, Johansen T. FKBP8 recruits LC3A to mediate parkin-independent mitophagy. EMBO Rep. 2017;18(6):947–61.
Wei Y, Chiang WC, Sumpter R Jr., Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168(1–2):224–38. e210.
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–8.
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25(3):302–5.
Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–60.
Deng H, Dodson MW, Huang H, Guo M. The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105(38):14503–8.
Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105(5):1638–43.
Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem. 2011;286(13):11649–58.
Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441(7097):1157–61.
Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441(7097):1162–6.
Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803.
Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem. 2011;117(5):856–67.
Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol. 2010;191(5):933–42.
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. 2010;8(1):e1000298.
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits parkin to damaged mitochondria and activates latent parkin for mitophagy. J Cell Biol. 2010;189(2):211–21.
Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates parkin E3 ligase activity by phosphorylating serine 65. Open Biol. 2012;2(5):120080.
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205(2):143–53.
Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460(1):127–39.
Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510(7503):162–6.
Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for parkin recruitment to damaged mitochondria. Nat Commun. 2012;3:1016.
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147(4):893–906.
Safiulina D, Kuum M, Choubey V, Gogichaishvili N, Liiv J, Hickey MA, Cagalinec M, Mandel M, Zeb A, Liiv M et al. Miro proteins prime mitochondria for parkin translocation and mitophagy. EMBO J. 2019. 10.15252/embj.201899384.
Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A. 2010;107(11):5018–23.
Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19(24):4861–70.
Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111(42):E4439–4448.
Yamano K, Youle RJ. Two different axes CALCOCO2-RB1CC1 and OPTN-ATG9A initiate PRKN-mediated mitophagy. Autophagy. 2020;16(11):2105–7.
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–14.
Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A. 2016;113(24):E3349–3358.
Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016;113(15):4039–44.
Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, Youle RJ. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell. 2019;74(2):347–362e346.
Yamano K, Kikuchi R, Kojima W, Hayashida R, Koyano F, Kawawaki J, Shoda T, Demizu Y, Naito M, Tanaka K, et al. Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J Cell Biol. 2020. https://doi.org/10.1083/jcb.201912144.
Rojansky R, Cha MY, Chan DC. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife. 2016. https://doi.org/10.7554/eLife.17896.
Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561(7722):258–62.
McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, Muqit MMK, Brooks SP, Ganley IG. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 2018;27(2):439–49. e435.
Hirota Y, Yamashita S, Kurihara Y, Jin X, Aihara M, Saigusa T, Kang D, Kanki T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy. 2015;11(2):332–43.
Chen J, Ren Y, Gui C, Zhao M, Wu X, Mao K, Li W, Zou F. Phosphorylation of parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T alpha-synuclein model of Parkinson’s disease. Cell Death Dis. 2018;9(6):700.
Deng R, Zhang HL, Huang JH, Cai RZ, Wang Y, Chen YH, Hu BX, Ye ZP, Li ZL, Mai J, et al. MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis. Autophagy. 2021;17(10):3011–29.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–903.
Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao WT, Ma HK, Jiang MD, Xu TT, Xu J, et al. HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020;36:101671.
Zhao JF, Rodger CE, Allen GFG, Weidlich S, Ganley IG. HIF1alpha-dependent mitophagy facilitates cardiomyoblast differentiation. Cell Stress. 2020;4(5):99–113.
Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS ONE. 2010;5(11):e15394.
Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41.
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–61.
Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun. 2017;8(1):548.
Hung CM, Lombardo PS, Malik N, Brun SN, Hellberg K, Van Nostrand JL, Garcia D, Baumgart J, Diffenderfer K, Asara JM, et al. AMPK/ULK1-mediated phosphorylation of parkin ACT domain mediates an early step in mitophagy. Sci Adv. 2021. https://doi.org/10.1126/sciadv.abg4544.
Iorio R, Celenza G, Petricca S. Mitophagy: molecular mechanisms, new concepts on parkin activation and the emerging role of AMPK/ULK1 axis. Cells. 2021. https://doi.org/10.3390/cells11010030.
Mannam P, Shinn AS, Srivastava A, Neamu RF, Walker WE, Bohanon M, Merkel J, Kang MJ, Dela Cruz CS, Ahasic AM, et al. MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;306(7):L604–619.
Scott I, Webster BR, Chan CK, Okonkwo JU, Han K, Sack MN. GCN5-like protein 1 (GCN5L1) controls mitochondrial content through coordinated regulation of mitochondrial biogenesis and mitophagy. J Biol Chem. 2014;289(5):2864–72.
Kang JW, Hong JM, Lee SM. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J Pineal Res. 2016;60(4):383–93.
Singh BK, Sinha RA, Tripathi M, Mendoza A, Ohba K, Sy JAC, Xie SY, Zhou J, Ho JP, Chang CY, et al. Thyroid hormone receptor and ERRalpha coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci Signal. 2018. https://doi.org/10.1126/scisignal.aam5855.
Yau WW, Singh BK, Lesmana R, Zhou J, Sinha RA, Wong KA, Wu Y, Bay BH, Sugii S, Sun L, et al. Thyroid hormone (T(3)) stimulates brown adipose tissue activation via mitochondrial biogenesis and MTOR-mediated mitophagy. Autophagy. 2019;15(1):131–50.
Zhou D, Zhou M, Wang Z, Fu Y, Jia M, Wang X, Liu M, Zhang Y, Sun Y, Lu Y, et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 2019;10(7):524.
Sin J, Andres AM, Taylor DJ, Weston T, Hiraumi Y, Stotland A, Kim BJ, Huang C, Doran KS, Gottlieb RA. Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy. 2016;12(2):369–80.
Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521(7553):525–8.
Dorn GW 2. Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res. 2010;3(4):374–83.
Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144(5):689–702.
Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, Jiang H, Kang SU, Andrabi SA, Dawson VL, et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci U S A. 2015;112(37):11696–701.
Pirooznia SK, Yuan C, Khan MR, Karuppagounder SS, Wang L, Xiong Y, Kang SU, Lee Y, Dawson VL, Dawson TM. PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol Neurodegener. 2020;15(1):17.
Kumar M, Acevedo-Cintron J, Jhaldiyal A, Wang H, Andrabi SA, Eacker S, Karuppagounder SS, Brahmachari S, Chen R, Kim H, et al. Defects in mitochondrial biogenesis drive mitochondrial alterations in PARKIN-deficient human dopamine neurons. Stem Cell Reports. 2020;15(3):629–45.
Wasner K, Smajic S, Ghelfi J, Delcambre S, Prada-Medina CA, Knappe E, Arena G, Mulica P, Agyeah G, Rakovic A, et al. Parkin deficiency impairs mitochondrial DNA dynamics and propagates inflammation. Mov Disord. 2022;37(7):1405–15.
Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, Matsumoto T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. 2006;15(6):883–95.
Gegg ME, Cooper JM, Schapira AH, Taanman JW. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE. 2009;4(3):e4756.
Kung-Chun Chiu D, Pui-Wah Tse A, Law CT, Ming-Jing Xu I, Lee D, Chen M, Kit-Ho Lai R, Wai-Hin Yuen V, Wing-Sum Cheu J, Wai-Hung Ho D, et al. Hypoxia regulates the mitochondrial activity of hepatocellular carcinoma cells through HIF/HEY1/PINK1 pathway. Cell Death Dis. 2019;10(12):934.
Moore TM, Cheng L, Wolf DM, Ngo J, Segawa M, Zhu X, Strumwasser AR, Cao Y, Clifford BL, Ma A, et al. Parkin regulates adiposity by coordinating mitophagy with mitochondrial biogenesis in white adipocytes. Nat Commun. 2022;13(1):6661.
Chen CCW, Erlich AT, Hood DA. Role of parkin and endurance training on mitochondrial turnover in skeletal muscle. Skelet Muscle. 2018;8(1):10.
Leduc-Gaudet JP, Reynaud O, Hussain SN, Gouspillou G. Parkin overexpression protects from ageing-related loss of muscle mass and strength. J Physiol. 2019;597(7):1975–91.
Rana A, Rera M, Walker DW. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci U S A. 2013;110(21):8638–43.
Ivankovic D, Chau KY, Schapira AH, Gegg ME. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem. 2016;136(2):388–402.
Liu L, Li Y, Wang J, Zhang D, Wu H, Li W, Wei H, Ta N, Fan Y, Liu Y, et al. Mitophagy receptor FUNDC1 is regulated by PGC-1alpha/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 2021;22(3):e50629.
Li W, Yin L, Sun X, Wu J, Dong Z, Hu K, Sun A, Ge J. Alpha-lipoic acid protects against pressure overload-induced heart failure via ALDH2-dependent Nrf1-FUNDC1 signaling. Cell Death Dis. 2020;11(7):599.
Vainshtein A, Tryon LD, Pauly M, Hood DA. Role of PGC-1alpha during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol. 2015;308(9):C710–719.
Riis S, Murray JB, O’Connor R. IGF-1 signalling regulates Mitochondria dynamics and turnover through a conserved GSK-3beta-Nrf2-BNIP3 pathway. Cells. 2020. https://doi.org/10.3390/cells9010147.
Lu Y, Fujioka H, Joshi D, Li Q, Sangwung P, Hsieh P, Zhu J, Torio J, Sweet D, Wang L, et al. Mitophagy is required for brown adipose tissue mitochondrial homeostasis during cold challenge. Sci Rep. 2018;8(1):8251.
Liang H, Ward WF. PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ. 2006;30(4):145–51.
Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018;25(6):1080–93.
Melser S, Chatelain EH, Lavie J, Mahfouf W, Jose C, Obre E, Goorden S, Priault M, Elgersma Y, Rezvani HR, et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013;17(5):719–30.
Choi JW, Jo A, Kim M, Park HS, Chung SS, Kang S, Park KS. BNIP3 is essential for mitochondrial bioenergetics during adipocyte remodelling in mice. Diabetologia. 2016;59(3):571–81.
Fujiwara M, Tian L, Le PT, DeMambro VE, Becker KA, Rosen CJ, Guntur AR. The mitophagy receptor bcl-2-like protein 13 stimulates adipogenesis by regulating mitochondrial oxidative phosphorylation and apoptosis in mice. J Biol Chem. 2019;294(34):12683–94.
Corona JC, Duchen MR. Impaired mitochondrial homeostasis and neurodegeneration: towards new therapeutic targets? J Bioenerg Biomembr. 2015;47(1–2):89–99.
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, Bohr VA. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15(10):565–81.
Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40(3):151–66.
Markaki M, Tavernarakis N. Mitochondrial turnover and homeostasis in ageing and neurodegeneration. FEBS Lett. 2020;594(15):2370–9.
Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res. 2012;111(8):1012–26.
Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW. 2nd: parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350(6265):aad2459.
Guo S, Zhang S, Zhuang Y, Xie F, Wang R, Kong X, Zhang Q, Feng Y, Gao H, Kong X, et al. Muscle PARP1 inhibition extends lifespan through AMPKalpha PARylation and activation in Drosophila. Proc Natl Acad Sci U S A. 2023;120(13):e2213857120.
Garrido-Maraver J, Paz MV, Cordero MD, Bautista-Lorite J, Oropesa-Avila M, de la Mata M, Pavon AD, de Lavera I, Alcocer-Gomez E, Galan F, et al. Critical role of AMP-activated protein kinase in the balance between mitophagy and mitochondrial biogenesis in MELAS disease. Biochim Biophys Acta. 2015;1852(11):2535–53.
Daskalaki I, Tavernarakis N. Mitochondrial biogenesis in organismal senescence and neurodegeneration. Mech Ageing Dev. 2020;191:111345.
Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine. 2017;21:7–13.
Fivenson EM, Lautrup S, Sun N, Scheibye-Knudsen M, Stevnsner T, Nilsen H, Bohr VA, Fang EF. Mitophagy in neurodegeneration and aging. Neurochem Int. 2017;109:202–9.
Cen X, Zhang M, Zhou M, Ye L, Xia H. Mitophagy regulates neurodegenerative diseases Cells. 2021. https://doi.org/10.3390/cells10081876.
Palikaras K, Tavernarakis N. Mitophagy in neurodegeneration and aging. Front Genet. 2012;3:297.
Prasuhn J, Davis RL, Kumar KR. Targeting mitochondrial impairment in Parkinson’s disease: challenges and opportunities. Front Cell Dev Biol. 2020;8:615461.
Acknowledgements
We wish to thank Professor Midgley Adam from Nankai University for his English editing of the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China 32230046 to QC, the National Natural Science Foundation of China (32293212 and 92254301) to LL, the Ministry of Science and Technology of China (2020YFA0803702) to LL, the Project for Young Scientists in Basic Research of the Chinese Academy of Sciences (YSBR-075) to LL, the National Natural Science Foundation of China (32170780) to YL, the Natural Science Foundation of Tianjin (20JCYBJC01210) to GC.
Author information
Authors and Affiliations
Contributions
LL and QC wrote the manuscript. YL and GC revised the manuscript. All authors provided intellectual input and read the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no conflicts of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, L., Li, Y., Chen, G. et al. Crosstalk between mitochondrial biogenesis and mitophagy to maintain mitochondrial homeostasis. J Biomed Sci 30, 86 (2023). https://doi.org/10.1186/s12929-023-00975-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-023-00975-7