
Abstract
The discovery of various noncoding RNAs (ncRNAs) and their biological implications is a growing area in cell biology. Increasing evidence has revealed canonical and noncanonical functions of long and small ncRNAs, including microRNAs, long ncRNAs (lncRNAs), circular RNAs, PIWI-interacting RNAs, and tRNA-derived fragments. These ncRNAs have the ability to regulate gene expression and modify metabolic pathways. Thus, they may have important roles as diagnostic biomarkers or therapeutic targets in various diseases, including neurodegenerative disorders, especially Parkinson’s disease. Recently, through diverse sequencing technologies and a wide variety of bioinformatic analytical tools, such as reverse transcriptase quantitative PCR, microarrays, next-generation sequencing and long-read sequencing, numerous ncRNAs have been shown to be associated with neurodegenerative disorders, including Parkinson’s disease. In this review article, we will first introduce the biogenesis of different ncRNAs, including microRNAs, PIWI-interacting RNAs, circular RNAs, long noncoding RNAs, and tRNA-derived fragments. The pros and cons of the detection platforms of ncRNAs and the reproducibility of bioinformatic analytical tools will be discussed in the second part. Finally, the recent discovery of numerous PD-associated ncRNAs and their association with the diagnosis and pathophysiology of PD are reviewed, and microRNAs and long ncRNAs that are transported by exosomes in biofluids are particularly emphasized.
Similar content being viewed by others

Background
After Alzheimer’s disease (AD), Parkinson’s disease (PD) is the second most common neurodegenerative disease worldwide. Gross pathology shows excessive loss of dopaminergic neurons in the substantia nigra (SN) at the midbrain and subsequent dopamine deficiency in the nerve terminals of the basal ganglia. Conventionally, if more than 50% of dopaminergic neurons are damaged [54], individuals show motor deficits such as bradykinesia (slow movement), limb rigidity, tremor at rest, gait disturbance (small stepped gait), and postural instability [147]. Currently, various brands of levodopa and dopamine agonists have been developed to alleviate motor symptoms, but none of them can slow the progression of the disease [55, 82]. Moreover, dopaminergic agents have little effect on controlling nonmotor symptoms of PD, such as anxiety, depression, sleep disorders, autonomic dysfunction, constipation, or cognitive decline [169]. The hallmark of the pathological findings of PD is the formation of eosinophilic inclusions called Lewy bodies (LBs) in the cytoplasm of dopaminergic neurons in the midbrain. In recent years, biochemical experiments have revealed the transformation of normal soluble α-syn monomers into pathological insoluble oligomer and fibril forms, resulting in large aggregates in the LBs. Increasing evidence suggests that these α-syn aggregates can spread from cell to cell through various transport routes [15, 64, 185] and extend remotely even from the gut to the brain via the highway-like vagus nerve or olfactory bulb [14].
In addition, α-syn pathology (synucleinopathy) is found in other central nervous system diseases. The deposition of α-syn in oligodendrocytes is the hallmark of multiple system atrophy (MSA), which is characterized by cerebellar ataxia, pyramidal signs, and autonomic dysfunction [53, 62]. Patients who suffer from dementia with Lewy bodies (DLB) were found to have early cognitive decline and prominent psychiatric symptoms such as hallucinations along with parkinsonism [122]. However, patients with tau-related pathological deposition in the central nervous system, termed tauopathy, also suffer from parkinsonism and other clinical manifestations, such as gaze palsy in progressive supranuclear palsy (PSP) [76, 205], or asymmetric parkinsonism and high cortical dysfunction in corticobasal degeneration (CBD) [6]. Those diseases that show atypical parkinsonism are overall classified as parkinsonism plus syndromes, as shown in Table 1 [206]. In clinical practice, how to precisely differentiate patients who have similar clinical features of parkinsonism is a major challenge to physicians. Hence, an objective, clear-cut and robust diagnostic biomarker of PD remains an unmet need. Although multiple biochemical and imaging biomarkers have been proposed for PD, none of them are convincing [107].
Recently, dysregulation of noncoding RNA (ncRNA) levels has been reported in several neurodegenerative disorders such as AD, PD and Huntington disease (HD) [179]. Although ncRNAs are not transcribed into proteins, ncRNAs still play several key roles in fundamental and complex biological processes such as development, inflammation, ageing, and degeneration [163]. Recent advances in sequencing technologies have further identified PD-associated ncRNAs and the function of ncRNAs in PD. In this review article, we will first introduce the biogenesis of different ncRNAs, including microRNAs (miRNAs), PIWI-interacting RNAs (piRNAs), circular RNAs (circRNAs), long noncoding RNAs (lncRNAs), and transfer RNA (tRNA)-derived fragments (tRFs). Then, we will discuss the pros and cons of the detection platforms and reproducibility of bioinformatic analytical tools due to their major impact on data consistency. In the final section, we will summarize the recent discovery of numerous PD-associated ncRNAs that may serve as novel biomarkers in the differential diagnosis of PD and play important roles in the pathophysiology of PD.
Classification of noncoding RNAs
A substantial variety of ncRNAs have been uncovered in recent decades. In ncRNA history, transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs) were first discovered in the 1950s and were also recognized as ncRNAs. The generic term ncRNA was not officially proposed until the discovery of small nuclear RNAs (snRNAs) and their neighbours, small nucleolar RNAs (snoRNAs), in the 1980s [20, 42]. However, in the early 2000s, the discovery of miRNAs finally attracted the attention of researchers worldwide because of their potential in orchestrating human gene expression mainly through post-transcriptional regulation [192]. Despite considerable scientific breakthroughs in the biological and medical aspects of miRNAs, our understanding of ncDNA continues to expand as ncRNAs are continuously identified. We recommend comprehensive reviews published in Cell in 2014 [27] and J Biomed Sci in 2020 [209]. Here, we focus on updated findings of miRNAs and recent studies introducing the biogenesis of novel categories of ncRNAs, including PIWI-interacting RNAs (piRNAs), circular RNAs (circRNAs), long noncoding RNAs (lncRNAs), and tRNA-derived fragments (tRFs), in the first section.
MicroRNA
MicroRNAs are small noncoding RNAs of approximately 21–25 nucleotides (nt) in length. The first miRNA, “lin-4 RNA”, was discovered in Caenorhabditis elegans and shown to participate in aberrant temporal regulation in the early developmental stage of C. elegans larvae [100, 204]. Since then, mounting evidence has demonstrated that miRNAs are expressed heterogeneously in different tissues [98] and translocate between subcellular compartments to mediate translational repression in numerous cellular processes, such as the regulation of cell differentiation, cell death and homeostasis [59].
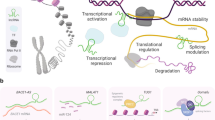
The biogenesis of miRNAs comprises a series of processes, generally including canonical and noncanonical pathways [135]. Both pathways are sometimes closely interlinked and use the same factors. In brief, as shown in Fig. 1A, miRNA genes are first transcribed into primary miRNAs (pri-miRNAs) and processed into precursor miRNAs (pre-miRNAs) [96]. Both the 5' and 3' arms of pre-miRNA can generate functional mature miRNAs. The passenger strand of miRNA (termed miRNA-star or miRNA* was once thought to be useless and doomed to be degraded. More recently, both miRNA and miRNA* were shown to coexist and function [139, 216]. Additionally, the ratio of 5p and 3p strands was found to be tissue-specific in mice [157]. Hence, the nomenclature of miR-#-5p or miR-#-3p is recommended. Since the mechanism of miRNA strand selection is still unclear [124], the role of miRNA*s in gene regulation is still under debate, and thus, the physiological relevance of some passenger strands has been assumed to be underestimated [139]. How the cell decides or selects which miRNA strands to be dominant, called “arm switching”, requires further study [67, 124]. Owing to the complexity of nomenclature, readers should note which target miRNA is selected from previous studies when assessing the biological function.

The biogenesis of miRNA, lncRNA, piRNA, circRNA and tRNA-derived fragments. A Several steps are required to produce mature miRNA, including A-to-I nucleotide editing by ADAR, 5’ and 3’ end trimming by Drosha and DGCR8, cleavage of double-stranded precursor miRNA (pre-miRNA) by Dicer through Dicer-dependent pathway or by AGO through Dicer-independent pathway, and finally, formation of miRNA-induced silencing complex (miRISC) with AGO proteins to bind other DNA or RNA targets. Some miRNAs will be modified by the adenosine deaminase (ADAR) proteins which act on double-stranded RNA to do A-to-I editing [148]. Next, two essential enzymes including protein DiGeorge syndrome critical region 8 (DGCR8) and Drosha forms the microprocessor complex and alternatively excise a pre-miRNA and remove the 5’ and 3’ terminals to produce a pre-miRNA duplex [105]. Pre-miRNA duplex in the nucleus is exported to the cytoplasm and is bound by Dicer to cleave the terminal loop to generate a mature miRNA duplex. Therefore, this process produces the isomirs, including 3p, 5p or polymorphic isomirs, which are classified by the arm of their pre-miRNAs [145]. A Genomes transcribed by RNA polymerase II (Pol II) produce mRNA but also lncRNA in both the sense and antisense directions. Most lncRNAs remain in the nucleus, while some of them are exported to the cytoplasm. C The piRNA clusters are a locus that can generate a family of piRNAs from the same single-stranded RNA transcript. PiRNA precursors are cleaved by PIWI proteins. Similar to the AGO protein in miRNAs, PIWI proteins also have three domains: MID, PIWI, and PAZ. D CircRNAs are preferentially generated by the noncanonical head-to-tail splicing of single or multiple exons, a mechanism called “alternative back-splicing (ABS)” by the depleted spliceosome. E Precursor tRNA (pre-tRNA) encoded from the tRNA gene is first trimmed by the endonucleases RNase P and RNase Z at its 5' and 3' ends, forming a cloverleaf-like tertiary structure composed of three stem loops (D loop, anticodon loop, and T loop), undergoing aminoacylation, and ultimately exporting out of the nucleus as a mature tRNA
In addition, the mechanisms by which miRNAs promote gene silencing through the inhibition of translation and the degradation of target mRNAs are continuously being discovered. Recent studies have focused on the mechanisms of miRNA-mediated post-transcriptional regulation [135], mostly by RNA binding proteins (RBPs) and other lncRNAs, the latter will be described in the next section. Since the regulation of miRNA function is still unclear and remains to be uncovered, we will not introduce all the cellular pathways and detailed mechanisms here. However, as miRNAs were also found to be secreted in extracellular fluids, probably through packing into exosomes [116], they have potential to serve as biomarkers for various diseases, including PD [80] (Table 2).
Long noncoding RNA
Similar to mRNAs in many ways, lncRNAs have a 5' terminal methyl guanosine (5'mG) cap and a 3' terminal polyadenylated tail, contain exons and introns, include several alternative splicing sites, and have no open reading frame (ORF) [145]. Genomes transcribed by RNA polymerase II (Pol II) produce mRNA but also lncRNA in both the sense and antisense directions (Fig. 1B). The length of lncRNAs is generally defined as longer than 200 nucleotides [77]. Most lncRNAs remain in the nucleus, while some of them are exported to the cytoplasm. Increasing evidence has shown the many biological functions of lncRNAs in cell biology, including direct DNA or mRNA binding, chromatin modifier regulation, post-transcriptional modification, and chromatin 3D structure formation, which have been thoroughly reviewed previously [183, 223].
Given that the mammalian brain contains up to 40% heavily transcribed lncRNAs [16], many aspects of lncRNAs are deeply involved in neuronal development and play a role in the pathophysiology of various neurological disorders, such as AD, PD, and HD [201]. Here, we briefly introduce lncRNAs and present the whole picture of the complex interplay of lncRNA-DNA and lncRNA-protein interactions and the complicated RNA machinery network, which is reviewed elsewhere [129]. Nonetheless, PD-related studies will be highlighted in the last section of this review.
PIWI-interacting RNA
Various researchers found a novel-at-the-time class of ncRNAs that is abundant in the fly [5] and mouse testes [3], later termed PIWI-interacting RNA (piRNA) [63]. A mature piRNA is typically 25–32 nucleotides in length after cleavage from a premature long sequence of a piRNA cluster. Increasing studies of piRNAs have revealed the delicate interactions of piRNAs with transposon elements (TEs), so-called jumping genes, which account for 90% of the repeated sequences (approximately 45% of the total genome) in noncoding regions and can stabilize the genome in a wide variety of species by regulating epigenetic modification and maintaining genome integrity [51]. Currently, our understanding of piRNA biology has substantially deepened. The piRNA clusters are a locus that can generate a family of piRNAs from the same single-stranded RNA transcript. Unlike miRNA processing with Dicer, piRNA precursors are cleaved by PIWI proteins. Similar to the AGO protein in miRNAs, PIWI proteins also have three domains: MID, PIWI, and PAZ (Fig. 1C). The difference between PIWI and AGO is that PIWI preferentially binds to the 3' terminal 2'-O-methyl group of piRNA, resulting in a closer link of PIWI to piRNA processing. PiRNAs can be roughly divided into two sets: one in gonad cells and another in somatic cells. Gonad-derived piRNAs typically follow the principle of “1U-10A” on their RNA transcripts, which enables these sets of piRNAs to undergo a unique self-amplification process named the “ping-pong cycle [4, 70] using transposon transcripts as templates. Notably, the unique 1U-10A template on the first 10th nucleotides of piRNA transcripts provides an opportunity to computerize and detect putative piRNAs by sequencing datasets. The second but relatively limited set of piRNAs is generated by “primary processing” of somatic cells.
In fact, the abundance of PIWI proteins and their homologues in animals, at least in pigs, is highest in the gonad, the kidney, and finally the brain [93]. Emerging data reveal the unexplored link of piRNAs in neural development, axonal regeneration, and memory formation, as described in a review [81], probably reflecting the fact that a set of TEs has a higher level in neuronal cells [128]. In recent decades, research on piRNAs has shifted from germline cell development to neurological disorders, including brain tumours [160], AD [151, 161], HD [49], and ageing, and this subject is increasing in popularity. These studies have only profiled piRNA identities in various neurological diseases. More issues merit a detailed investigation, including complete piRNA databases across various species and cells and the time- and space-specific expression pattern in tissues. All these works and valuable efforts can help elucidate aberrant piRNA biological networking in diseases with unmet medical needs, including PD.
Circular RNA
Circular RNAs were first reported as a plant viroid RNA structure in 1976 [164]. Typically, the eukaryotic proteome is translated from canonical linear mRNA transcribed by Pol II, while some isoforms of protein products can be generated by alternative splicing [133]. It has been proposed that when downstream mRNA processing is limited under certain conditions, circRNAs are preferentially generated by the noncanonical head-to-tail splicing of single or multiple exons, a mechanism called “alternative back-splicing (ABS)” by the depleted spliceosome [105] (Fig. 1D).
The first study describing the biological function of circRNAs in human brain development was published in 2013, proposing that a circRNA that is antisense to cerebellar degeneration-related protein 1 (CDR1as) can sponge many miRNAs, particularly miR-7, and therefore achieve post-translational gene regulation [125]. Since then, increasing evidence has shown that circRNAs are abundant in human tissues, particularly in the brain [162]. Furthermore, a comprehensive analysis revealed that circRNAs are highly enriched in the frontal cortex, hippocampus, and cerebellum [65], and the expression of circRNAs is highly spatially and temporally dynamic in the central nervous system (CNS) [230]. Accumulating data have promoted interest in studying the transcriptomic regulatory networks of circRNAs in various neuropsychiatric disorders [115]. To date, most circRNAs are being studied in the field of cancer [217], with some in cardiovascular diseases or dementia, but these molecules have rarely been explored in the field of PD. For more information on the functional impact of circRNAs, some comprehensive reviews are worth reading [30, 214].
Transfer RNA-derived fragments
Transfer RNA is transcribed by RNA polymerase III and highly abundant, accounting for up to 15% of all RNA in tissues and cells [144]. Precursor tRNA (pre-tRNA) encoded from the tRNA gene is first trimmed by the endonucleases RNase P and RNase Z at its 5' and 3' ends, forming a cloverleaf-like tertiary structure composed of three stem loops (D loop, anticodon loop, and T loop), undergoing aminoacylation, and ultimately exporting out of the nucleus as a mature tRNA (Fig. 1E). The demographic summary in a recent review is worth reading [186]. After export out of the nucleus, tRNA canonically pairs triplet anticodons with the complementary codon on mRNA and facilitates peptide elongation and protein translation.
Accumulating evidence has revealed that fragments of mature and precursor tRNAs, including tRFs and tiRNAs, once considered redundant in cell biology, have additional noncanonical functions, such as miRNA gene silencing, post-translational modification of mRNA, protein interactions, epigenetic modification, cellular stress responses, and sperm maturation [172]. However, most studies on disease-relevant tRNA-derived fragments are in the field of cancer. We refer readers to some excellent reviews [211]. In PD, tRFs are still a growing field waiting to be explored.
Techniques for noncoding RNA detection and analysis
Total RNA, when originally isolated, is composed of multiple RNA species, including rRNA, precursor messenger RNA (pre-mRNA), messenger RNA (mRNA), and several types of ncRNAs. Noncoding RNAs are highly heterogeneous in terms of their length and conformation. These molecules can be separated into 3 categories: (1) small ncRNAs (< 50 nt), including microRNAs (miRNAs; 19–25 nt), small interfering RNAs (19–29 nt), PIWI-interacting RNAs (piRNAs, 25–31 nt), and other functional small RNAs, such as transcription initiation RNAs (tiRNAs, 17–18 nt), tRNA-derived fragments (tRFs, 14–36 nt), snoRNA-derived RNAs (17–24 nt or > 27 nt), and sectional ribosomal RNA-derived fragments (rRFs, 15–81 nt); (2) intermediate-sized ncRNAs (50–500 nt), including 5S rRNAs (~ 120 nt), 5.8S rRNA (~ 150 nt), tRNAs (76–90 nt), snoRNAs (60–300 nt), and small nuclear RNAs (snRNAs, ~ 150 nt); and (3) long noncoding transcripts greater than 500 nt, including linear lncRNAs and circular circRNAs [189]. Over 90% of the total RNA molecules present in a cell are rRNAs and tRNAs, while small RNAs account for ∼1% or less [235]. These ncRNAs can regulate gene expression by directly or indirectly binding to specific DNA or RNA sequences.
Because many of these ncRNAs are tissue- and disease-specific, the expression profile (the RNA sequences and expression levels) of these ncRNAs can be used as biomarkers for diseases. Different platforms can provide different information about the sequence and expression level of ncRNAs. Sometimes a combination of these platforms could be used to obtain a more comprehensive view of the expression profiles of multiple RNAs. In this part of the review, the basic principle of five common platforms, microarray, RT-qPCR, Illumina NGS, PacBio and Nanopore, will be discussed. We will focus on how each platform can be used to generate the expression profiles of ncRNAs, including known and unknown transcripts. Non-coding RNAs include small RNAs (miRNAs, piRNAs and tRFs) and long RNAs (lncRNAs and circRNA).
Small ncRNAs
When total RNA is extracted from biological materials (e.g., cells or tissues), subsets of RNA molecules need to be isolated or enriched using specific protocol, such as a ribodepletion protocol to remote ribosomal RNAs or size selection by electrophoresis, to filter out unwanted transcripts [97]. RNA isolation and library preparation strongly affect the detection of target species of ncRNAs [189].
With a mixture of small RNAs, since miRNAs (19–25 nt), piRNAs (25–31 nt), and tRFs (14–36 nt) are all similar in size, they cannot be easily distinguished by examining their sizes. Luckily, most of the time these small RNAs could be identified in the database that contains previously reported transcripts. In some cases when the database or reference sequences are not available, specific characteristics of miRNAs, piRNAs and tRFs need to be considered to aid the identification of the novel transcripts.
Characteristics of miRNAs, piRNAs and tRFs
Typical miRNAs have the following characteristics: (1) their length distribution is narrow and often between 21 and 23 nt; (2) their precursor forms a hairpin structure; therefore, the genomic sequencing flanking a miRNA sequence contains a highly complementary 20- to 30-nt segment; (3) in most cases, pre-miRNA processing results in asymmetric strand accumulation; (4) a miRNA 5' end is most often uridine; (5) a 3' end is usually variable and, at low frequency, can be post-transcriptionally modified by the addition of adenosine or uridine; and (6) mature miRNAs, and often pre-miRNA sequences, are often conserved in closely related species; (7) in most cases, miRNAs originate from nonrepetitive genomic sequences [95]. During tRNA maturation, the 3′- trailer sequences are removed from pre-tRNA, which results in the production of 1-tRF [60]. The other two classes of tRFs are generated from mature tRNAs: 5′-tRF as produced by cleavage of the 5′ end in the D-loop and 3′- tRF as produced through cleavage of the 3′ end in the T-loop [227]. Since tRFs are fragments of tRNAs, a tRF should partially resemble to the tRNA sequence [196]. There is no common features for all piRNAs yet. Therefore multiple features if often employed when identifying piRNA. For example, several K-mers based features had been proposed [109]. Some position-specific properties include a preference for uridine at the 5′ end (75.81%), which is a main characteristic of primary piRNAs, and an A-bias at the 10th nucleotide position (40.61%) [40]. Since there is no consistent characteristics of all piRNAs, the analysis of piRNAs is often done after the depletion of other types of small RNAs [108].
Microarray
To determine the expression profile of these small RNAs, researchers can use different platforms. Microarray is often chosen if the sequences of the sample are known and we want to determine the differential expression.
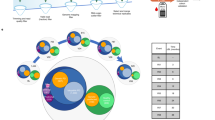
Microarrays are a major high-throughput (reads per run) tool that can simultaneously provide the relative concentration or expression levels of hundreds of target RNA templates [18] based on the computed scores of image intensity. The array platform consists of antisense DNA oligonucleotide probes, spotted or printed onto glass slides or nylon membranes, are designed to hybridize with the appropriate RNAs (small or long RNAs) [95]. Microarray data can immediately identify target molecules with up- or downregulated expression. Nonetheless, array technology has several limitations. For example, background hybridization limits the accuracy of expression measurements, particularly for transcripts present in low abundance. Furthermore, cross-hybridization, nonspecific hybridization and limited detection range of individual probes may occur. There are also issues associated with probe redundancy and annotation [233]. Specificity and sensitivity can be compromised in small RNAs with high sequence similarity due to the nature of short hybridization sequences [177]. Array hybridization methods normally require relatively large amounts (micrograms) of RNA therefore not sufficiently sensitive to profile miRNA expression in a single cell or few cells. Theoretically, reverse transcription quantitative real-time PCR (RT-qPCR) can provide a more sensitive method of profiling [95]. For the pros and cons of microarray, please refer to Fig. 2 and Additional file 1.

RNA sequencing technologies and workflows of qPCR, microarray, Illumina NGS, PacBio and ONT (Oxford Nanopore Technology). A cDNA is synthesized by reverse transcription of extracted RNAs. Fluorescent signals are emitted and detected by the qPCR instruments during PCR. The figure shows the basic principle of SYBR green detection. The quantitative real-time PCR amplification plot is shown at the bottom. The number of PCR cycles is shown on the x-axis, and the fluorescence from the amplification reaction, which is proportional to the amount of amplified product in the tube, is shown on the y-axis. B After the extracted RNA targets are converted to cDNA /cRNA templates, they can hybridize with the cDNA probes on the chips. The cDNA / cRNA samples would carry signals and are labeled with fluorescent tags. When target cDNA / cRNA templates bind to the complementary oligonucleotide probes, the signals will be released. The signal intensities correspond to the abundance of cDNA / cRNA binds to the probes. Each dot observed in the bottom figure represents a cumulative hybridization reaction. Red color denotes higher expression levels of experimental groups while green color denotes higher expression levels of the control group. cDNA arrays typically involve two channels (two colors in the B), but single channel (one color) is also available [178]. C After cDNA library preparation, individual cDNA molecules are clustered on a flow cell. Illumina NGS detects the sequences by synthesis using fluorescent labelled nucleotides. In each small step of sequencing, the growing DNA strand will emit signals from one of the four fluorophores when the nucleotide has been incorporated (images are modified from [202]. The emission wavelength and intensity are used to identify the bases. D After PacBio SMRT-adaptor ligation, circularized cDNA molecule is formed. The individual molecules are loaded into a sequencing chip, where they bind to a polymerase immobilized at the bottom of a nanowell. As each of the fluorescently labelled nucleotides is incorporated into the growing strand, the fluorescent signals are emitted and detected by the PacBio instrument (images are modified from [202]. cDNA sequencing on the PacBio platform enables full-length sequencing from 5’ cap to the 3’ RNA cleavage site. (E) After library preparation, individual molecules attached with motor protein during adaptor ligation are loaded into a flow cell. The motor protein controls the translocation of the RNA strand through the nanopore, causing a change in current that is characteristic for the subsequent bases and will serve as the basis for basecalling. The figure at the bottom shows the corresponding electrical currents (in the pA-range) to nucleotides. The pros and cons of all platforms are listed in the Additional file 1. The numbers of 1, 2, 3 correspond to very good, good and fair performance of each platform. N.A. non-available
RT-qPCR
RT-qPCR is another platform that has often been used to analyse RNA or DNA expression profiles. Because RT-qPCR can provide high sensitivity and specificity for transcripts with various sizes, it is suitable for quantitative study. This method is also widely used in most research labs and by researchers. In fact, RT-qPCR is currently the gold standard method to verify data obtained by microarrays or next generation sequencing (NGS) approaches [177]. However, several drawbacks exist. Because primers are required for RT-qPCR, nonspecific primer design, inconsistent data analysis and normalization can negatively affect the reproducibility of this method [177]. Like microarrays, RT-qPCR can only interrogate a limited set of variants with known sequences and has limited discovery power. The capacity for the number of reactions it can perform each time is also limited. For the pros and cons of RT-qPCR, please refer to Fig. 2 and Additional file 1.
Illumina Next Generation Sequencing (NGS)
NGS is a good tool if we do not know the sequence of the target RNAs or want to study minor mutations or polymorphisms. We will focus on Illumina NGS since it is the major NGS platform currently in the market. NGS has been widely tested and used by many lapidaries, and the bias and error profiles are well understood. A large catalogue of compatible methods and computational workflows is also available. RNA-seq has considerable advantages for examining transcriptome fine structure, such as the detection of novel transcripts, allele-specific expression and splice junctions [233].
NGS is a very high throughput (reads per run) platform and currently provides 100–1000 times more reads per run than long-read platforms. NGS can not only profile the expression of known RNA sequences but is also suitable for identifying various unknown variants [177] and can read degraded or truncated RNA. These fragments or short transcripts will be turned into cDNA libraries through cDNA synthesis. Fragmentation is not required when sequencing small RNAs. Because each fragment can be sequenced repeatedly, NGS has the highest accuracy in detecting the sequence of the transcripts. Low error rates are particularly important for sequencing miRNAs, whose relatively small sizes could result in misalignment or loss of reads if error rates are too high. Because of its high throughput, this method is also suitable for minor variant detection, human axon and genomic sequencing, genome-wide association studies, and gene expression studies [2]. For detection of RNA transcripts with moderate to high abundance, 30–40 million reads are required to accurately quantify their expressions. For coverage over complex transcript libraries, including rare and lowly expressed transcripts, up to 500 million reads are required [56]. The unbiased data acquisition, sequence coverage and depths of NGS are unparalleled by any other available method, and this technique is the only discovery-based approach allowing the identification of novel small RNAs [177].
RNA-seq analysis is vulnerable to the general biases and errors inherent in the NGS technology. Because library preparation includes sequence fragmentation, adaptor ligation, cDNA synthesis and PCR amplification, errors can occur [175]. Uneven read coverage, complex splicing and potential sequencing bias could complicate the sequencing even more [138, 233]. The fragments are not uniformly sampled and sequenced, as there is variability in sequencing depth across the transcriptome due to preferential sites of fragmentation and variable primer and tag nucleotide composition effects [73, 121]. For the pros and cons of Illumina NGS, please refer to Fig. 2 and Additional file 1.
Expression profile by NGS
The expression level of each RNA unit is measured by the number of sequenced fragments that map to the reference transcript, which is expected to correlate directly with its abundance level. RNA-seq data (counts of mapped reads) are fundamentally different from microarray data (computed scores of image intensity) in terms of the expression level analysis. In RNA-seq, the expression signal of a transcript is dependent on the sequencing depth and the expression levels of other transcripts, whereas in array-based methods, probe intensities are independent of each other [153]. Normalized RNA-seq data, therefore, must be modeled statistically using discrete distributions.
The nature of the features encompassed by read count data depends on what the mapping reference (database) is and what type of aligner tool was used. Some types of analytical software are designed to handle isoform differences [101, 191], and others analyse generic features of the transcriptome [153]. To accurately estimate gene expression, read counts must be normalized to correct for systematic variability, such as library fragment size and read depth [97, 140, 158]. This issue, as well as other technical differences, has motivated the development of a growing number of statistical algorithms that implement a variety of approaches for normalization and differential expression (DE) detection [153].
Long RNAs
There are currently three main platforms for long RNA sequencing and expression profiling: Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT). For long RNA sequencing, including lncRNA and circRNA sequencing, similar to short-read RNA sequencing, the target ncRNAs will firstly be specifically enriched as much as possible to reduce interference signals from other transcripts [66, 189]. Library preparation is first dependent on rRNA depletion methods, then reverse transcription (RT) with random primers and size selection by gel electrophoresis, leading to the deficiency of small ncRNAs [193]. Depletion of linear RNAs can be done by RNase R treatment, leading to the enrichment of circRNAs.
Characteristics of lncRNA and circRNA
Like small RNAs, databases are available for most of the lncRNAs and circRNAs. If certain lncRNAs or circRNAs cannot be found in the database, specific characteristics of lncRNAs and circRNAs should be considered to aid the identification of novel lncRNAs and circRNAs.
CircRNAs are a type of long RNA formed by covalent binding of the 3′ and 5′ ends after reverse splicing [207, 208]. The junction between two related exons in the opposite order is called the back-sliced junction (BSJ), which represent a molecular signature of circRNA [28, 57]. Many tools recognize circRNA by identifying the BSJ read. Most algorithms embedded in tools are based on splitting the reads (reads spanning BSJs are split into segments and are aligned to the reference sequence in reverse order) (called segmented-read-based), while several other tools are based on a pre-defined BSJ and flanking sequence of a circRNA (pseudo-sequences to recognize BSJ reads). These tools then map the read directly to that pseudo-reference for discovering a BSJ [28, 31, 57].
LncRNAs are non-coding RNAs with lengths greater than 200 nt [207, 208]. LncRNAs can be preliminarily defined as long RNA transcripts that are capable of autonomous transcription and are longer than 200 nt but lack the capacity to encode proteins [152]. The prediction of new lncRNAs includes two steps: basic screening and potential coding ability screening. Transcript lengths, exon numbers, ORF length and expression level are considered in the basic screening step [218]. Next, all transcripts with protein-coding potential are filtered out (CPC, CNCI, CPAT and Pfam software) [83].
NGS
Although microarrays and RT-qPCR can be used in long RNA expression analysis [102], since the primer or probe binding region is relatively short in the full-length transcript, some information, such as minor mutations, isoforms and structural variances, can be overlooked. NGS can also be used to determine the expression profiles of long RNAs. One major benefit of ensemble-based platforms if low sequencing error rates (< 1%) dominated by single mismatches [ref]. The long RNAs are fragmented, reverse transcribed into cDNA by random primers, and undergo end repair, sequencing adaptor ligation, and size selection for subsequent sequencing. In this way, not only lncRNAs but also mRNAs, circRNAs, and some intermediate-sized ncRNAs can be analysed by NGS. However, the reconstruction of transcripts from short-read data is difficult. Short RNA-seq reads capture only small fragments of transcripts. RNA-seq data, therefore, lack clear isoform data, leading to the inference of many erroneous isoforms. Most algorithms can identify discrete transcript components, but the assembly of complete transcript structures remains a major challenge [97, 184]. On the other hand, long-read full-length cDNA captures transcripts end-to-end, making isoform inference unambiguous [21].
PacBio
The two main long-read platforms are PacBio and Oxford Nanopore Technology (ONT). Long-read sequencing is more accurate and sensitive in structural variation and isoform detection [1]. Because it loads full-length cDNA sequences, this technique can capture many full-length transcripts (1–50 kb), including new isoform structural variations. Because reading results do not need to be reassembled, the computational methods for de novo transcriptome analysis are simplified. These methods are also suitable for studies of minor variations, novel assembly, epigenetics, and RNA isoforms [2]. However, the technology features low to medium throughput: currently, only 500,000 to 10 million reads per run (NGS obtains 100–1000 times more reads per run) [182]. Thus, this method requires high throughput to reach the same accuracy in terms of short-scale variations or low-frequency detections.
PacBio single-molecule real-time (SMRT) isoform sequencing (Iso-Seq) can capture the full length of transcripts, thereby presenting an easier and more accurate method for gene annotation, isoform identification, and lncRNA discovery [39]. Due to the large amount of template required for PacBio sequencing, large-volume PCR is performed. cDNA size selection is optional but highly recommended, as PacBio has weak power in detecting short RNA sequences or degraded RNA templates. After PCR end repair and PacBio SMRT-adaptor ligation, long-read sequencing is performed [182]. Another advantage of this sequencing approach is the ability to produce extraordinarily long reads (average lengths of 4200 to 8500 bp), which greatly improves the identification of novel transcript structures [7, 97, 173]. The extremely long reads generated by the PacBio platform are ideal for de novo transcriptome assembly in which the reads are not aligned to a reference transcriptome. Longer reads can facilitate the accurate detection of alternative splice isoforms, which may not be discovered with shorter reads.
The limitation of lower throughput and higher error rate is most obvious when performing the large-scale differential expression analysis. In these studies, the expression of ncRNA needs to be precisely profiled to attain sufficient statistical power to have confidence in the transcriptome fold changes. For the pros and cons of PacBio, please refer to Fig. 2 and Additional file 1.
Oxford Nanopore Technology (ONT)
ONT sequencers measure changes in ionic current when the DNA fragments translocate through protein nanopores in a semisynthetic insulated membrane; this process does not require enzyme-based nucleotide incorporation or detection of fluorescence signals [39].
Since PCR amplification is optional and direct cDNA sequencing is possible, some errors during DNA polymerase could be avoided (e.g., incorrect nucleotides might be added to the sequence during polymerase), leading to higher-quality results. However, the sequencing yields (numbers of reads) were higher for PCR-amplified cDNA libraries. If PCR is performed, users could start with much smaller amounts of input RNA [182]. Similar to that of PacBio, because it does not go through RNA fragmentation, the structure of the noncoding RNA is retained and can avoid computational algorithm error during template assembly. Thus, this method is suitable for detecting structural variations and isoforms (e.g., [188]. Because ONT can process cRNA directly without cDNA synthesis, RNA modification and epigenetic information are also retained, similar to methylation and other modifications [1, 182]. Expression is directly approximated by the number of reads that mapped on a given transcript [170]. Oikonomopoulos et al. [137] suggested that the expression levels of long RNAs are better captured by ONT, while cDNA-seq (Illumina) appears to be more biased. In general, ONT still has higher sequencing error rate than NGS and PacBio [202]. For the pros and cons of ONT, please refer to Fig. 2 and Additional file 1.
PacBio vs. ONT
For PacBio and ONT Pc data, we found that short read lengths (< 500 bp) had low alignment rates. This finding is likely due to a larger portion of adapter and linker sequences in this short-length data bin [39]. In practice, PacBio and ONT sequencing have their own merits and demerits [39]. One of the greatest advantages of ONT compared to PacBio sequencing is that it can estimate transcript expression levels [168]. In the present study, Cui et al. [39] analysed the correlation between Illumina and ONT data of each replicate sample and found correlations > 0.8 for all groups. The high correlation suggests that ONT can quantify transcript expression levels well. Briefly, PacBio was superior in identifying alternative splicing events, whereas ONT Pc could estimate transcript expression levels [39].
PacBio technology is now widely used for the characterization of cancer transcriptomes, where detection of novel isoforms and fusion transcript is superior to that of short-read technologies. This approach surpasses mapping-based or assembly-based approaches. The effectiveness of MinION in the accurate quantification of transcripts, in the detection of transcript variants and fused genes, in transcript-based haplotype phasing and allele-specific expression and in single-cell expression profiling has been shown in multiple studies [168]. In addition, full-length transcript sequence information is very useful for both genome annotation and gene function studies [137].
Brief Summary
Each platform has its strengths and weaknesses and can be used to address different research questions in RNA expression profiling. There is currently no one-size-fits-all approach for all aspects of RNA profiling. Microarrays and RT-qPCR provide direct quantification of target RNAs, but they are unable to identify novel RNA sequences and can only work with known sequences. Long-read sequencing platforms (such as PacBio and ONT) can be used to detect and analyse complete full-length RNA sequences and identify new RNA transcripts, structural variations and RNA isoforms [2], but because they have a higher error rate than short-sequencing platforms (such as NGS), they are not suitable for analysis that requires accurate sequencing, e.g., expression profiling. This method is not effective for analysis of truncated, degraded or small RNA transcripts. Short-read sequencing can provide indirect quantification, can identify novel RNAs, has high throughput (reads per run), and has many analytical tools. However, it is less accurate in analysing structural variations, RNA isoforms and comprehensive RNA transcriptomes [1]. Most (> 90%) ncRNAs are miRNAs, and in conclusion, different platforms are suitable for different levels of specificity and sensitivity and are often complementary to each other. For comprehensive noncoding RNA profiling, a combination of methods is recommended.
The role of noncoding RNAs in PD
Recent advances in sequencing technologies have enabled high-resolution RNA profiling and thus has provided insights into PD-associated ncRNAs. In the final section, the role of each RNA (miRNA, lncRNA, circRNA, pRNA and tRNA) in serving as potential biomarkers for PD is summarized. Their functional aspects in the pathogenesis of PD are also discussed separately.
MiRNAs in PD
Many studies and review articles emphasize the orchestrated role of miRNAs in the pathogenesis, differential diagnosis, and prognosis of PD. Previous studies of microRNAs associated with PD are summarized in Table 2. A recent meta-analysis thoroughly summarized the identities of differentially expressed miRNAs in PD [165]. Theoretically, an ideal study design requires large-scale, longitudinal, international cohorts using multidisciplinary methods, including NGS, microarray, and RT-qPCR, to detect new types of ncRNAs, validate the regulatory direction of differentially expressed ncRNAs, and succeed in investigating their biological functions in the pathogenesis or prognosis of PD in translational studies. Kern at el. recently provided a good example that fulfils almost all these requirements [87]. These researchers reported circulating small ncRNA profiles, primarily (~ 93%) miRNAs, in two large-scale longitudinal cohorts (Parkinson’s Progression Markers Initiative (PPMI) and Luxembourg Parkinson’s Study (NCER-PD)) using NGS and microarray datasets, respectively. Comparing total PD groups (genetic and idiopathic) and control groups (healthy and unaffected) with some cases that were serially followed, they first identified both diagnostic and prognostic miRNA biomarkers in the PPMI cohort. Between total PD and controls, five miRNAs were differentially expressed in PD (upregulated expression of miR-6836-3p and miR-6777-3p and downregulated expression of miR-487b-3p, miR-493-5p, and miR-15b-5p). Only miR-15b-5p was differentially expressed in plasma-derived exosomes from PD patients [210], and the other 4 differentially expressed miRNAs have never been identified in PD before. Moreover, only downregulated expression of miR-487-3p and miR-15b-5p was validated between the PPMI and NCER-PD cohorts. A similar study was performed by the same group in a smaller cohort (106 PD patients versus 91 healthy controls (HCs)) [43], while 5 differentially expressed miRNAs at that time were not able to be reproduced in this larger cohort. The differences between the two studies, including sample size (ten times larger in Kern et al.’s study, sequencing platform (Illumina Solexa by Ding et al. NextSeq by Kern et al. and validated platform (microarray by Ding et al. RT-qPCR by Kern et al. may be responsible for the discrepancy between the two studies. Regarding prognostic miRNAs correlated with the worsening of motor symptoms and accompanied by the network analysis of miRNA-to-mRNA interactions, a total of 8 miRNAs with downregulated expression (let-7b-5p, miR-140-3p, miR-574-5p, miR-769-5p, miR-3157-3p, miR-3960, miR-5690, miR-6734-5p were highlighted in patients with a progressive course. However, the progression of other nonmotor symptoms, such as cognitive impairment or psychiatric symptoms, should also be important. Last, most of the participants in the PPMIR and NCER-PD cohorts were Caucasians, hindering universal application worldwide. This large-scale study noted the difficulties we found in PD-associated ncRNA studies, including lack of standard sequencing methods, few reproducible targets, and lack of multiethnic populations. The use of other sources of samples (e.g., blood, urine, or saliva,free circulating form or extracellular vesicle-derived form and enrichment of miRNA origins (e.g., brain or other organs; neurons or other neuronal cell types would have a greater impact on the final list of differentially expressed miRNA profiles. In fact, miRNAs could be further categorized according to their biological pathways associated with mitochondria, autophagy, inflammation, and PD-related genes, including SNCA, the gene encoding alpha-synuclein. Herein, we highlight some novel and innovative mechanisms of miRNAs that will shed light on future works in this section.
MiRNAs in biofluids other than blood
MiRNAs can be extracted from various biofluids, including blood, serum, plasma and other sources. Since a recent study reported that the levels of miR-153 and miR-223 were decreased in the saliva of 83 PD patients versus 77 HCs, as detected by quantitative RT-qPCR [38], salivary miRNAs are now noninvasive and easy-to-access sources. Intriguingly, a Chinese study revealed increased levels of miR-874 and miR-145-3p in 30 PD patients compared to 30 HCs by RT-qPCR [33]. This discrepancy probably reflects the difference in ethnicity or technical preparation of the samples.
Brain-derived miRNAs in PD
Many studies extend their interest from circulating free-form miRNAs to tissue-specific miRNAs, such as brain-derived miRNAs. Circulating brain-enriched miRNAs would enable us to detect CNS signals from the peripheral bloodstream. For example, a combined group of 12 brain-enriched miRNAs, including miR-7, miR-124, miR-129, miR-139, and miR-431, could help us to differentiate individuals with PD from healthy individuals, although the discrimination using the aforementioned 4 miRNAs was moderate, with an area under the curve of 0.705 [154]. Intriguingly, Ravanidis et al. tested their 12 brain-derived miRNAs in another independent cohort with 109 idiopathic PD patients and 92 HCs [155]. This time, fewer miRNAs remained significantly different (increased miR-22-3p, miR-139-5p, miR-154-5p, miR-330-5p) between PD patients and HCs, and the highest discrimination accuracy was 0.730 by the pooled miRNAs including different miRNA candidates (miR-7-5p, miR-136-3p, and miR-409-3p). People may be curious about the accuracy of discrimination power when applying miRNA profiling in the real world. Nonetheless, we should always consider another critical issue: how can we assure that these miRNAs are definitely generated within the CNS?
Exosomal miRNAs in PD
Researchers have attempted to identify techniques that not only determine miRNA identities but also show the specific tissues, cells, organelles, or extracellular vesicles (EVs) from which they are derived. The characteristics of EVs, such as surface markers, size, and cargo, are extremely complicated and interwoven. Hence, the effort to categorize various EVs is an emerging field of science worldwide. By size exclusion, exosomes are one of the categories of EVs characterized by lipid bilayers and sizes of 40–160 nm [86]. Exosomes contain various cargos, including mRNAs, ncRNAs, proteins, and metabolites. The enrichment of miRNA in exosomes, abbreviated as “exo-miR” [13], is considered to participate in cell-to-cell signalling [136]. More importantly, exosomes can easily cross through the blood–brain barrier (BBB) and thus can be detected in the peripheral bloodstream and reflect homeostasis in the CNS [10].
Several studies have described the diagnostic role of exo-miR in PD [146]. Gui et al. was the first and only team to uncover a distinct pattern of exo-miRNAs in cerebrospinal fluid (CSF) of PD patients compared to HCs with decreased levels of ex-miR-1 and ex-miR-19b-3p and increased levels of ex-let-7-c-3p, ex-miR-10a-5p, ex-miR-153, and ex-miR-409-3p [69]. In a comparison with AD patients, Gui et al. also disclosed that a fraction of selected CSF-derived exo-miRNAs could be used to differentiate healthy individuals from those with PD with AD by miRNA array using RT-qPCR. Caldi et al. employed high-throughput (reads per run) small RNA sequencing by NGS to perform an unsupervised survey of CSF-derived exo-miR between PD and HC groups, and they identified another group of exo-miR, including miR-99a-5p, miR-126-5p, and miR-501-3p, as diagnostic markers of PD [23]. In a comparison of profiles of exo-miR and free-form miRNAs in CSF, there was no overlap when we examined circulating CSF-derived miRNAs, and miR-24 and miR-205 were identified as diagnostic markers separating PD patients from controls [119]. It is yet too early to determine the superiority of free-form miRNAs and exo-miRs from different biofluids in the diagnosis of PD. Major challenges also remain for selecting and extracting cell-specific exosomes (neurons, oligodendrocytes, microglia, or astrocytes in the CSF or bloodstream [78, 143].
In addition to exo-miR’s high permeability across biological membranes and blood–brain barriers, its low immunogenicity with low rejection response compared to intracranial or intravenous stem cell therapy and tissue- or cell-specific targeting ability also make it a good candidate for therapeutic druggable targets of PD. However, further research is required to confirm the therapeutic potential of exosome-based therapy in clinical applications. One of the hot topics that seems promising is using stem cell-derived exosomes. Crude exosome pellets extracted from mesenchymal stem cells (MSCs) were found to be beneficial to the outcome of traumatic brain injury [219] and cognitive deficiency [32], probably via miRNA transfer [150]. A recent study revealed that exo-miR-188-3p injection into the substantia nigra pars compacta (SNpc) of mice with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD could inhibit cell division protein kinase 5 (CDK5)-induced apoptosis and NACHT leucine-rich repeat protein 3 (NLRP3) inflammasome-induced pyroptosis [104]. These researchers transfected adipose-derived MSCs (ADSCs) with a vector containing miR-188-3p, and then, exo-miR-188-3p was abundantly secreted by ADSCs. After extraction of exo-miR-188-3p-enriched exosomes, PD-related pathogenesis, such as apoptosis and pyroptosis, could be reversed in the animal model. Nonetheless, Li et al. used intracranial injection of exo-miRs rather than the peripheral route, hindering its use in routine clinical practice. Notably, the massive generation of recombinant exosomes is still in its immature stage [61], raising questions over scientists claiming how many fractions of exosomes contain selected exo-miRs. The purity of exosomal pellets is also a major challenge because it is dramatically influenced by existing methods of isolation [92]. This issue should always be kept in mind for future experiments.
Pathogenic miRNAs in PD
The pathogenesis of PD comprises multiple mechanisms and can be generally classified into several categories: increased a-syn production, decreased a-syn clearance (owing to lysosome-autophagosome impairment), mitochondrial dysfunction, enhanced neuroinflammation, reduced neuronal survival, and dysregulated PD-related genes such as SNCA, PTEN-induced putative kinase 1 (PINK1), leucine-rich repeat kinase 2 (LRRK2), glucosylceramidase beta (GBA), or Parkin RBR E3 ubiquitin protein ligase (PRKN). The targeted genes of miRNAs could directly deactivate the transcription of protein-coding genes and subsequently inhibit their protein products, which is the earliest concept we know about miRNAs. For example, miR-16–1 decreases alpha-synuclein clearance by inhibiting heat shock protein 70 (hsp70) and inducing lysosomal dysfunction [232] in an experimental setting. Decreases in miR-16 or its passenger strands (miR-16–2-3p) in human leukocytes [181] or plasma [19] might be a survival strategy of neuronal cells as well as a general effect on other body tissues.
Neuroinflammation: miR-29c
The anti-neuroinflammatory effect of miR-29c has been extensively demonstrated in a series of studies published by Wang R. et al. in an MPTP-treated mouse model of PD and neuronal cell lines. In brief, miR-29c reduces the levels of proinflammatory interleukins (IL-1b, IL-18) [195], miR-29c-3p inhibits NLRP3 inflammasome activation by targeting nuclear factor of activated T cell 5 (NFAT5) [197] and inhibits autophagy by targeting ten-eleven translocation 2 (TET2) [200]. Several human studies have also reported consistently decreased expression of miR-29c in human serum [9, 111, 120], except one study [141] showing that miR-29c expression increased in human serum. Interestingly, no miR-29c was differentially expressed in studies using human plasma, suggesting that sample origins and the preparation protocol could make a major difference in the final list of differentially expressed miRNAs.
Autophagy-related alpha-synuclein clearance: let-7
The autophagosome-lysosome network is a major intracellular degradation system that digests unwanted proteins, including α-syn, in cells [225]. Genetic PD due to GBA or LRRK2 mutations presents dysregulation of intracellular vesicle transportation among autophagosomes, endosomes, and lysosomes. Aberrant let-7 miRNA levels can affect autophagy through proteins and lncRNAs. For example, increased levels of Igg-1 and atg-13 proteins can be induced by knockdown of let-7 in a C. elegans PD model [171]. The involvement of the mTORC1 protein pathway [48] in primary cortical neurons from transgenic mice and other lncRNAs (H19 and Lin28) in breast cancer cell lines [213] warrants further studies to elucidate the underlying mechanism between let-7 and autophagy. In humans, the let-7 family is very large [159], and different isoforms have been discovered in association studies in plasma (downregulated let-7a and let-7f expression [33]), serum (upregulated let-7d expression [11]), CSF (upregulated let-7c-3p [69] and let-7f-5p expression [46]), and CSF-derived exosomes (upregulated let-7 g-3p expression [19]) (Table 2). Determination of the most PD-relevant let-7 isoforms will require further study.
SNCA accumulation: miR-7 and miR-34b/c
The dysregulation of miR-7, which binds to the 3' UTR of the SNCA gene, may induce excessive α-syn generation and accumulation [47, 84]. Interestingly, two studies revealed opposite changes in the direction of miR-7 in different human sources, upregulated expression in plasma [154] and downregulated expression in the brain [123]. This kind of paradoxical phenomenon is also found in miR-34b/c, which has multiple biological functions in inhibiting SNCA transcription [85], dopaminergic neuronal survival, and mitophagy [126]. The decrease in miR-34b alone or combined with miR-34b and miR-34C is relatively consistent in postmortem findings [126, 194], albeit with its incremental change in human plasma being found only in plasma-derived exosomes (exo-miR-34b-5p actually) in one study [68] rather than free-form isoforms in numerous other studies.
The precise mechanism behind this intriguingly paradoxical phenomenon is still unclear. Taking traumatic brain injury as an example, direct release [99] or transportation via extracellular vesicles [195, 197,221,199] of target miRNA (miR-9 here) outside the BBB has been reported, as we found for miR-34b/c isoforms. These paradoxical biological alterations might also be relevant to patients with neurodegenerative diseases. Moreover, we should question why a decrease in miR-7 in the brain, which normally inhibits SNCA transcription, occurs in PD patients and should theoretically inhibit excessive α-syn production under normal conditions. Another study of TBI and miR-9 also suggested that altered levels of certain miRNAs in neurons might have additional benefits or unexpected harm to the growth of other types of neuronal cells, such as astrocytes [207, 208] or microglia [222]. In the near future, we need to solve the puzzle of complicated network shifts of single or multiple miRNAs in neurons and glial cells in the brain, probably with the help of advanced techniques such as single-cell RNA-seq [112]. We strongly recommend recent reviews for details of each PD-associated miRNA and related studies [132, 190]. Herein, putative miRNA- and other ncRNA-associated pathogenic pathways in PD are graphically summarized in Fig. 3.

The overview of ncRNAs in the pathogenesis of PD. Several mechanisms involved in PD-relevant pathogenesis and associated ncRNAs are listed. The impairment of lysosome and autophagy systems through defects of GBA, LRRK2 genes, or LAMP2A and Hsp70 proteins reduce the clearance of α-syn. The excessive production of α-syn could be induced by hyperactive SNCA transcription associated with miRNA and lncRNA (SNCA-AS1). Aberrant α-syn metabolism results in accumulation and aggregation of phosphorylated α-syn. Several causative genes of PD such as LRRK22, PRKN, PINK1, DJ-1 are also modulated by miRNAs and largely induce mitochondrial impairment. Other groups of miRNAs also impair mitophagy or mitochondrial proteins (Bax). Together with the enhanced neuroinflammation (through pro-inflammatory cytokines and NLRP3-inflammasome) and reduced neurotrophic factors (via BDNF, FGF20), neuronal survival is damaged in the end. The interactions of miRNA-lncRNA (miR-223 and lncRNA GAS5) and miRNA-circRNA (miR-128 and cirSLC8A1) are highlighted by asterisks (*). BDNF Brain-derived neurotrophic factor (BDNF), DJ-1 Protein deglycase DJ-1 / Parkinson disease protein 7; FGF20 Fibroblast Growth Factor 20; GBA Glucosylceramidase Beta, Hsp70 70 kilodalton heat shock proteins; LAMP2A Lysosome-associated membrane protein 2, LRRK2 Leucine Rich Repeat Kinase 2, NLRP3 NLR Family Pyrin Domain Containing 3, PINK1 PTEN-induced kinase 1, PRKN parkin RBR E3 ubiquitin protein ligase, SNCA Synuclein Alpha
MiRNA-lncRNA interactions
The modulatory property of miRNAs in the pathogenesis of PD not only have direct but also indirect effects. Another essential mechanism of indirect modulation is via the “sponge effect of lncRNA”, that is, one lncRNA can bind a large set of miRNAs and alter their activities. For example, Chen et al. [31] found that the lncRNA nuclear enriched abundant transcript 1 (NEAT1) could upregulate phosphodiesterase 4B (PDE4B) expression to accelerate the progression of PD by sponging miRNA-124-3p [31] and miR-374c-5p [45]. A well-known lncRNA, RNA X-inactive-specific transcript (XIST), exhibits anti-inflammatory properties by regulating the aforementioned miR-29c-3p, altering NFAT protein levels and overactivating the NLRP3 inflammasome in a rat model of epilepsy and a neuronal cell culture [229]. Another lncRNA, GAS5, demonstrated its proinflammatory effect in rotenone-induced PD mice and lipopolysaccharide (LPS)-treated microglial cells by competitively binding to miR-223-3p and then activating NLRP3 [215]. The concentration and dilution of miRNAs via lncRNAs and their sophisticated interactions are another hot topic.
LncRNAs in PD
Studies centred on lncRNAs in PD typically emphasize several aspects, including α-syn aggregation and clearance, dopaminergic neuron degeneration, neuroinflammation, and PD-related genes, as reviewed in a recent article encompassing human, animal and cellular studies [110, 212]. In this previous review, most studies used MPTP-injected mice or neuronal SH-SY5Y cells to explore the pathogenetic role of each lncRNA interacting with these pathways. Given that one lncRNA can be multitargeted to a wide variety of downstream miRNAs, mRNAs, and PD-related genes and proteins, we expect that future studies will reveal more inspiring findings if various RNAs obtained from postmortem tissues and patient-derived dopaminergic cells are analysed in parallel [50].
Diagnostic lncRNAs in PD
Several human studies that used lncRNAs as diagnostic biomarkers are summarized (Table 3). Elkouris et al. identified six sense and/or antisense lncRNA genes targeting PD-related genes, including SNCA, PINK-1, UCHL-1, and MAPT, and proved their abundant existence in healthy iPSC-derived dopaminergic neurons in comparison with iPSCs and fibroblasts [50]. Next, they measured the RNA expression of six lncRNAs in the human brain from both the SN and cerebellum (9 PD and 8 HC) in human peripheral blood monocytic cells (PBMCs) (20 PD and 20 HC samples) and found that four out of six lncRNAs, SNCA-AS1, MAPT-AS1, AK127687 and AX747125, were detected in CSF-derived exosomes (2 HCs). Through sophisticated experiments, Fan’s team ultimately demonstrated that these lncRNAs are enriched in dopaminergic neurons and detected in both peripheral and central tissues; thus, they are suitable as diagnostic markers of PD.
Using a microarray, Fan and his colleagues also found that four differentially expressed lncRNAs (AC131056.3–001, HOTAIRM1, lnc-MOK-6:1, and RF01976.1–201) had upregulated expression in leukocytes of PD patients [52]. The dysregulation of two lncRNAs (AC131056.3–001 and HOTAIRM1) promoted apoptosis in dopaminergic neurons. Other researchers have focused on PD and other neurodegenerative diseases. Garofalo and his colleagues explored the issue by comparing the whole picture of RNA metabolism in three neurodegenerative diseases, PD, AD, and amyotrophic lateral sclerosis (ALS) [58]. High-throughput RNA-seq was exploited to identify ncRNAs in PBMCs of patients. Four lncRNAs were found to be differentially expressed in 6 PD patients versus 6 controls, with one lncRNA (SCARNA2) with upregulated expression and three with downregulated expression (2 ncRNAs: RP1-29C18.9, RP1-29C18.8; 1 protein-coding RNA: TBC1D3). Among them, only RP1-29c18.8 could not be validated by RT-qPCR in PBMCs. However, further functional and pathway analyses remain largely unexplored.
Exosomal lncRNAs in PD
Exosomal lncRNAs are an emerging field, as we found with miRNAs. In the plasma of 93 PD patients and 85 controls, Zou et al. isolated likely CNS-derived exosomes containing L1CAM by a microbead-based method, screened the lncRNA profile by microarray, and emphasized only one lncRNA, lnc-POU3F3, which showed highly upregulated expression in PD [236]. Although the whole picture of the differentially expressed lncRNA profile was not disclosed, the researchers highlighted a significant inverse correlation of exosomal lnc-POU3F3 levels with lysosomal enzyme β-glucocerebrosidase (GCase) activity, which is encoded by the GBA1 gene. In addition, both exosomal lnc-POU3F3 and GCase activity in PD were significantly correlated with disease severity but not exosomal α-syn levels. Therefore, this study concluded that increased exosomal lnc-POU3F3 plus decreased GCase activity in PD could serve not only as a diagnostic biomarker but also as a therapeutic target of PD.
Some studies screened for lncRNA targets by higher-throughput RNA-seq. A recent study published by Wang et al. revealed that in 7 PD patients and 7 HCs, RNA-seq identified 15 PD-relevant exosomal lncRNAs with upregulated expression and 24 with downregulated expression by ultracentrifugation of isolated exosomes (Wang Q et al. 2020). Among those differentially expressed lncRNAs, MSTRG.336210.1 and lnc‐MKRN2‐42:1 were highly expressed among controls, while MSTRG.242001.1 and MSTRG.169261.1 were highly expressed in patients. The researchers focused on motor severity-correlated lnc-MKRN2-42:1 because some of its targeted genes also had downregulated expression in the plasma of 24 PD patients and 11 HCs. Indeed, future works are still needed to validate the results in a larger cohort and clarify the biological functions of lnc-MKRN2-42:1 in animal or cell models. However, it is worth noting that Wang et al. also sequenced circRNAs and uncovered 62 circRNAs with upregulated expression and 37 with downregulated expression in their cohort, but a detailed description of these molecules is not available. We look forward to further progress in the field of lncRNAs and other ncRNAs, including circRNAs, piRNAs, and tRFs, as we will discuss in the following sections.
CircRNAs in PD
Given that circRNAs are brain-enriched [162] and predominantly located at synapses and dendrites [226], an age-dependent accumulation of circRNAs in the CNS has been discovered in model organisms such as flies, worms, and mice [37, 203]. There are two studies focused on circRNAs in PD, one in the bloodstream and another in postmortem tissues (Table 3).
Blood-derived circRNAs in PD
A recent study investigated the RNA-seq profiles of circRNAs along with mRNAs and miRNAs in peripheral blood from a small number of participants, with only 4 patients with PD and 4 HCs [91]. A total of 129 circRNAs with upregulated expression and 282 with downregulated expression were found in PD samples compared to controls. Most differentially expressed circRNAs were associated with PD, AD or HD-related pathways analysed by the Kyoto Encyclopedia of Genes and Genomes. The researchers also highlighted 2 circRNAs with upregulated (chr11:5225503–5226657: + , hsa_circ_0036353) and 8 with downregulated (hsa_circ_0000690, hsa_circ_0001535, hsa_circ_0001451, hsa_circ_0004870, hsa_circ_0000605, hsa_circ_0014606, hsa_circ_0001801, hsa_circ_0001772) expression and disclosed that source genes (HBB, SIN3A, FBXW7, ITGAL, SIN3A) of the highlighted circRNAs were predominantly linked to functions of homeostasis and oxidative stress response, indicating the central role of reactive oxidative stress and dyshomeostasis in PD [41].
Brain-derived circRNAs in PD
In 2020, Hanan et al. studied circRNAs in the human brains of PD patients and examined brain tissues from three distinct regions, the amygdala, substantia nigra (SN), and mesial temporal gyrus, from 42 PD patients and 27 healthy individuals using pooled existing databases [72]. In addition to abundant mRNAs, approximately 0.02% of circRNAs were also uncovered in each sample by deep RNA-seq at 50 million reads per se. Interestingly, an age-related increase in circRNAs was similarly found in the amygdala and mesial temporal gyrus of PD patients and HCs. However, SN-derived circRNA levels tended to increase with age only in the HCs. Nevertheless, SN-derived circRNAs decreased in PD versus HC groups, which might, according to their hypothesis, be due to the loss of dopaminergic neurons in the SN of PD patients. Next, among 24 differentially expressed circRNAs found between the PD and HC groups (not clearly elaborated in the article), Hanan and her colleagues focused on a circRNA called circSLC8A1, which showed upregulated expression in the SN of PD patients. The SLC8A1 gene encodes a sodium/calcium exchanger [88]. Interestingly, miR-128-targeted mRNAs also showed upregulated expression in the SN of PD brains. A cell model was then constructed using 293HEK cells with shRNA knockdown of circSLC8A1 alone, albeit with mRNA transcripts of SLC8A1. RNA-seq eventually identified 24 out of 110 genes with upregulated expression that were targeted by miR-128 between 293HEK KO cells and controls. Taken together, the results of this study suggest that neuron-derived circSLC8A1 may modulate the functions of miR-128 and play a certain role in the pathophysiology of PD.
Another recent study utilized the most fundamental method, rt-qPCR, to screen known brain-drived circRNAs in human PBMCs [156], where they identified 48 out of 87 circRNAs. Among them, six circRNAs (MAPK9_circ_0001566, HOMER1_circ_0006916, SLAIN1_circ_0000497, DOP1B_circ_0001187, RESP1_circ_0004368, and PSEN1_circ_0003848) were significantly down-regulated in 60 PD versus 60 HC but no circRNA was found upregulated. Nevertheless, after a stepwise logistic regression selection model, there were four circRNAs with highest discrimination power between PD and HC, including SLAIN1_circ_0000497, SLAIN2_circ_0126525, ANKRD12_circ_0000826, and PSEN1_circ_0003848. Only SLAIN1_circ_0000497 and PSEN1_circ_0003848 were overlapped. Clearly, more investigation is warranted in the future.
PiRNAs in PD
To date, only two published papers have reported their findings of specific piRNA profiles in PD based on cell and worm models, as well as postmortem tissues. The first study of piRNAs in PD was published by Schulze et al. in 2018, comparing transcriptomic and epigenomic analysis using RNA-seq between 15 lines of fibroblasts (9 PD, 6 HC), 24 lines of iPSCs (6 PD, 6 HC), and 10 lines of differentiated neurons (5 PD, 5 HC) [166]. There were no differentially expressed genes between fibroblasts, fibroblast-derived iPSCs and iPSC-differentiated midbrain neurons from the two groups, except in PD-derived neurons, where WNT3 expression was upregulated and pathways involving NOS1, CREB, and PGC1alpha were inactivated. Intriguingly, small RNA sequencing via NGS found deregulated miRNA and piRNA patterns between groups. Moreover, they replicated the protocol in the cingulate gyrus from 8 PD and 8 HC samples, aberrant expression of piRNAs, including 561 piRNAs with upregulated and 553 with downregulated expression, was also disclosed. Most targeted genomic regions were transposable elements that showed highly downregulated expression in a disease-specific manner. As a result, dysregulated piRNA features were likely due to the impact of the pathophysiology of PD itself. One more issue of concern about is the bioinformatic pipeline they used. Schulze et al. defined canonical piRNAs, or piRNA-like molecules in their context, by a nucleotide length within 24–32 bp, slightly longer than 22 bp of miRNAs, which excluded possible overlap with snoRNAs. However, many works remain to be clarified given that nearly a hundred piRNA targets await functional curation.
PiRNAs in a PD nematode model
Another study published by Shen et al. [174] used a worm model. Transgenic C. elegans nematodes overexpressing human α-syn wild type (WT) and the A53T mutant (HASNWT OX andHASNA53T OX) were crossbred with C. elegans with knockout (KO) of the human TDP-43-like protein tdp-1 (tdp-1 KO). Interestingly, among 6 various genotypes, various ncRNAs were differentially expressed between HASNA53T OX and WT, including 32 miRNAs and 112 piRNAs. However, the differentially expressed ncRNAs between HASNWT OX and WT only included 8 miRNAs and no piRNAs. More strikingly, a major difference in 31 miRNAs and 440 piRNAs was also uncovered when comparing HASNWT OX and HASNA53T OX.
Unknown effect of alpha-synuclein on transposon elements in PD
Apparently, there is still no convincing experimental evidence demonstrating that WT and A53T α-syn have diverse impacts in influencing TEs through distinct piRNA alterations. However, the dramatic change of piRNAs in A53T mutant nematodes suggests that maybe it is mutant strain rather wild type α-syn that should be investigated in relation to the piRNA—TE loop deregulation. Another misfolded protein commonly associated with neurodegenerative disease, tau, is a hot topic in studying tau-depleted piRNA and dysregulated TE patterns in AD [71] and tauopathies [187]. More studies to discover the interplay between the α-syn-piRNA-TE axis are warranted in the near future.
tRNA fragments in PD
A recent review summarizing the discovery of tRFs in neurodegenerative diseases, including PD, is available [148]. Unfortunately, there is only one human study that revealed altered tRF patterns in patients with PD [113]. Three existing RNA-seq samples from the prefrontal cortex, CSF, and serum of PD patients and controls were collected and reanalysed to identify differentially expressed tRFs between groups. The discrimination of patients and healthy subjects by selective tRF profiles further yielded a high sensitivity and specificity despite sex-dependent tRF expression. These preliminary findings warrant more validation studies with a larger sample size or more types of parkinsonism syndromes to determine the true power of tRFs in the differential diagnosis of PD.
Another study focused on brain-derived tRFs in mice called senescence-accelerated mouse prone 8 (SAMP8) [231]. Intriguingly, tRFs with a miRNA-like pattern were found to primarily target a causative gene of PD, Park2, or Parkin. In 43 PD-related PARK families, autosomal-recessive early onset PD induced by Parkin mutation is the most common genetic cause of familial PD worldwide [75]. However, SAMP8 mice are typically not considered a model of PD but ageing or early AD [142], characterized by autophagic deficits, mitochondrial dysfunction, excessive oxidative stress, and, most importantly, tau protein aggregation. It would be more convincing for us to determine the pathoetiology of tRFs in PD if similar findings were replicated in transgenic mouse models of PD.
Summary
The discovery of diagnosis, treatment, prognosis-related ncRNAs in PD is challenging, largely owing to the complexity of disease itself, the variability of sample origins (blood-, brain-, urine-, saliva-, or exosome-derived), the non-standardized method using distinct RNA-seq platforms and software pipelines, and inconsistent results of ncRNA candidates in human association studies. Validations of robust ncRNAs relevant to the differential diagnosis between PD, HC or disease controls, and disease progression in motor, psychiatric, cognitive domains should be further clarified with the highest interest. To date, only a short list of ncRNA candidates can be replicated in mechanisms-based experiments using cell or animal models. Most of pathogenic miRNAs in PD which also have significant alteration in human studies were highlighted as aforementioned. Discoveries of safer virus- [17] and novel exosome-based [114] delivery platform associated with highly efficient knockdown system [74] are increasing, mostly related to the use of miRNA [12] and circRNA [8]. Briefly, ncRNA candidates are more promising in the translation from disease-relevant biomarkers to treatment-based druggable targets [36]. The advance in RNA-based therapy from the recent experience of mRNA-based COVID vaccines can certainly make a giant step forward to the development of ncRNA-based disease-modifying treatments in the future.
Concluding remarks
The discovery of novel ncRNAs with diverse biological functions within different species and cell types makes ncRNAs one of the most exciting scientific topics. In this review, we have introduced many ncRNAs, including miRNAs, lncRNAs, piRNAs, circRNAs, and tRFs. MicroRNAs, lncRNAs and circRNAs can serve as diagnostic biomarkers, while miRNAs are the most promising therapeutic targets of PD, especially coupled with exosomal transportation. However, the evidence for piRNAs and tRFs is less convincing at this stage.
There are still many new classes of ncRNA sequences to be found by higher-throughput, longer-read, and extensive sequencing platforms. A complementary approach using multiple platforms is often recommended when generating RNA profiles or analysing RNA expression. However, their biological functions still warrant further clarification by functional analysis in the pathophysiology of PD.
The complex network of ncRNAs with DNA, proteins, and other ncRNAs makes this puzzle hard to unravel. With the help of collaborative sequencing and analytical methods and proper selection of model organisms, we can expect to gain clearer insight to identify disease-relevant ncRNAs as diagnostic biomarkers or therapeutic targets of PD in all biological aspects in the near future.
Availability of data and materials
Not applicable.
Abbreviations
- ABS:
-
Alternative back-splicing
- AD:
-
Alzhimer’s disease
- ADAR:
-
Adenosine deaminase
- ADSC:
-
Adipose-derived MSCs
- AGO:
-
Argonaute
- AGO2:
-
Argonaute 2
- ALS:
-
Amyotrophic lateral sclerosis
- BBB:
-
Blood–brain barrier
- CBD:
-
Corticobasal degeneration
- CCS:
-
Circular consensus sequence
- CDK5:
-
Cell division protein kinase 5
- CDR 1:
-
Cerebellar degeneration-related protein 1
- circRNA:
-
Circular RNA
- CNS:
-
Central nervous system
- Cq:
-
Quantitative cycle
- DGCR8:
-
DiGeorge syndrome critical region 8
- DLB:
-
Dementia with Lewy bodies
- eRNA:
-
Enhancer RNA
- exo-miR:
-
MiRNA in exosomes
- GCase:
-
β-Glucocerebrosidase
- GBA:
-
Glucosylceramidase Beta
- HD:
-
Huntington disease
- hsp70:
-
Heat shock protein 70
- iPSC:
-
Induced pluripotent cell
- LB:
-
Lewy bodies
- lncRNA:
-
Long non-coding RNA
- LRRK2:
-
Leucine-rich repeat kinase 2
- MID:
-
The middle domain
- miRISC:
-
MiRNA-induced silencing complex
- miRNA:
-
MicroRNA
- mRNA:
-
Messenger RNA
- MSA:
-
Multiple system atrophy
- MSC:
-
Mesenchymal stem cell
- ncRNA:
-
Non-coding RNA
- NEAT1:
-
Nuclear enriched abundant transcript 1
- NLRP3:
-
NAcht Leucine-rich repeat protein 3
- nt:
-
Nucleotides
- ONT:
-
Oxford Nanopore Technologies
- ORF:
-
Open reading frame
- lncRNA:
-
Long non-coding RNA
- PAZ:
-
Piwi–Argonaute–Zwille domain
- PBMC:
-
Peripheral blood monocytic cell
- PD:
-
Parkinson disease
- PDE4B:
-
Phosphodiesterase 4B
- PINK1:
-
PTEN-induced putative kinase 1
- piRNA:
-
PIWI-interacting RNA
- PIWI:
-
The P-element induced wimpy testes domain
- Pol II:
-
RNA polymerase II
- pre-miRNA:
-
Precursor microRNA
- pre-tRNA:
-
Precursor tRNA
- pri-miRNA:
-
Primary microRNA
- PRKN:
-
Parkin RBR E3 Ubiquitin Protein Ligase
- PSP:
-
Progressive supranuclear palsy
- qPCR:
-
Real-time quantitative PCRs
- RBP:
-
RNA binding proteins
- ROI:
-
Read of insert
- rRNA:
-
Ribosomal RNA
- tRNA:
-
Transfer RNA
- RT:
-
Reverse transcription
- sitRNA-5:
-
Stress-induced tRNA-5
- SN:
-
Substantia nigra
- SNpc:
-
Substantia nigra pars compacta
- TE:
-
Transposon element
- TF:
-
Transcription factor
- tiRNA:
-
TRNA halves
- tRF:
-
TRNA-derived fragment
- tRNA:
-
Transfer RNA
- trRNA:
-
TRNA-derived small RNAs
- UMI:
-
Unique molecular identifiers
- WT:
-
Wild type
- α-syn:
-
Alpha-synuclein
References
Aganezov S, Goodwin S, Sherman RM, Sedlazeck FJ, Arun G, Bhatia S, et al. Comprehensive analysis of structural variants in breast cancer genomes using single-molecule sequencing. Genome Res. 2020;30(9):1258–73.
Ameur A, Kloosterman WP, Hestand MS. Single-molecule sequencing: towards clinical applications. Trends Biotechnol. 2019;37(1):72–85.
Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442(7099):203–7.
Aravin AA, Hannon GJ, Brennecke J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science. 2007;318(5851):761–4.
Aravin AA, Naumova NM, Tulin AV, Vagin VV, Rozovsky YM, Gvozdev VA. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr Biol. 2001;11(13):1017–27.
Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496–503.
Au KF, Sebastiano V, Afshar PT, Durruthy JD, Lee L, Williams BA, et al. Characterization of the human ESC transcriptome by hybrid sequencing. Proc Natl Acad Sci. 2013;110:E4821–30.
Bai H, Lei K, Huang F, Jiang Z, Zhou X. Exo-circRNAs: a new paradigm for anticancer therapy. Mol Cancer. 2019. https://doi.org/10.1186/s12943-019-0986-2.
Bai X, Tang Y, Yu M, Wu L, Liu F, Ni J, et al. Downregulation of blood serum microRNA 29 family in patients with Parkinson’s disease. Sci Rep. 2017;7(1):1–7.
Banks WA, Sharma P, Bullock KM, Hansen KM, Ludwig N, Whiteside TL. Transport of extracellular vesicles across the blood-brain barrier: Brain pharmacokinetics and effects of inflammation. Int J Mol Sci. 2020;21(12):4407.
Barbagallo C, Mostile G, Baglieri G, Giunta F, Luca A, Raciti L, et al. Specific signatures of serum miRNAs as potential biomarkers to discriminate clinically similar neurodegenerative and vascular-related diseases. Cell Mol Neurobiol. 2020;40(4):531–46.
Baumann V, Winkler J. miRNA-based therapies: strategies and delivery platforms for oligonucleotide andnon-oligonucleotide agents. Future Med Chem. 2014;6(17):1967–84.
Bhome R, Del Vecchio F, Lee G-H, Bullock MD, Primrose JN, Sayan AE, et al. Exosomal microRNAs (exomiRs): small molecules with a big role in cancer. Cancer Lett. 2018;420:228–35.
Borghammer P, Van Den Berge N. Brain-first versus gut-first Parkinson’s disease: a hypothesis. J Parkinsons Dis. 2019;9(s2):S281–95.
Brás IC, Outeiro TF. Alpha-synuclein: mechanisms of release and pathology progression in synucleinopathies. Cells. 2021;10(2):375.
Briggs JA, Wolvetang EJ, Mattick JS, Rinn JL, Barry G. Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron. 2015;88(5):861–77.
Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G. Viral vector platforms within the gene therapy landscape. Sig Transduct Target Ther. 2021. https://doi.org/10.1038/s41392-021-00487-6.
Bumgarner R. Overview of DNA microarrays: types, applications, and their future. Curr Protoc Mol Bio. 2013;101(1):22.
Burgos K, Malenica I, Metpally R, Courtright A, Rakela B, Beach T, et al. Profiles of extracellular miRNA in cerebrospinal fluid and serum from patients with Alzheimer’s and Parkinson’s diseases correlate with disease status and features of pathology. PLoS ONE. 2014;9(5):e94839.
Busch H, Reddy R, Rothblum L, Choi YC. SnRNAs, snRNPs, and RNA processing. Annu Rev Biochem. 1982;51:617–54.
Byrne A, Cole C, Volden R, Vollmers C. Realizing the potential of full-length transcriptome sequencing. Phil Trans R Soc B. 2019;374:20190097.
Caggiu E, Paulus K, Mameli G, Arru G, Sechi GP, Sechi LA. Differential expression of miRNA 155 and miRNA 146a in Parkinson’s disease patients. Eneurologicalsci. 2018;13:1–4.
Caldi Gomes L, Roser AE, Jain G, Pena Centeno T, Maass F, Schilde L, et al. MicroRNAs from extracellular vesicles as a signature for Parkinson’s disease. Clin J Transl Med. 2021;11(4):e357.
Cao X-Y, Lu J-M, Zhao Z-Q, Li M-C, Lu T, An X-S, et al. MicroRNA biomarkers of Parkinson’s disease in serum exosome-like microvesicles. Neurosci Lett. 2017;644:94–9.
Cardo LF, Coto E, de Mena L, Ribacoba R, Moris G, Menéndez M, et al. Profile of microRNAs in the plasma of Parkinson’s disease patients and healthy controls. J Neurol. 2013;260(5):1420–2.
Cardo LF, Coto E, Ribacoba R, Menéndez M, Moris G, Suárez E, et al. MiRNA profile in the substantia nigra of Parkinson’s disease and healthy subjects. J Mol Neurosci. 2014;54(4):830–6.
Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77–94.
Chen L, Wang C, Sun H, Wang J, Liang Y, Wang Y, et al. The bioinformatics toolbox for circRNA discovery and analysis. Brief Bioinform. 2021;22(2):1706–28.
Chen L, Yang J, Lü J, Cao S, Zhao Q, Yu Z. Identification of aberrant circulating mi RNA s in Parkinson’s disease plasma samples. Brain Behav. 2018;8(4):e00941.
Chen L-L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol. 2020;21(8):475–90.
Chen M-Y, Fan K, Zhao L-J, Wei J-M, Gao J-X, Li Z-F. Long non-coding RNA nuclear enriched abundant transcript 1 (NEAT1) sponges microRNA-124-3p to up-regulate phosphodiesterase 4B (PDE4B) to accelerate the progression of Parkinson’s disease. Bioengineered. 2021;12(1):708–19.
Chen SY, Lin MC, Tsai JS, He PL, Luo WT, Chiu IM, et al. Exosomal 2’, 3’-CNP from mesenchymal stem cells promotes hippocampus CA1 neurogenesis/neuritogenesis and contributes to rescue of cognition/learning deficiencies of damaged brain. Stem Cells Transl Med. 2020;9(4):499–517.
Chen Y, Zheng J, Su L, Chen F, Zhu R, Chen X, et al. Increased salivary microRNAs that regulate DJ-1 gene expression as potential markers for Parkinson’s disease. Front Aging Neurosci. 2020;12:210.
Chi L-M, Wang L-P, Jiao D. Identification of differentially expressed genes and long noncoding RNAs associated with Parkinson’s disease. Parkinson’s Disease. 2019. https://doi.org/10.1155/2019/6078251.
Cho HJ, Liu G, Jin SM, Parisiadou L, Xie C, Yu J, et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum Mol Genet. 2013;22(3):608–20.
Correddu D, Leung IKH. Targeting mRNA translation in Parkinson's disease. Drug Discov Today. 2019;4(6):1295–303.
Cortés-López M, Gruner MR, Cooper DA, Gruner HN, Voda A, Linden AM, et al. Global accumulation of circRNAs during aging in Caenorhabditis elegans. BMC Genomics. 2018. https://doi.org/10.1186/s12864-017-4386-y.
Cressatti M, Juwara L, Galindez JM, Velly AM, Nkurunziza ES, Marier S, et al. Salivary microR-153 and microR-223 levels as potential diagnostic biomarkers of idiopathic Parkinson’s disease. Mov Disord. 2020;35(3):468–77.
Cui J, Shen N, Lu Z, Xu G, Wang Y, Jin B. Analysis and comprehensive comparison of PacBio and nanopore-based RNA sequencing of the Arabidopsis transcriptome. Plant Methods. 2020;16:85.
Czech B, Hannon GJ. One loop to rule them all: the ping-pong cycle and piRNA-guided silencing. Trends Biochem Sci. 2016;41(4):324–37.
Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis. 2013;3(4):461–91.
Dieci G, Preti M, Montanini B. Eukaryotic snoRNAs: a paradigm for gene expression flexibility. Genomics. 2009;94(2):83–8.
Ding H, Huang Z, Chen M, Wang C, Chen X, Chen J, et al. Identification of a panel of five serum miRNAs as a biomarker for Parkinson’s disease. Parkinsonism Relat Disord. 2016;22:68–73.
Dong H, Wang C, Lu S, Yu C, Huang L, Feng W, et al. A panel of four decreased serum microRNAs as a novel biomarker for early Parkinson’s disease. Biomarkers. 2016;21(2):129–37.
Dong L, Zheng Y, Gao L, Luo X. lncRNA NEAT1 prompts autophagy and apoptosis in MPTP-induced Parkinson’s disease by impairing miR-374c-5p. Acta Biochim Biophys Sin. 2021;53(7):870–82.
Dos Santos MCT, Barreto-Sanz MA, Correia BRS, Bell R, Widnall C, Perez LT, et al. miRNA-based signatures in cerebrospinal fluid as potential diagnostic tools for early stage Parkinson’s disease. Oncotarget. 2018;9(25):17455.
Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285(17):12726–34.
Dubinsky AN, Dastidar SG, Hsu CL, Zahra R, Djakovic SN, Duarte S, Esau CC, Spencer B, Ashe TD, Fischer KM, MacKenna DA, Sopher BL, Masliah E, Gaasterland T, Chau BN, Pereira de Almeida L, Morrison BE, La Spada AR. Let-7 coordinately suppresses components of the amino acid sensing pathway to repress mTORC1 and induce autophagy. Cell Metab. 2014;20(4):626–38.
Dubois C, Kong G, Tran H, Li S, Pang TY, Hannan AJ, et al. Small non-coding RNAs are dysregulated in Huntington’s disease transgenic mice independently of the therapeutic effects of an environmental intervention. Mol Neurobiol. 2021;58(7):3308–18.
Elkouris M, Kouroupi G, Vourvoukelis A, Papagiannakis N, Kaltezioti V, Matsas R, et al. Long non-coding RNAs associated with neurodegeneration-linked genes are reduced in Parkinson’s disease patients. Front Cell Neurosci. 2019;13:58.
Ernst C, Odom DT, Kutter C. The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat Commun. 2017;8(1):1–10.
Fan Y, Li J, Yang Q, Gong C, Gao H, Mao Z, et al. Dysregulated long non-coding RNAs in Parkinson’s disease contribute to the apoptosis of human neuroblastoma cells. Front Neurosci. 2019;13:1320.
Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med. 2015;372(3):249–63.
Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114(5):2283–301.
Fox SH, Katzenschlager R, Lim SY, Barton B, De Bie RM, Seppi K, et al. International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2018;33(8):1248–66.
Fu GK, Xu W, Wilhelmy J, Mindrinos MN, Davis RW, Xiao W, et al. Molecular indexing enables quantitative targeted RNA sequencing and reveals poor efficiencies in standard library preparations. Proc Natl Acad Sci U S A. 2014;111(5):1891–6.
Gao Y, Zhao F. Computational strategies for exploring circular RNAs. Trends Genet. 2018;34(5):389–400.
Garofalo M, Pandini C, Bordoni M, Pansarasa O, Rey F, Costa A, et al. Alzheimer’s, Parkinson’s disease and amyotrophic lateral sclerosis gene expression patterns divergence reveals different grade of RNA metabolism involvement. Int J Mol Sci. 2020;21(24):9500.
Gebert LF, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20(1):21–37.
Gebetsberger J, Polacek N. Slicing tRNAs to boost functional ncRNA diversity. RNA Biol. 2013;10(12):1798–806.
Geeurickx E, Tulkens J, Dhondt B, Van Deun J, Lippens L, Vergauwen G, et al. The generation and use of recombinant extracellular vesicles as biological reference material. Nat Commun. 2019;10(1):1–12.
Gilman S, Wenning G, Low PA, Brooks D, Mathias C, Trojanowski J, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71(9):670–6.
Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442(7099):199–202.
Goedert M, Eisenberg DS, Crowther RA. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci. 2017;40:189–210.
Gokool A, Anwar F, Voineagu I. The landscape of circular RNA expression in the human brain. Biol Psychiat. 2020;87(3):294–304.
Gong S, Gaccioli F, Dopierala J, Socio U, Cook E, Volders PJ, et al. The RNA landscape of the human placenta in health and disease. Nat Commun. 2021;12:2639.
Griffiths-Jones S, Hui JHL, Marco A, Ronshaugen M. MicroRNA evolution by arm switching. EMBO Reports. 2011;12:172–7.
Grossi I, Radeghieri A, Paolini L, Porrini V, Pilotto A, Padovani A, et al. MicroRNA-34a-5p expression in the plasma and in its extracellular vesicle fractions in subjects with Parkinson’s disease: an exploratory study. Int J Mol Med. 2021;47(2):533–46.
Gui Y, Liu H, Zhang L, Lv W, Hu X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget. 2015;6(35):37043.
Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, et al. A slicer-mediated mechanism for repeat-associated siRNA 5’end formation in Drosophila. Science. 2007;315(5818):1587–90.
Guo C, Jeong H-H, Hsieh Y-C, Klein H-U, Bennett DA, De Jager PL, et al. Tau activates transposable elements in Alzheimer’s disease. Cell Rep. 2018;23(10):2874–80.
Hanan M, Simchovitz A, Yayon N, Vaknine S, Cohen-Fultheim R, Karmon M, et al. A Parkinson’s disease CircRNAs resource reveals a link between circSLC8A1 and oxidative stress. EMBO Mol Med. 2020;12(9):e11942.
Hansen KD, Brenner SE, Dudoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010;38:e131.
He AT, Liu J, Li F, Yang BB. Targeting circular RNAs as a therapeutic approach: current strategies andchallenges. Sig Transduct Target Ther. 2021. https://doi.org/10.1038/s41392-021-00569-5.
Hedrich K, Eskelson C, Wilmot B, Marder K, Harris J, Garrels J, et al. Distribution, type, and origin of Parkin mutations: review and case studies. Mov Disord. 2004;19(10):1146–57.
Höglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. 2017;32(6):853–64.
Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16(2):71–84.
Hornung S, Dutta S, Bitan G. CNS-derived blood exosomes as a promising source of biomarkers: opportunities and challenges. Front Mol Neurosci. 2020;13:38.
Hoss AG, Labadorf A, Beach TG, Latourelle JC, Myers RH. microRNA profiles in Parkinson’s disease prefrontal cortex. Front Aging Neurosci. 2016;8:36.
Huang W. MicroRNAs: biomarkers, diagnostics, and therapeutics. In: Huang J, Borchert GM, Dou D, Huan J, Lan W, Tan M, Bin Wu, editors. Bioinformatics in MicroRNA Research. Springer: New York; 2017. p. 57–67.
Huang X, Wong G. An old weapon with a new function: PIWI-interacting RNAs in neurodegenerative diseases. Transl Neurodegener. 2021;10(1):1–21.
Jankovic J, Tan EK. Parkinson’s disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry. 2020;91(8):795–808.
Ji H, Niu C, Zhan X, Xu J, Lian S, Xu B, et al. Identification, functional prediction, and key lncRNA verification of cold stress-related lncRNAs in rats liver. Sci Rep. 2020;10:521.
Junn E, Lee K-W, Jeong BS, Chan TW, Im J-Y, Mouradian MM. Repression of α-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci. 2009;106(31):13052–7.
Kabaria S, Choi DC, Chaudhuri AD, Mouradian MM, Junn E. Inhibition of miR-34b and miR-34c enhances a-synuclein expression in Parkinson’s disease. FEBS Lett. 2015;589:319–25.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020. https://doi.org/10.1126/science.aau6977.
Kern F, Fehlmann T, Violich I, Alsop E, Hutchins E, Kahraman M, et al. Deep sequencing of sncRNAs reveals hallmarks and regulatory modules of the transcriptome during Parkinson’s disease progression. Nat Aging. 2021;1:309–22.
Khananshvili D. The SLC8 gene family of sodium-calcium exchangers (NCX) - structure, function, and regulation in health and disease. Mol Aspects Med. 2013;34(2–3):220–35.
Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, et al. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317(5842):1220–4.
Kim W, Lee Y, McKenna ND, Yi M, Simunovic F, Wang Y, et al. miR-126 contributes to Parkinson’s disease by dysregulating the insulin-like growth factor/phosphoinositide 3-kinase signaling. Neurobiol Aging. 2014;35(7):1712–21.
Kong F, Lv Z, Wang L, Zhang K, Cai Y, Ding Q, et al. RNA-sequencing of peripheral blood circular RNAs in Parkinson disease. Medicine. 2021;100(23):e25888.
Konoshenko MY, Lekchnov EA, Vlassov AV, Laktionov PP. Isolation of extracellular vesicles: general methodologies and latest trends. BioMed Res Int. 2018. https://doi.org/10.1155/2018/8545347.
Kowalczykiewicz D, Pawlak P, Lechniak D, Wrzesinski J. Altered expression of porcine Piwi genes and piRNA during development. PLoS ONE. 2012;7(8):e43816.
Kraus TF, Haider M, Spanner J, Steinmaurer M, Dietinger V, Kretzschmar HA. Altered long noncoding RNA expression precedes the course of Parkinson’s disease—a preliminary report. Mol Neurobiol. 2017;54(4):2869–77.
Krichevsky AM. MicroRNA profiling: from dark matter to white matter, or identifying new players in neurobiology. Sci World J. 2007;7:780248.
Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610.
Kukurba KR, Montgomery SB. RNA sequencing and analysis. Cold Spring Harb Protoc. 2015;11:951–69.
Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129(7):1401–14.
Laterza OF, Lim L, Garrett-Engele PW, Vlasakova K, Muniappa N, Tanaka WK, et al. Plasma MicroRNAs as sensitive and specific biomarkers of tissue injury. Clin Chem. 2009;55(11):1977–83.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–54.
Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BMG, et al. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29(8):1035–43.
Li L, Wang L, Li H, Han X, Chen S, Yang B, et al. Characterization of LncRNA expression profile and identification of novel LncRNA biomarkers to diagnose coronary artery disease. Atherosclerosis. 2018;275:359–67.
Li N, Pan X, Zhang J, Ma A, Yang S, Ma J, et al. Plasma levels of miR-137 and miR-124 are associated with Parkinson’s disease but not with Parkinson’s disease with depression. Neurol Sci. 2017;38(5):761–7.
Li Q, Wang Z, Xing H, Wang Y, Guo Y. Exosomes derived from miR-188-3p-modified adipose-derived mesenchymal stem cells protect Parkinson’s disease. Mol Ther Nucleic Acids. 2021;23:1334–44.
Liang D, Tatomer DC, Luo Z, Wu H, Yang L, Chen LL, et al. The output of protein-coding genes shifts to circular RNAs when the pre-mRNA processing machinery is limiting. Mol Cell. 2017;68(5):940-954.e943.
Liao XY, Wang WW, Yang ZH, Wang J, Lin H, Wang QS, et al. Microarray analysis of transcriptome of medulla identifies potential biomarkers for Parkinson’s disease. Int J Genomics. 2013;2013:606919.
Lin CH, Wu RM. Biomarkers of cognitive decline in Parkinson’s disease. Parkinsonism Relat Disord. 2015;21:431–43.
Liu P, Dong Y, Gu J, Puthiyakunnon S, Wu Y, Chen XG. Developmental piRNA profiles of the invasive vector mosquito Aedes albopictus. Parasites Vectors. 2016;9:524.
Liu Y, Li A, Xie G, Liu G, Hei X. Computational methods and online resources for identification of piRNA-related molecules. Interdiscip Sci Comput Life Sci. 2021;13:176–91.
Lv Q, Wang Z, Zhong Z, Huang W. Role of long noncoding RNAs in Parkinson’s disease: putative biomarkers and therapeutic targets. Parkinson’s Disease. 2020;2020:5374307.
Ma W, Li Y, Wang C, Xu F, Wang M, Liu Y. Serum miR-221 serves as a biomarker for Parkinson’s disease. Cell Biochem Funct. 2016;34(7):511–5.
Ma SX, Lim SB. Single-Cell RNA Sequencing in Parkinson’s Disease. Biomedicines. 2021;9(4):368.
Magee R, Londin E, Rigoutsos I. TRNA-derived fragments as sex-dependent circulating candidate biomarkers for Parkinson’s disease. Parkinsonism Relat Disord. 2019;65:203–9.
Maheshwari R, Tekade M, Gondaliya P, Kalia K, D'Emanuele A, Tekade RK. Recent advances in exosome-based nanovehicles as RNA interference therapeutic carriers. Nanomedicine. 2017;12(21):2653–75.
Mahmoudi E, Fitzsimmons C, Geaghan MP, Weickert CS, Atkins JR, Wang X, et al. Circular RNA biogenesis is decreased in postmortem cortical gray matter in schizophrenia and may alter the bioavailability of associated miRNA. Neuropsychopharmacology. 2019;44(6):1043–54.
Makarova JA, Shkurnikov MU, Wicklein D, Lange T, Samatov TR, Turchinovich AA, et al. Intracellular and extracellular microRNA: an update on localization and biological role. Prog Histochem Cytochem. 2016;51(3–4):33–49.
Mangul S, Yang HT, Strauli N, Gruhl F, Porath HT, Hsieh K, et al. ROP: dumpster diving in RNA-sequencing to find the source of 1 trillion reads across diverse adult human tissues. Genome Biol. 2018;19(36):1–12.
Margis R, Margis R, Rieder CR. Identification of blood microRNAs associated to Parkinson’s disease. J Biotechnol. 2011;152(3):96–101.
Marques TM, Kuiperij HB, Bruinsma IB, Van Rumund A, Aerts MB, Esselink RA, et al. MicroRNAs in cerebrospinal fluid as potential biomarkers for Parkinson’s disease and multiple system atrophy. Mol Neurobiol. 2017;54(10):7736–45.
Martins M, Rosa A, Guedes LC, Fonseca BV, Gotovac K, Violante S, et al. Convergence of miRNA expression profiling, α-synuclein interaction and GWAS in Parkinson’s disease. PLoS ONE. 2011;6(10):e25443.
McIntyre LM, Lopiano KK, Morse AM, Amin V, Oberg AL, Young LJ, et al. RNAseq: technical variability and sampling. BMC Genomics. 2011;12:293.
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88–100.
McMillan KJ, Murray TK, Bengoa-Vergniory N, Cordero-Llana O, Cooper J, Buckley A, et al. Loss of microRNA-7 regulation leads to α-synuclein accumulation and dopaminergic neuronal loss in vivo. Mol Ther. 2017;25(10):2404–14.
Medley JC, Panzade G, Zinovyeva AY. microRNA strand selection: Unwinding the rules. WIREs RNA. 2021;12:
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–8.
Miñones-Moyano E, Porta S, Escaramís G, Rabionet R, Iraola S, Kagerbauer B, et al. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet. 2011;20(15):3067–78.
Mo M, Xiao Y, Huang S, Cen L, Chen X, Zhang L, et al. MicroRNA expressing profiles in A53T mutant alpha-synuclein transgenic mice and Parkinsonian. Oncotarget. 2017;8(1):15.
Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435(7044):903–10.
Nair L, Chung H, Basu U. Regulation of long non-coding RNAs and genome dynamics by the RNA surveillance machinery. Nat Rev Mol Cell Biol. 2020;21(3):123–36.
Neilsen CT, Goodall GJ, Bracken CP. IsomiRs–the overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012;28(11):544–9.
Ni Y, Huang H, Chen Y, Cao M, Zhou H, Zhang Y. Investigation of long non-coding RNA expression profiles in the substantia nigra of Parkinson’s disease. Cell Mol Neurobiol. 2017;37(2):329–38.
Nies YH, Mohamad Najib NH, Lim WL, Kamaruzzaman MA, Yahaya MF, Teoh SL. MicroRNA Dysregulation in Parkinson's Disease: A Narrative Review. Front Neurosci. 2021;15:
Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463(7280):457–63.
Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 2016;17(2):83–96.
O’Brien J, Hayder H, Zayed Y, Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne). 2018;9:402.
O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol. 2020;21(10):585–606.
Oikonomopoulos S, Bayega A, Fahiminiya S, Djambazian H, Berube P, Ragoussis J. Methodologies for transcript profiling using long-read technologies. Front Genet. 2020;11:606.
Oikonomopoulos S, Wang YC, Djambazian H, Badescu D, Ragoussis J. Benchmarking of the Oxford Nanopore MinION sequencing for quantitative and qualitative assessment of cDNA populations. Sci Rep. 2016;6(1):1–13.
Okamura K, Phillips M, Tyler D, Duan H, Chou AT, Lai ET. The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat Struct Mol Biol. 2008;2009(15):354–63. https://doi.org/10.1038/nsmb.1409.
Oshlack A, Wakefield MJ. Transcript length bias in RNA-seq data confounds systems biology. Biol Direct. 2009;4:14.
Ozdilek B, Demircan B. Serum microRNA expression levels in Turkish patients with Parkinson’s disease. Int J Neurosci. 2020. https://doi.org/10.1080/00207454.2020.1784165.
Pallàs M. Senescence-accelerated mice P8: a tool to study brain aging and Alzheimer's disease in a mouse model. International Scholarly Research Notices. 2012;2012.
Pistono C, Bister N, Stanová I, Malm T. Glia-derived extracellular vesicles: role in central nervous system communication in health and disease. Front Cell Dev Biol. 2020;8:623771.
Plescia OJ, Palczuk NC, Cora-Figueroa E, Mukherjee A, Braun W. Production of antibodies to soluble RNA (sRNA). Proc Natl Acad Sci USA. 1965;54(4):1281.
Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136(4):629–41.
Porro C, Panaro MA, Lofrumento DD, Hasalla E, Trotta T. The multiple roles of exosomes in Parkinson’s disease: an overview. Immunopharmacol Immunotoxicol. 2019;41(4):469–76.
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30(12):1591–601.
Prehn JH, Jirström E. Angiogenin and tRNA fragments in Parkinson’s disease and neurodegeneration. Acta Pharmacol Sin. 2020;41(4):442–6.
Qin LX, Tan JQ, Zhang HN, Tang JG, Jiang B, Shen XM, et al. Preliminary study of hsa-mir-626 change in the cerebrospinal fluid in Parkinson’s disease. Neurol India. 2021;69(1):115.
Qiu G, Zheng G, Ge M, Wang J, Huang R, Shu Q, et al. Mesenchymal stem cell-derived extracellular vesicles affect disease outcomes via transfer of microRNAs. Stem Cell Res Ther. 2018;9(1):1–9.
Qiu W, Guo X, Lin X, Yang Q, Zhang W, Zhang Y, et al. Transcriptome-wide piRNA profiling in human brains of Alzheimer’s disease. Neurobiol Aging. 2017;57:170–7.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47–62.
Rapaport F, Khanin R, Liang Y, Pirun M, Krek A, Zumbo P, et al. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013;14:3158.
Ravanidis S, Bougea A, Papagiannakis N, Koros C, Simitsi AM, Pachi I, et al. Validation of differentially expressed brain-enriched microRNAs in the plasma of PD patients. Ann Clin Transl Neurol. 2020;7(9):1594–607.
Ravanidis S, Bougea A, Papagiannakis N, Maniati M, Koros C, Simitsi AM, et al. Circulating brain-enriched microRNAs for detection and discrimination of idiopathic and genetic Parkinson’s disease. Mov Disord. 2020;35(3):457–67.
Ravanidis S, Bougea A, Karampatsi D, Papagiannakis N, Maniati M, Stefanis L, et al. Differentially expressed circular RNAs in peripheral blood mononuclear cells of patients with Parkinson’s disease. Mov Disord. 2021;36(5):1170–9.
Ro S, Park C, Sanders KM, McCarrey JR, Yan W. Cloning and expression profiling of testis-expressed microRNAs. Develop Biol. 2007;311(2):592–602.
Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22.
Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008;18(10):505–16.
Roy J, Mallick B. Investigating piwi-interacting RNA regulome in human neuroblastoma. Genes Chromosom Cancer. 2018;57(7):339–49.
Roy J, Sarkar A, Parida S, Ghosh Z, Mallick B. Small RNA sequencing revealed dysregulated piRNAs in Alzheimer’s disease and their probable role in pathogenesis. Mol BioSyst. 2017;13(3):565–76.
Rybak-Wolf A, Stottmeister C, Glažar P, Jens M, Pino N, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58(5):870–85.
Salta E, De Strooper B. Noncoding RNAs in neurodegeneration. Nat Rev Neurosci. 2017;18(10):627.
Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci. 1976;73(11):3852–6.
Schulz J, Takousis P, Wohlers I, Itua IO, Dobricic V, Rücker G, et al. Meta-analyses identify differentially expressed microRNAs in Parkinson’s disease. Ann Neurol. 2019;85(6):835–51.
Schulze M, Sommer A, Plötz S, Farrell M, Winner B, Grosch J, et al. Sporadic Parkinson’s disease derived neuronal cells show disease-specific mRNA and small RNA signatures with abundant deregulation of piRNAs. Acta Neuropathol Commun. 2018;6(1):1–18.
Schwienbacher C, Foco L, Picard A, Corradi E, Serafin A, Panzer J, et al. Plasma and white blood cells show different miRNA expression profiles in Parkinson’s disease. J Mol Neurosci. 2017;62(2):244–54.
Seki M, Katsumata E, Suzuki A, Sereewattanawoot S, Sakamoto Y, Mizush-ima-Sugano J, et al. Evaluation and application of RNA-Seq by MinION. DNA Res. 2018;26(1):55–65.
Seppi K, Ray Chaudhuri K, Coelho M, Fox S, Katzenschlager R, Perez Lloret S, the collaborators of the Parkinson’s Disease Update on Non-Motor Symptoms Study Group on behalf of the Movement Disorders Society Evidence-Based Medicine Committee, et al. Update on treatments for nonmotor symptoms of Parkinson’s disease-an evidence-based medicine review. Mov Disord. 2019;34(2):180–98.
Sessegolo C, Cruaud C, Da Silva C, Cologne A, Dubarry M, Derrien T, et al. Transcriptome profiling of mouse samples using nanopore sequencing of cDNA and RNA molecules. Sci Rep. 2019;9:14908.
Shamsuzzama KL, Nazir A. Modulation of α-synuclein expression and associated effects by MicroRNAlet-7 in transgenic C. elegans. Front Mol Neurosci. 2017;13(10):328.
Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351(6271):391–6.
Sharon D, Tilgner H, Grubert F, Snyder M. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol. 2013;31:1009–14.
Shen L, Wang C, Chen L, Wong G. Dysregulation of MicroRNAs and PIWI-interacting RNAs in a Caenorhabditis elegans Parkinson’s disease model overexpressing human α-synuclein and influence of tdp-1. Front Neurosci. 2021;15:134.
Shi H, Zhou Y, Jia E, Pan M, Bai Y, Ge Q. Bias in RNA-seq library preparation: current challenges and solutions. Biomed Res Int. 2021;2021:6647597.
Shu Y, Qian J, Wang C. Aberrant expression of microRNA-132-3p and microRNA-146a-5p in Parkinson’s disease patients. Open Life Sciences. 2020;15(1):647–53.
Siddika T, Heinemann IU. Bringing MicroRNAs to Light. Methods for MicroRNA quantification and visualization in live cells. Front Bioeng Biotechnol. 2021;8:1534.
Slonim DK, Yanai I. Getting started in gene expression microarray analysis. PLoS Comput Biol. 2009;5(10):e1000543.
Sonntag KC. MicroRNAs and deregulated gene expression networks in neurodegeneration. Brain Res. 2010;1338:48–57.
Soreq L, Guffanti A, Salomonis N, Simchovitz A, Israel Z, Bergman H, et al. Long non-coding RNA and alternative splicing modulations in Parkinson’s leukocytes identified by RNA sequencing. PLoS Comput Biol. 2014;10(3):e1003517.
Soreq L, Salomonis N, Bronstein M, Greenberg DS, Israel Z, Bergman H, et al. Small RNA sequencing-microarray analyses in Parkinson leukocytes reveal deep brain stimulation-induced splicing changes that classify brain region transcriptomes. Front Mol Neurosci. 2013;6:10.
Stark R, Grzelak M, Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet. 2019;20(11):631–56.
Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021;22:96–118.
Steijger T, Abril JF, Engstrom PG, Kokocinski F, Consortium R, Hubbard TJ, et al. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods. 2013;10:1177–84.
Steiner JA, Angot E, Brundin P. A deadly spread: cellular mechanisms of α-synuclein transfer. Cell Death Differ. 2011;18(9):1425–33.
Su Z, Wilson B, Kumar P, Dutta A. Noncanonical roles of tRNAs: tRNA fragments and beyond. Annu Rev Genet. 2020;54:47–69.
Sun W, Samimi H, Gamez M, Zare H, Frost B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat Neurosci. 2018;21(8):1038–48.
Sun YH, Wang A, Song C, Shankar G, Srivastava RK, Au KF. Single-molecule long-read sequencing reveals a conserved intact long RNA profile in sperm. Nat Commun. 2021;12(1):1–12.
Sun YM, Chen YQ. Principles and innovative technologies for decrypting noncoding RNAs: from discovery and functional prediction to clinical application. J Hematol Oncol. 2020;13:109.
Titze-de-Almeida SS, Soto-Sánchez C, Fernandez E, Koprich JB, Brotchie JM, Titze-de-Almeida R. The promise and challenges of developing miRNA-based therapeutics for Parkinson’s disease. Cells. 2020;9(4):841.
Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotech. 2013;31:46–53.
Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. 2019;20:5–20.
Veneziano D, Di Bella S, Nigita G, Lagana A, Ferro A, Croce CM. Noncoding RNA: current deep sequencing data analysis approaches and challenges. Hum Mutat. 2016;37:1283–98.
Villar-Menéndez I, Porta S, Buira SP, Pereira-Veiga T, Díaz-Sánchez S, Albasanz JL, et al. Increased striatal adenosine A2A receptor levels is an early event in Parkinson’s disease-related pathology and it is potentially regulated by miR-34b. Neurobiol Dis. 2014;69:206–14.
Wang P, Ma H, Zhang Y, Zeng R, Yu J, Liu R, et al. Plasma exosome-derived MicroRNAs as novel biomarkers of traumatic brain injury in rats. Int J Med Sci. 2020;17(4):437–48.
Wang Q, Lee I, Ren J, Ajay SS, Lee YS, Bao X. Identification and Functional Characterization of tRNA-derived RNA Fragments (tRFs) in Respiratory Syncytial Virus Infection. Mol Ther. 2013;21(2):368–79.
Wang Q, Han CL, Wang KL, Sui YP, Li ZB, Chen N. Integrated analysis of exosomal lncRNA and mRNA expression profiles reveals the involvement of lnc-MKRN2-42: 1 in the pathogenesis of Parkinson’s disease. CNS Neurosci Ther. 2020;26(5):527–37.
Wang R, Yang Y, Wang H, He Y, Li C. MiR-29c protects against inflammation and apoptosis in Parkinson’s disease model in vivo and in vitro by targeting SP1. Clin Exp Pharmacol Physiol. 2020;47:372–82.
Wang R, Li Q, He Y, Yang Y, Ma Q, Li C. miR-29c-3p inhibits microglial NLRP3 inflammasome activation by targeting NFAT5 in Parkinson’s disease. Genes Cells. 2020;25(6):364–74.
Wang R, Yao J, Gong F, Chen S, He Y, Hu C, et al. miR-29c-3p regulates TET2 expression and inhibits autophagy process in Parkinson’s disease models. Genes Cells. 2021;26(9):684–97.
Wei CW, Luo T, Zou SS, Wu AS. The role of long noncoding RNAs in central nervous system and neurodegenerative diseases. Front Behav Neurosci. 2018;12:175.
Weirather JL, de Cesare M, Wang Y, Piazza P, Sebastiano V, Wang XJ, et al. Comprehensive comparison of pacific biosciences and oxford nanopore technologies and their applications to transcriptome analysis. F1000Res. 2017;3(6):100.
Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, et al. Genomewide analysis of Drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014;9(5):1966–80.
Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75(5):855–62.
Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8(3):270–9.
Williams DR, Litvan I. Parkinsonian syndromes. Continuum (Minneap Minn). 2013;19:1189–2122.
Wu J, Li H, He J, Tian X, Luo S, Li J, et al. Downregulation of microRNA-9-5p promotes synaptic remodeling in the chronic phase after traumatic brain injury. Cell Death Dis. 2021;12:9.
Wu Y, Zhao T, Deng R, Xia X, Li B, Wang X. A study of differential circRNA and lncRNA expressions in COVID-19-infected peripheral blood. Sci Rep. 2021;11:7991.
Wu YY, Kuo HC. Functional roles and networks of non-coding RNAs in the pathogenesis of neurodegenerative diseases. J Biomed Sci. 2020;27:1–23.
Xie S, Niu W, Xu F, Wang Y, Hu S, Niu C. Differential expression and significance of miRNAs in plasma extracellular vesicles of patients with Parkinson’s disease. Int J Neurosci. 2020. https://doi.org/10.1080/00207454.2020.1835899.
Xie Y, Yao L, Yu X, Ruan Y, Li Z, Guo J. Action mechanisms and research methods of tRNA-derived small RNAs. Signal Transduct Target Ther. 2020;5(1):1–9.
Xin C, Liu J. Long Non-coding RNAs in Parkinson’s Disease. Neurochem Res. 2021;46:1031–42.
Xiong H, Shen J, Chen Z, Yang J, Xie B, Jia Y, et al. H19/let-7/Lin28 ceRNA network mediates autophagy inhibiting epithelial-mesenchymal transition in breast cancer. Int J Oncol. 2020;56(3):794–806.
Xu K, Zhang Y, Li J. Expression and function of circular RNAs in the mammalian brain. Cell Mol Life Sci. 2021;78(9):4189–200.
Xu W, Zhang L, Geng Y, Liu Y, Zhang N. Long noncoding RNA GAS5 promotes microglial inflammatory response in Parkinson's disease by regulating NLRP3 pathway through sponging miR-223-3p. Int Immunopharmacol. 2020;85:
Yang JS, Phillips MD, Betel D, Mu P, Ventura A, Siepel AC, et al. Widespread regulatory activity of vertebrate microRNA* species. RNA. 2011;17:312–26.
Yang Q, Li F, He A, Yang BB. Circular RNAs: expression, localization, and therapeutic potentials. Mol Ther. 2021;29(5):1683–702.
Yang S, Yang H, Luo Y, Deng X, Zhou Y, Hu B. Long non-coding RNAs in neurodegenerative diseases. Neurochem Int. 2021;148:105096.
Yang Y, Ye Y, Su X, He J, Bai W, He X. MSCs-derived exosomes and neuroinflammation, neurogenesis and therapy of traumatic brain injury. Front Cell Neurosci. 2017;11:55.
Yang Z, Li T, Cui Y, Li S, Cheng C, Shen B, et al. Elevated plasma microRNA-105-5p level in patients with idiopathic Parkinson’s disease: a potential disease biomarker. Front Neurosci. 2019;13:218.
Yang Z, Li T, Li S, Wei M, Qi H, Shen B, et al. Altered expression levels of microRNA-132 and Nurr1 in peripheral blood of Parkinson’s disease: potential disease biomarkers. ACS Chem Neurosci. 2019;10(5):2243–9.
Yao H, Ma R, Yang L, Hu G, Chen X, Duan M, et al. MiR-9 promotes microglial activation by targeting MCPIP1. Nat Commun. 2014;5:4386.
Yao RW, Wang Y, Chen LL. Cellular functions of long noncoding RNAs. Nat Cell Biol. 2019;21(5):542–51.
Yao Y, Qu M, Li G, Zhang F, Rui H. Circulating exosomal miRNAs as diagnostic biomarkers in Parkinson’s disease. Eur Rev Med Pharmacol Sci. 2018;22:5278–83.
Yim WWY, Mizushima N. Lysosome biology in autophagy. Cell Discov. 2020;6:6.
You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci. 2015;18(4):603–10.
Yu M, Lu B, Zhang J, Ding J, Liu P, Lu Y. tRNA-derived RNA fragments in cancer: current status and future perspectives. J Hematol Oncol. 2020;13:121.
Zhang LW, Zhang J, Wang K, Wang RB. Serum microRNA-30c-5p and microRNA-373 expressions as potential biomarkers for Parkinson’s disease. All Life. 2020;13(1):194–200.
Zhang M, Yang H, Chen Z, Hu X, Wu T, Liu W. Long noncoding RNA X-inactive-specific transcript promotes the secretion of inflammatory cytokines in LPS stimulated astrocyte cell via sponging miR-29c-3p and regulating nuclear factor of activated T cell 5 expression. Front Endocrinol (Lausanne). 2021;12:573143.
Zhang P, Zhang XO, Jiang T, Cai L, Huang X, Liu Q, et al. Comprehensive identification of alternative back-splicing in human tissue transcriptomes. Nucleic Acids Res. 2020;48(4):1779–89.
Zhang S, Li H, Zheng L, Li H, Feng C, Zhang W. Identification of functional tRNA-derived fragments in senescence-accelerated mouse prone 8 brain. Aging (Albany NY). 2019;11(22):10485.
Zhang Z, Cheng Y. miR-16–1 promotes the aberrant α-synuclein accumulation in parkinson disease via targeting heat shock protein 70. Sci World J. 2014;2014:938348.
Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE. 2014;9(1):e78644.
Zhou Y, Gu C, Li J, Zhu L, Huang G, Dai J, et al. Aberrantly expressed long noncoding RNAs and genes in Parkinson’s disease. Neuropsychiatr Dis Treat. 2018;14:3219.
Zhuang F, Fuchs RT, Robb GB. Small RNA expression profiling by high-throughput sequencing: implications of enzymatic manipulation. J Nucleic Acids. 2012;2012:360358.
Zou J, Guo Y, Wei L, Yu F, Yu B, Xu A. Long noncoding RNA POU3F3 and α-synuclein in plasma L1CAM exosomes combined with β-glucocerebrosidase activity: potential predictors of Parkinson’s disease. Neurotherapeutics. 2020;17(3):1104–19.
Acknowledgements
All figures are created by Biorender.com. A sincere thank you for Meng-Ling Chen who provided Biorender.com for figures drawing and Teh-Cheng Wang for his assistance in literature of the experiments of ncRNA. We also thank Chia-Lang Hsu of the Department of Medical Research, National Taiwan University Hospital for great advice in drawing the Figure 2.
Funding
This article is funded by National Taiwan University Hospital and grant from the Ministry of Science and Technology (Taiwan R.O.C.) under grant numbers: MOST 109-2314-B-002-120-MY3.
Author information
Authors and Affiliations
Contributions
MCK assisted in literature review, drafting the manuscript, composing the figures and reviewing the final paper. SCHL assisted in literature review, drafting the part of Techniques of non-coding RNA detection and composing the table and the figure. YFH assisted in literature review and drafting the introduction of miRNA. RMW contributed to the concept and design of this article, assessed the literatures and revised the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have agreed to publish this manuscript. All materials and images are original. No consent needs to declare.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Appendix. Pros and cons of RNA sequencing platforms.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kuo, MC., Liu, S.CH., Hsu, YF. et al. The role of noncoding RNAs in Parkinson’s disease: biomarkers and associations with pathogenic pathways. J Biomed Sci 28, 78 (2021). https://doi.org/10.1186/s12929-021-00775-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-021-00775-x