
Abstract
The neuronal ceroid lipofuscinoses (NCLs), commonly known as Batten disease, belong to a family of neurological disorders that cause blindness, seizures, loss of motor function and cognitive ability, and premature death. There are 13 different subtypes of NCL that are associated with mutations in 13 genetically distinct genes (CLN1-CLN8, CLN10-CLN14). Similar clinical and pathological profiles of the different NCL subtypes suggest that common disease mechanisms may be involved. As a result, there have been many efforts to determine how NCL proteins are connected at the cellular level. A main driving force for NCL research has been the utilization of mammalian and non-mammalian cellular models to study the mechanisms underlying the disease. One non-mammalian model that has provided significant insight into NCL protein function is the social amoeba Dictyostelium discoideum. Accumulated data from Dictyostelium and mammalian cells show that NCL proteins display similar localizations, have common binding partners, and regulate the expression and activities of one another. In addition, genetic models of NCL display similar phenotypes. This review integrates findings from Dictyostelium and mammalian models of NCL to highlight our understanding of the molecular networking of NCL proteins. The goal here is to help set the stage for future work to reveal the cellular mechanisms underlying the NCLs.
Similar content being viewed by others


Background
Neuronal ceroid lipofuscinosis
The neuronal ceroid lipofuscinoses (NCLs), commonly known as Batten disease, are devastating forms of neurodegeneration that affect the global population [1]. Mutations have been documented in 13 genetically distinct genes (CLN1-CLN8, CLN10-CLN14), each of which causes a specific subtype of the disease (e.g., mutations in CLN3 cause CLN3 disease) [2, 3]. While the NCLs affect all ages and ethnicities, the disease is recognized as the most common form of childhood neurodegeneration [1]. Clinical manifestations of the NCLs include seizures, vision loss, reduced mental capacity, decline in motor function, and a shortened lifespan [4]. At the cellular level, ceroid lipofuscin accumulates in neurons, as well as other cell types outside of the central nervous system, due to aberrant lysosomal function [5]. Unfortunately, there is currently no cure for the NCLs, in large part due to our poor understanding of the proteins associated with the disease. As a result, other than Brineura, which is an enzyme replacement therapy specific for only one subtype of the disease (CLN2 disease), there are currently no effective treatments to prevent or delay the NCLs [6].
Dictyostelium as a model organism for NCL research
Many animal and mammalian cell models have been used to study the pathological mechanisms underlying the NCLs [7, 8]. However, these systems present some limitations, such as the costs associated with maintaining small experimental animals (e.g., mice) and the limited number of phenotypes that can be screened in immortalized or patient-derived human cells. Not surprisingly, model organisms such as yeast, worm, fruit fly, and the social amoeba Dictyostelium discoideum have been used to study the roles of NCL proteins within the cell and in the context of a whole organism [8]. Collectively, this work has provided valuable insight into the localizations and functions of NCL proteins in human cells.
Dictyostelium discoideum, hereafter referred to simply as Dictyostelium, has emerged as an exceptional model system for not only studying the NCLs, but also a variety of other human neurological disorders [9, 10]. In addition to NCL proteins, Dictyostelium has been used to study the functions of proteins associated with Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, prion diseases, epilepsy, and lissencephaly [11,12,13,14,15,16]. The 34 Mb haploid genome of Dictyostelium encodes an estimated 12,500 proteins, which includes homologs of 11 of the 13 human NCL proteins [9]. The life cycle of Dictyostelium, which has been studied for over 85 years, is comprised of unicellular and multicellular phases that allow a diversity of fundamental cellular and developmental processes to be examined in rich detail [17]. During the unicellular growth phase, cells feed on a food source such as bacteria, and proliferate via mitosis. Once the food source is depleted, cells enter a 24-h multicellular developmental programme that concludes with the formation of a fruiting body. The fruiting body is composed a slender stalk that supports a mass of viable spores that germinate and restart the life cycle once a food source becomes available. In humans, diseases that affect the nervous system share common cellular features such as the accumulation of autophagic vacuoles and mitochondrial dysfunction. These processes, along with other fundamental cellular and developmental processes including cell proliferation, chemotaxis, and differentiation, can be thoroughly studied in Dictyostelium using robust and high-throughput assays [17, 18]. In addition, the metazoan-like behaviour of Dictyostelium cells ensures a high probability that findings from the organism can be successfully translated to human cells. However, as with any model organism, there are some limitations to consider. For example, Dictyostelium lacks a central nervous system and has a limited number of cell types, which may prevent the translation of findings to specific tissues or organs in mammalian systems. Despite these caveats, Dictyostelium provides exceptional opportunities to test hypotheses related to NCL protein function in a simple eukaryotic organism that can complement and possibly inform research in mammalian systems.
The molecular networking of NCL proteins
Since mutations in NCL proteins cause the accumulation of ceroid lipofuscin within cells, and result in broadly similar clinical manifestations between the different subtypes, the proteins are thought to have shared functions, regulate similar processes, and/or participate in shared or converging pathways [19]. In support of this hypothesis, model systems used to study the NCLs show common phenotypes [8]. At the cellular level, NCL proteins display similar localizations, have common binding partners, and regulate the expression and activities of one another. Understandably, much of this work was performed in mammalian models. However, as noted above, valuable insights have also been obtained from Dictyostelium. For example, many of the NCL protein homologs in Dictyostelium localize to the macropinocytic pathway, which is responsible for bringing fluid-phase material into the cell [20]. In addition, cell lines lacking NCL proteins display similar phenotypes [10]. This work, coupled with observations in mammalian models of NCL, suggests that NCL proteins are connected at the molecular level. These findings, and others, will be described in the sections below to help shed light on the networking of NCL proteins within the eukaryotic cell.
Main text
NCL proteins localize to intracellular trafficking pathways
Mammals
Early work in mammalian cells revealed similar subcellular localizations of some of the NCL proteins suggesting that a common cellular process may be disrupted in the NCLs [21]. Concerted research efforts have now placed the NCL proteins within intracellular trafficking pathways (e.g., endoplasmic reticulum, Golgi complex, lysosomes, vesicles), which is consistent with the lysosome dysfunction and ceroid lipofuscin accumulation observed in many cellular models of NCL (Fig. 1) [22]. Palmitoyl protein thioesterase 1 (PPT1)/CLN1, tripeptidyl peptidase 1 (TPP1)/CLN2, ceroid lipofuscinosis neuronal 5 (CLN5), cathepsin D (CTSD)/CLN10, and CTSF/CLN13 localize to the lysosome and function as soluble enzymes [23,24,25,26,27,28]. CLN5 and CTSD/CLN10 also localize extracellularly [25, 29]. CLN3 localizes to the Golgi complex and lysosomal membrane [30, 31]. Major facilitator superfamily domain-containing protein 8 (MFSD8)/CLN7 and ATPase cation-transporting 13A2 (ATP13A2)/CLN12 localize to the lysosomal membrane, while CLN6 and CLN8 are endoplasmic reticulum (ER) membrane proteins [32,33,34,35]. DnaJ homolog subfamily C member 5 (DNAJC5)/CLN4 and potassium channel tetramerization domain-containing protein 7 (KCTD7)/CLN14 are peripheral membrane proteins, while progranulin (PGRN)/CLN11 is secreted [36,37,38].

The localizations of NCL proteins in mammalian cells. Note that proteins present in more than one group are underlined
Dictyostelium
Nine of the thirteen human NCL proteins have clear homologs in Dictyostelium, which includes homologs of PPT1/CLN1 (Ppt1), TPP1/CLN2 (see next sentence), CLN3 (Cln3), DNAJC5/CLN4 (Dnajc5), CLN5 (Cln5), MFSD8/CLN7 (Mfsd8), CTSD/CLN10 (CtsD), PGRN/CLN11 (Grn), and ATP13A2/CLN12 (Kil2) [9]. Six proteins in Dictyostelium share sequence similarity with human TPP1/CLN2 (Tpp1A, Tpp1B, Tpp1C, Tpp1D, Tpp1E, Tpp1F) [39]. However, of the six proteins, Tpp1A and Tpp1B are closest in size to human TPP1/CLN2 (600 and 598 amino acids, respectively, compared to 563 amino acids) and have the highest sequence similarity. The expression of tpp1B is markedly higher than the other tpp1 genes during growth and the early stages of multicellular development [8]. tpp1F is also highly expressed during growth relative to tpp1A, tpp1C, tpp1D, and tpp1E. Upon starvation, the expression of tpp1B and tpp1F decrease dramatically reaching their lowest levels once the multicellular aggregate has formed and remain low during the mid-to-late stages of development. In contrast, the expression of tpp1A increases dramatically upon starvation, and remains high during the mid-to-late stages of multicellular development. During this time, the expression levels of tpp1B, tpp1C, tpp1D, tpp1E, and tpp1F are comparatively lower. Together, these expression profiles suggest that Tpp1B and Tpp1F may be the dominant Tpp1 proteins during growth, while Tpp1A may be the dominant Tpp1 protein during development. Since one TPP1/CLN2 protein has been identified in mammalian cells, it will be important for future work to reveal the true homolog of human TPP1/CLN2 in Dictyostelium. In addition, the Dictyostelium genome encodes several proteins that share similarity with human CTSF/CLN13 and KCTD7/CLN14 [8]. Specific proteins have been identified as the likely homologs of human CTSF/CLN13 and KCTD7/CLN14, however they have not been experimentally validated. Finally, the Dictyostelium genome does not encode homologs of human CLN6 or CLN8 [9]. Interestingly, these are the only ER-resident proteins among the family of NCL proteins. Current knowledge indicates that the Dictyostelium ER shares several functions with the ER in mammalian cells including calcium storage, lipid biosynthesis, and the unfolded protein response [40]. Furthermore, Dictyostelium is the only microorganism known to encode homologs of human calnexin and calreticulin [41]. So, it is somewhat surprising that Dictyostelium does not contain homologs of CLN6 and CLN8. Their absence in Dictyostelium could indicate a specialized role for these proteins in the ER of mammalian cells that is either non-existent or dispensable in Dictyostelium. These observations suggest that future work on CLN6 and CLN8 in mammalian cells may consider focusing on specialized or highly evolved ER-dependent processes that are not present in evolutionarily lower eukaryotes; processes that may or may not be cell type-dependent.
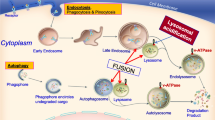
A proteomics-based analysis revealed that 8 of the 9 clear NCL protein homologs in Dictyostelium localize to the macropinocytic pathway, which is responsible for bringing fluid-phase material into the cell [8, 20]. In addition, some of the proteins that share similarity with human CTSF/CLN13 and KCTD7/CLN14 also localize to this pathway [8, 20]. More directed studies have also provided support for the localization of NCL proteins to intracellular trafficking pathways in Dictyostelium. For example, the Dictyostelium homologs of human TPP1/CLN2 (Tpp1A, Tpp1F), CLN3 (Cln3), MFSD8/CLN7 (Mfsd8), and CTSD/CLN10 (CtsD) all localize to the late endosome/lysosome suggesting they play a role in the digestion of ingested material [39, 42,43,44,45,46] (Fig. 2). In addition to the late endosome/lysosome, Tpp1F also localizes to the ER and Golgi complex indicating that the protein is likely synthesized in the ER, processed in the Golgi complex, and then trafficked to the lysosome [39]. Like Tpp1F, Tpp1B and Cln3 also localize to the Golgi complex [39, 47].

The networking of NCL proteins in Dictyostelium. (1) Material is taken up by the cell and incorporated into an endosome, which matures into a lysosome. Tpp1A, Tpp1F, Cln3, Mfsd8, and CtsD localize to the late endosome/lysosome. Tpp1F also localizes extracellularly as does Ppt1 and Tpp1B. (2) Tpp1B localizes to the Golgi complex and binds the Golgi pH regulator (GPHR) (in addition to extracellularly, see #1). Tpp1F localizes to the Golgi complex and endoplasmic reticulum (ER) (in addition to late endosome/lysosome and extracellularly, see #1) and binds the GPHR. Cln3 localizes to the Golgi complex (in addition to the late endosome/lysosome, see #1). Cln5 is glycosylated in the ER and then trafficked to the cell cortex and contractile vacuole (CV) system. (3) Cln3 localizes to the CV system (in addition to the late endosome/lysosome and Golgi complex, see #1 and #2). During starvation, loss of cln3 alters the intracellular activity of alpha-mannosidase (ManA), the expression of beta-glucosidase (gluA), the intracellular activity of GluA, and the expression of N-acetylglucosaminidase (nagA). Cln5 interacts with ManA, GluA, and NagA. Loss of cln3 alters the expression of nagB and the secretion of NagB during starvation. Finally, loss of cln3 alters the extracellular activities of ManA, GluA, and Nag during starvation. (4) Cln5 is secreted during starvation. Secretion of Cln5 is regulated by autophagic mechanisms (i.e., autophagy inhibition decreases secretion) and Cln3 (i.e., cln3-deficiency alters secretion). Inside the cell, Cln5 interacts with Tpp1B. (5) Loss of cln3 alters the intracellular and extracellular activity of Tpp1 during starvation. cln3-deficiency alters the expression of tpp1F and the secretion of Tpp1F during starvation. Loss of cln3 increases the expression of tpp1A during hypertonic stress and alters the expression of tpp1D and grn during starvation. cln3-deficiency alters the expression of ctsD, the intracellular and extracellular activity of CtsD, and the secretion of CtsD during starvation. (6) Loss of cln3 alters the expression of aprA and the intracellular amount of AprA during starvation. cln3-deficiency alters the secretion of AprA during growth and starvation. Loss of cln3 alters the secretion of CfaD during growth and the amount of CadA in conditioned starvation buffer. Cln5 interacts with AprA, CfaD, CadA, and CtsD. (7) Loss of cln3 alters the expression of cprD, cprG, and bip2 (luminal-binding protein 2, DDB0233663) during starvation. cln3-deficiency increases the expression of cprE during hypotonic stress. Loss of cln3 alters the secretion of CprD, CprE, CprG, and Bip2 during starvation. Cln5 interacts with CprD, CprE, CprG, and Bip2. (8) Loss of cln3 alters the expression of cprF during hypotonic stress and starvation. cln3-deficiency alters the secretion of CprA, CprB, and CprF during starvation. (9) Like Cln5, Mfsd8 interacts with CtsD, CadA, and CfaD (see #6). Like cln3− cells (see #4 and #5), loss of mfsd8 alters the secretion of Cln5 and CtsD during starvation
Cln5 is glycosylated in Dictyostelium, and not surprisingly, the protein is detected in the ER [26, 48] (Fig. 2). Cln5 also localizes to the cell cortex and contractile vacuole (CV) system [26, 48]. During the early stages of multicellular development, Cln5 is detected extracellularly, which aligns with observations in mammalian cells [25, 26, 48,49,50]. The secretion of Cln5 in Dictyostelium appears to be regulated by autophagic mechanisms and Cln3, which also localizes to the CV system [43, 45, 47, 48]. While the main function of the CV system in Dictyostelium is osmoregulation, the organelle has also been linked to unconventional protein secretion [51]. Interestingly, experimental evidence suggests that regulated intracellular transport might occur between the CV and endo-lysosomal systems, which provides further support for the localization of NCL proteins to intracellular trafficking pathways [52,53,54]. Consistent with the localization of GFP-Cln3 to the CV system in Dictyostelium, hypertonic stress dramatically increases the expression of human myc-tagged CLN3 mRNA and protein in baby hamster kidney cells [55]. Finally, like Cln5, Ppt1, Tpp1B, Tpp1F, and CtsD are also secreted during Dictyostelium development suggesting that NCL proteins may play an important role outside the cell [42, 47, 56].
Together, these findings show that homologs of NCL proteins in Dictyostelium may play an important role in regulating intracellular trafficking mechanisms. These findings are consistent with observations in mammalian systems and support the notion that aberrant processing of intracellular material is a key component of NCL pathology.
Protein interactomes shed light on the interactions between NCL proteins
Mammals
Modern omics technologies have the potential to provide significant new insight into protein interactomes, potential biomarkers, and biological pathways affected by the NCLs [57]. For example, proteomics-based analyses in SH-SY5Y human neuroblastoma cells revealed that PPT1/CLN1, CLN3, and CLN5 interact with 23, 58, and 31 proteins, respectively [58, 59]. A comparison of the three interactomes revealed several proteins that may serve important functions in the NCL-causing pathway (Table 1). PPT1/CLN1 and CLN3 share four common interactors (ATP1A1, DBH, SLC25A1, SLC25A13). Three of those proteins (DBH, SLC25A1, SLC25A13) also interact with CLN5. DBH (dopamine β-hydroxylase) is a copper-containing oxygenase that catalyzes the conversion of dopamine to norepinephrine [60]. Not surprisingly, altered dopamine signalling has been linked to the NCLs, as well as other neurological disorders including Alzheimer’s disease, Parkinson’s disease, and schizophrenia [61,62,63,64,65]. Mutations in SLC25A1 are associated with impaired neuromuscular transmission [66]. SLC25A1 encodes a mitochondrial tricarboxylate transport protein that regulates the exchange of mitochondrial citrate for cytosolic malate and contributes to fatty acid and sterol biosynthesis [67]. Intriguingly, mutations in NCL genes alter lipid homeostasis [68,69,70]. Finally, SLC25A13 is a calcium-binding carrier protein that catalyzes the exchange of cytoplasmic glutamate with aspartate across the inner mitochondrial membrane [71, 72]. Mutations in SLC25A13 cause citrullinemia, an autosomal recessive disease characterized by elevated levels of ammonia in the blood, which leads to neuropsychiatric symptoms such as abnormal behaviour, memory loss, seizures, and even death [73]. Importantly, mitochondrial dysfunction has been reported in the NCLs and there is evidence supporting altered ammonia homeostasis in at least one subtype of the disease (e.g., CLN3 disease) [74,75,76]. Together, these findings suggest that further investigation into the involvement of DBH, SLC25A1, and SLC25A13 in NCL pathology is warranted.
Proteomics-based analyses have also provided evidence that NCL proteins interact with each other (Table 2). For example, PPT1/CLN1 was shown to interact with CTSD/CLN10 in SH-SY5Y human neuroblastoma cells, possibly due to their common function as lysosomal enzymes [23, 27, 59]. In addition, co-immunoprecipitation and in vitro binding assays revealed that CLN5 polypeptides interact directly with TPP1/CLN2 and CLN3 in COS-1 cells [77]. These interactions were later confirmed in a study that also identified PPT1/CLN1, CLN6, and CLN8 as CLN5-interactors in mouse brain extract, COS-1 cells, and HeLa cells [78]. These findings are significant since they highlight a conserved interaction between CLN5 and these NCL proteins across multiple models. Interestingly, work has shown that the intracellular trafficking of CLN5 in HeLa cells is regulated by PPT1/CLN1 and may involve F1-ATPase, a common interactor of PPT1/CLN1 and CLN5 [78]. The association of CLN5 with PPT1/CLN1 and TPP1/CLN2 could reflect their common function as lysosomal enzymes, albeit with distinctly different biological activities [23, 24, 26] (Fig. 1). The interaction between CLN3 and CLN5 could be due to their common link to Rab7 and the retromer complex, while the binding of CLN5 to CLN6 and CLN8 could indicate that these proteins play a role in processing CLN5 to its mature form in the ER [84,85,86]. Finally, CTSD/CLN10 has been shown to interact with PGRN/CLN11 in HEK293T cells, which could possibly be explained by the observations that both proteins are secreted [29, 38, 79, 80] (Table 2, Fig. 1). Together, these results suggest that the networking of NCL proteins within the cell may involve direct interactions with each other.
Dictyostelium
Recent work in Dictyostelium reported the Cln5 and Mfsd8 protein interactomes [26, 46]. In addition to identifying the interactors of each protein, these studies also provided insight into the molecular interactions between NCL protein homologs in Dictyostelium. For example, this work showed that Cln5 interacts with Tpp1B and CtsD [26] (Fig. 2). In mammalian cells, CLN5 interacts with TPP1/CLN2, and increased TPP1/CLN2 activity has been observed in fibroblasts from a CLN5 disease patient [77, 78] (Table 2, Table 3). In addition to Cln5, CtsD was also reported to bind to Mfsd8 in Dictyostelium, thus establishing a molecular connection between Cln5, Mfsd8, and CtsD in this model organism [46] (Fig. 2). The binding of MFSD8/CLN7 to CTSD/CLN10 has not been reported in mammalian cells but results from Dictyostelium suggest they may interact with each other.
As discussed previously, CLN5 interacts with CLN3 in mammalian cells [77, 78]. Several pieces of evidence also support a connection between Cln3 and Cln5 in Dictyostelium. During starvation, cln3-deficiency alters the expression of N-acetylglucosaminidase (nagA) and beta-glucosidase (gluA) [94] (Fig. 2). cln3-deficiency also alters the intracellular and extracellular activity of alpha-mannosidase (ManA) and beta-glucosidase (GluA) during starvation, as well as the extracellular activity of N-acetylglucosaminidase (Nag) [94]. Intriguingly, Cln5 interacts with ManA, GluA, and NagA, thus establishing a network involving Cln3, Cln5, and these lysosomal enzymes in Dictyostelium [26]. In support of these findings, recent work reported abnormal enzyme amounts in lysosomal fractions from cerebellar cells derived from a Cln3Δex7/8 mouse [90] (Table 3).
In Dictyostelium, cln3-deficiency increases cell proliferation during growth and reduces cell adhesion during the early stages of multicellular development [43, 45]. To help explain these phenotypes, previous work showed that cln3-deficiency alters the secretion of autocrine proliferation repressor A (AprA) and counting factor-associated protein D (CfaD) during growth, the expression and secretion of AprA during starvation, and the extracellular amount of calcium-dependent cell adhesion protein A (CadA) during starvation [43, 45, 94] (Fig. 2). The connection between Cln3 and Cln5 in Dictyostelium is strengthened by the finding that AprA, CfaD, and CadA have all been identified as Cln5-interactors [26]. As discussed earlier, CtsD was identified in both the Cln5 and Mfsd8 interactomes [26, 46]. Not surprisingly, CfaD and CadA were also identified as Mfsd8-interactors further supporting the molecular connection between Cln5 and Mfsd8 in Dictyostelium [46].
The Dictyostelium genome encodes several cysteine proteinases (Cpr) that share sequence similarity with human CTSF/CLN13. Three of these proteins, CprD, CprE, and CprG, were identified in the Cln5 interactome [26] (Fig. 2). In keeping with the connection between Cln3 and Cln5 in Dictyostelium, loss of cln3 alters the expression of cprD, cprF, and cprG during starvation and increases the expression of cprE and cprF during hypotonic stress [94, 95]. cln3-deficiency also alters the secretion of CprA, CprB, CprD, CprE, CprF, and CprG [47]. These results suggest that future research in mammalian models of NCL may consider examining the molecular interactions between CLN3, CLN5, and CTSF/CLN13.
In Dictyostelium, Cln5 interacts with luminal-binding protein 2 (Bip2, DDB0233663), whose expression and secretion are affected by the loss of cln3 during starvation [26, 47, 94] (Fig. 2). Bip2 is massively over-secreted by cln3− cells compared to WT cells during starvation and is one of the most significantly affected proteins [47]. Bip2 is the Dictyostelium homolog of human glucose-regulated protein 78 (GRP78)/BiP, which plays an important role in cancer progression and neurodegeneration [96]. Intriguingly, CLN3 interacts with GRP78/BiP in COS-1 cells [97]. Together, this research has set the stage for future work in mammalian models of NCL to explore the molecular connection between CLN3 and CLN5.
Finally, as discussed above, DBH, SLC25A1, and SLC25A13 all interact with PPT1/CLN1, CLN3, and CLN5 in SH-SY5Y human neuroblastoma cells [58, 59]. While the Dictyostelium genome does not encode a homolog of DBH, it does encode several homologs of human SLC25A1 and SLC25A13 (www.dictybase.org). Homologs of SLC25A1 and SLC25A13 have been identified as Mfsd8-interactors in Dictyostelium (mitochondrial substrate carrier family protein, McfG, McfN, McfT, McfZ) [46]. In addition, several Mcf proteins are differentially expressed in cln3− cells compared to WT during starvation (McfC, McfF, McfG, McfM, McfR) [94]. Therefore, Dictyostelium presents an excellent system to dissect the roles of SLC25A1 and SLC25A13 in NCL biologys.
Loss or mutation of individual NCL genes affects other NCL genes/proteins
Several studies in Dictyostelium and mammalian models of NCL have shown that loss or mutation of a single NCL gene can affect the expression, amounts, and/or enzymatic activities of other NCL genes/proteins. These studies will be described in the sections below to highlight the links between individual NCL genes and other NCL genes/proteins.
Loss of PPT1/CLN1 affects CTSD/CLN10
Brain tissues and cells from Ppt1/Cln1−/− mice display increased expression of CtsD/Cln10 [87] (Table 3). Loss of PPT1/CLN1 function in mice also disrupts the maturation of CTSD/CLN10 to its enzymatically active form, which leads to an accumulation of undegraded cargo in the lysosome [87]. These observations are further supported by the finding that CTSD/CLN10 interacts with PPT1/CLN1 in SH-SY5Y human neuroblastoma cells, which may reflect their common function as lysosomal enzymes [23, 27, 59] (Fig. 1, Table 2). Together, these results suggest a link between PPT1/CLN1 and CTSD/CLN10.
Loss of CLN3 affects PPT1/CLN1, TPP1/CLN2, CLN5, CTSD/CLN10, and PGRN/CLN11
Lysosomes isolated from the cortex of a Cln3Δex7/8 mouse brain, as well as fibroblasts from CLN3 disease patients, progressively accumulate autofluorescent ceroid and contain significantly reduced levels of PPT1/CLN1, which corelate with reduced PPT1/CLN1 enzymatic activity [89] (Table 3). The significance of these observations is strengthened by their detection in different model systems. Interestingly, a study published in parallel reported an increased amount and activity of PPT1/CLN1 in lysosomes from cerebellar cells isolated from the same mouse model [90]. While the explanation for this discrepancy is not immediately clear, it may reflect a cell type dependent effect of mutated Cln3 on the expression and activity of PPT1/CLN1 in mice (e.g., cortex vs. cerebellum). Along with PPT1/CLN1, Schmidtke et al. [90] also reported an increased amount of TPP1/CLN2 in lysosomal fractions from cerebellar cells derived from a Cln3Δex7/8 mouse. Results from Dictyostelium are consistent with these observations since loss of cln3 alters intracellular and extracellular Tpp1 activity during starvation [94] (Fig. 2). cln3-deficiency also alters the expression of tpp1A during hypertonic stress, the expression of tpp1D during starvation, and the expression and secretion of Tpp1F during starvation [47, 94, 95]. Therefore, data from Dictyostelium and mammals support a molecular interaction between PPT1/CLN1, TPP1/CLN2, and CLN3, which may be explained the proposed role of CLN3 in the trafficking of lysosomal enzymes [85, 90].
Lysosomal fractions from cerebellar cells derived from a Cln3Δex7/8 mouse also display decreased amounts of CLN5 and CTSD/CLN10, as well as other lysosomal enzymes such as hexosaminidase A, hexosaminidase B, and alpha-mannosidase [90] (Table 3). In baby hamster kidney cells, hypertonic stress increases the expression of human myc-tagged CLN3 mRNA and protein [55]. Under these conditions, human myc-tagged CLN3 co-localizes with CTSD/CLN10 and decreases the activity of CTSD/CLN10 but has no effect on the amount of CTSD/CLN10 protein [82] (Table 2). In Dictyostelium, loss of cln3 alters the expression of ctsD, the intracellular and extracellular activity of CtsD, and the secretion of Cln5 [48, 94] (Fig. 2). cln3-deficiency also alters the expression of the Dictyostelium homologs of hexosaminidase A and B (nagA and nagB) [94]. Intriguingly, Cln5 interacts with NagA and alpha-mannosidase, which are affected by the loss of CLN3 function in mice [26, 90]. Together, these results establish links between CLN3, CLN5, and CTSD/CLN10. Like PPT1/CLN1 and TPP1/CLN2, the connection between these proteins may be explained by the proposed role of CLN3 in the trafficking of lysosomal enzymes [85, 90]. Finally, loss of cln3 in Dictyostelium affects the expression of grn during starvation, which could be linked to the role of Cln3 in protein secretion (i.e., PGRN/CLN11 is secreted in mammalian cells) [47]. Importantly, this finding suggests there may be a molecular interaction between CLN3 and PGRN/CLN11 in mammalian cells.
Loss of DNAJC5/CLN4 affects PPT1/CLN1
In brain tissue obtained from CLN4 disease patients, mutations in DNAJC5/CLN4 increase the amount of PPT1/CLN1 but reduce its enzymatic activity causing it to mis-localize [81] (Table 2, Table 3). Consistent with these findings, PPT1/CLN1 isolated from HEK293T cell media was shown to de-palmitoylate DNAJC5/CLN4 isolated from mouse brain [81]. While the link between the two proteins is not entirely clear, the data suggest that PPT1/CLN1 may regulate DNAJC5/CLN4 function through de-palmitoylation. Also, since DNAJC5/CLN4 has been linked to the trafficking of synaptic vesicles, it is possible that DNAJC5/CLN4 may regulate the trafficking of PPT1/CLN1 [36]. While additional work is required to clarify the link between DNAJC5/CLN4 and PPT1/CLN1, these observations suggest a molecular association may exist between the two proteins.
Loss of CLN5 affects TPP1/CLN2 and CTSD/CLN10
CLN5 disease patient fibroblasts display increased TPP1/CLN2 activity [77] (Table 3). In support of these findings, CLN5 has been shown to interact with TPP1/CLN2 in mouse brain extract, COS-1 cells, and HeLa cells (Table 2) [77, 78]. In Dictyostelium, Cln5 interacts with Tpp1B, which is the most highly expressed Tpp1 protein in the organism [8, 26] (Fig. 2). Recently, whole-exome sequencing of Alzheimer’s disease families revealed a missense variant in CLN5 that segregated with Alzhemier’s disease, which supports the hypothesis that there may be similarities between NCL pathology and other forms of neurodegeneration, including Alzheimer’s disease, Parkinson’s disease, and frontotemporal dementia [80, 83, 92, 98, 99]. This variant displayed glycosylation defects in CLN5 that caused it to be retained in the ER of HeLa cells, which coincided with reduced trafficking to the endo-lysosomal compartment [83]. The variant also showed reduced processing of CTSD/CLN10 and decreased levels of full-length amyloid precursor protein, thus providing evidence linking the function of CLN5 to CTSD/CLN10 (Table 2). The binding of Cln5 to CtsD in Dictyostelium is consistent with these observations [26] (Fig. 2). Together, these results establish links between TPP1/CLN2, CLN5, and CTSD/CLN10, which are likely reflective of their common function as lysosomal enzymes, albeit with different activities [24, 26, 27].
Loss of MFSD8/CLN7 affects CLN5
A SILAC-based quantitative analysis of lysosomes from Mfsd8/Cln7-deficient mouse embryonic fibroblasts (MEFs) derived from a Mfsd8/Cln7−/− mouse revealed that loss of Mfsd8/Cln7 significantly depletes the amount of lysosomal CLN5, as well as other lysosomal enzymes [91] (Table 3). The depletion of CLN5 was shown to be due to increased proteolytic degradation by cysteine proteases in the lysosomes of Mfsd8/Cln7−/− MEFs [91]. These findings are also supported by work in Dictyostelium, which showed that Mfsd8 regulates the secretion of Cln5 during the early stages of multicellular development [46] (Fig. 2). In total, these data suggest that depletion of CLN5 may contribute to the pathogenesis associated with CLN7 disease and that MFSD8/CLN7 may play an important role in the trafficking of lysosomal enzymes.
Is CTSD/CLN10 a common pathogenic link for all NCLs?
The findings described above suggest that loss of CTSD/CLN10 activity may be a common pathogenic link between several subtypes of NCL (i.e., links between CTSD/CLN10 and PPT1/CLN1, CLN3, and CLN5). Based on these observations, it is not surprising that loss of TPP1/CLN2, PGRN/CLN11, or ATP13A2/CLN12 also affects the function of CTSD/CLN10. In brain tissues from a Tpp1/Cln2−/− mouse model, loss of Tpp1/Cln2 increases the expression of CtsD/Cln10 [88] (Table 3). Loss of Tpp1/Cln2 in this mouse model also increases the expression of Pgrn/Cln11 suggesting there may be a molecular connection between TPP1/CLN2 and PGRN/CLN11. In liver, spleen, and brain (cerebrum, cerebellum, midbrain) lysates from PGRN/CLN11-deficient mice, the activity of CTSD/CLN10 is significantly reduced, without any changes in the level of CTSD/CLN10 protein [79]. These observations were independently validated in fibroblasts and induced pluripotent stem cell (iPSC)-derived cortical neurons from patients with frontotemporal dementia (FTD, carry heterozygous mutations in PGRN/CLN11), thus highlighting the conserved effect of PGRN/CLN11 on CTSD/CLN10 activity in multiple models [80, 92]. In support of these findings, FLAG-tagged CTSD/CLN10 was shown to interact directly with untagged PGRN/CLN11 in HEK293T cells [79] (Table 2). A parallel study confirmed the interaction between CTSD/CLN10 and PGRN/CLN11 and showed that PGRN/CLN11 of human origin can increase the activity of CTSD/CLN10 from bovine spleen [80]. Together, these findings suggest that PGRN/CLN11 activates CTSD/CLN10, which provides a possible explanation for the overlapping pathology observed in individuals with mutated CTSD/CLN10 or PGRN/CLN11. Finally, in SH-SY5Y human neuroblastoma cells, knockdown of ATP13A2/CLN12 reduces the amount and activity of CTSD/CLN10 [93] (Table 3). In summary, these findings show that at least one aspect of CTSD/CLN10 function (e.g., expression, amount, processing, activity) is linked to the expression and/or activity of six NCL genes/proteins (e.g., PPT1/CLN1, TPP1/CLN2, CLN3, CLN5, PGRN/CLN11, ATP13A2/CLN12), which constitutes half of all NCL genes/proteins.
NCL gene expression during mammalian development and cancer
There is also evidence supporting the potential networking of NCL genes during mammalian development and cancer. During brain development in mice, Tpp1/Cln2, Cln3, and Cln5 are spatially and temporally co-expressed [100]. Specifically, these genes are expressed in regions associated with the formation of new neurons including the germinal epithelium, ventricle regions, hippocampus, and cerebellum. In addition, the genes are temporally co-expressed in neuronal progenitor cells suggesting they may play an important role in early embryonal neurogenesis. Gene expression profiling of the cortex from Cln1 and Cln5 knockout mice revealed that loss of either Ppt1/Cln1 or Cln5 affects the expression of genes that regulate neuronal growth cone stabilization [101]. Intriguingly, the three major genes comprising this pathway (adenylate cyclase-associated protein 1, protein tyrosine phosphatase receptor type F, and protein tyrosine phosphatase 4a2) all cluster to the same locus as Ppt1/Cln1, further supporting a functional relationship between the gene products. In addition, immunofluorescence experiments performed on cortical neurons isolated from these mice display abnormal distribution of the cytoskeletal proteins, actin and beta-tubulin, as well as growth cone associated proteins GAP-43, synapsin, and Rab3. NCL gene expression has also been linked to various forms of cancer [102,103,104,105]. In one study, a close correlation between PPT1/CLN1 and TPP1/CLN2 expression was reported in the progression and metastasis associated with colorectal cancer [103]. In total, NCL gene expression during cancer and mammalian development suggests a shared regulation of these genes.
NCL proteins are secreted during Dictyostelium development
Many lysosomal enzymes have been detected extracellularly and their roles outside the cell are gaining increased attention with respect to disease development and progression [106]. With that in mind, observations first made in Dictyostelium suggest that secretion may play an important role in the pathology underlying the NCLs. Proteomic-based analyses revealed that 5 of the 11 NCL protein homologs are secreted during Dictyostelium multicellular development including Ppt1, Tpp1B and Tpp1F, Cln5, CtsD, and proteins that share sequence similarity with human CTSF/CLN13 (CprA, CprB, CprD, CprE, CprF, CprG, uncharacterized protein DDB0252831) [47, 56] (Fig. 2). While CLN5 and CTSD/CLN10 have been detected extracellularly in mammalian systems, the observations in Dictyostelium are significant since they provided the first evidence in any system that PPT1/CLN1, TPP1/CLN2, and CTSF/CLN13 may function outside the cell. However, the precise functions of NCL proteins outside the cell are not known. Since they function as enzymes intracellularly, it is possible they may have enzymatic activity outside the cell, but this remains to be determined. In support of extracellular enzyme activity, previous work in Dictyostelium detected Tpp1 and CtsD activity in conditioned starvation buffer [94]. However, additional work is required to determine if these activities can be attributed to secreted NCL proteins and if these extracellular activities can be detected in mammalian models of NCL. In support of the molecular networking of NCL proteins, loss of cln3 alters protein secretion during development, including homologs of NCL proteins (Cln5, CtsD, CprA, CprB, CprD, CprE, CprF, CprG) and lysosomal enzymes such as N-acetylglucosaminidase and cathepsin B [47] (Fig. 2). Secretion of Cln5 and CtsD is also affected by the loss of mfsd8 [46]. These discoveries indicate that aberrant secretion may play an important role in NCL pathology. In fact, this work has informed research in mammalian systems. For example, aberrant secretion was subsequently reported in glia and neurons isolated from Cln3-deficient mice, and brain and cerebrospinal fluid from NCL patients (CLN1, CLN2, and CLN3 disease) [107, 108]. Together, these findings highlight the importance of determining the impact of altered protein secretion in the NCLs.
Common phenotypes in mammalian and Dictyostelium models of NCL
As discussed above, there is evidence at the molecular level linking the NCL proteins to a common biological pathway (e.g., similar localizations, binding and interactome studies, expression analyses). Importantly, these observations are supported by work at the cellular and organismal level, which shows that loss or mutation of individual NCL genes causes similar phenotypes in Dictyostelium and mammalian models of NCL. These studies will be discussed below to highlight the possible role of NCL proteins in regulating a common cellular pathway.
Mammals
In CLN3 disease patient fibroblasts, the V0a1 subunit of v-ATPase, which regulates lysosomal acidification, is mis-localized to the plasma membrane instead of its normal location on the lysosomal membrane [89]. This defect dysregulates lysosomal acidification, which has also been reported in Cln1−/− mice. In addition, lysosomal fractions from mouse models of CLN3 and CLN7 disease display altered amounts of lysosomal enzymes [90, 91]. Previous work also showed that the aberrant growth and apoptosis of CLN5 disease patient fibroblasts can be corrected by re-introducing the CLN8 protein [109]. Interestingly, like CLN5 disease patient fibroblasts, ceramide synthase activity is also decreased in CLN8−/− cells derived from a naturally occurring mouse model for CLN8 disease suggesting that CLN5 and CLN8 may function in the same biological pathway [109]. Additionally, several NCL disease models display aberrant autophagy, which has also been reported in other forms of neurodegeneration [110]. In fact, previous work has linked the functions of 10 of the 13 NCL proteins to autophagic mechanisms (PPT1/CLN1, TPP1/CLN2, CLN3, CLN5, CLN6, MFSD8/CLN7, CTSD/CLN10, PGRN/CLN11, ATP13A2/CLN12, KCTD7/CLN14) [111,112,113,114,115,116,117,118,119,120].
Another possibility for the networking of NCL proteins involves the proteins functioning along converging pathways. In mice, loss of both Cln1 and Cln5 causes more severe phenotypes and leads to an earlier onset of the disease than in mice where only one of Cln1 or Cln5 is knocked out [121]. While fertile, the double knockout mice showed a slight decrease in expected breeding ratios as well as impaired embryoid body formation by induced pluripotent stem cells derived from the mice. These results suggest that the enhanced pathological phenotypes caused by the simultaneous loss of both Cln1 and Cln5 may be caused by the disruption or dysregulation of converging pathogenic pathways.
Dictyostelium
Several gene-deficiency models have been generated in Dictyostelium that have enhanced our knowledge of NCL protein function in this model organism (tpp1A−, cln3−, cln5−, and mfsd8− cells) (Table 4). Autofluorescent material has been detected in tpp1A−, cln3−, and cln5− cells and these knockout cells display common phenotypes [44, 48, 94, 122]. For example, cln3− cells display increased cell proliferation during growth [43]. Loss of tpp1 also affects cell proliferation, but the effect is reduced growth, not increased, suggesting that while a common growth-regulating pathway may be affected in all three cell lines, the proteins may serve different functions in this pathway [122]. Knockdown of tpp1 increases phagocytosis and micropinocytosis, the latter of which has also been reported for cln3− cells [43, 122]. During multicellular development, both cln3− and cln5− cells display reduced adhesion [45, 48]. Finally, loss of tpp1 or cln3 causes cells to develop precociously and tpp1 knockdown cells form smaller slugs and fruiting bodies [43, 44, 122]. Together, findings from Dictyostelium and mammalian models of NCL suggest that similar downstream secondary events may occur in the cells of NCL patients. However, the specific mechanisms underlying these events in the individual NCL subtypes remain to be determined.
Future avenues for exploring the molecular networking of NCL proteins
Since there is currently only one approved therapy for the NCLs, which is specific for one subtype of the disease, additional work is needed to determine the cellular pathways affected by mutations in NCL genes. As discussed in this review, important information can be obtained by examining the localizations of NCL proteins, assessing their interactions with one another and common binding partners, and examining the effects of individual NCL gene loss or mutation on other NCL genes/proteins. Continued work in these areas is vital for defining the cellular roles of NCL proteins, the pathways they regulate, and the conditions that impair their functions. To supplement this research, future work that systematically examines NCL gene expression in specific models of the disease has the potential to elucidate new interactions between NCL genes/proteins. For example, most of the NCL proteins fall into one of two general classes, enzymes (e.g., TPP1/CLN2, CTSD/CLN10) or putative channels/transporters (e.g., CLN3, MFSD8/CLN7). Therefore, it may be possible to suppress phenotypes in certain subtypes of the disease by modulating the expression of other NCL genes. When considering approaches for treating patients with NCL, we must consider that it may not be possible to restore the normal function of the mutated protein. With that in mind, RNA sequencing can be used to identify cellular pathways affected by NCL gene loss or mutation. A comparison of the datasets from different models can then provide insight into the cellular pathways affected by the NCLs, as well as potentially reveal proteins associated with multiple subtypes of the disease, which may lead to the identification of new molecular targets. Therapies could theoretically be designed against these new targets to potentially suppress clinical symptoms in multiple NCL subtypes.
Future potential of Dictyostelium for NCL research
While mammalian and patient-derived cells have the potential to enhance our understanding of the pathological mechanisms underlying the NCLs, the ability of Dictyostelium to inform research in these systems holds much promise, especially since the genome encodes more homologs of NCL proteins than other eukaryotic model organisms (e.g., yeast, worm, and fruit fly) [9]. Future work that studies the localizations of the uncharacterized NCL protein homologs in Dictyostelium will broaden our understanding of the pathways regulating NCL protein function and will allow us to identify specific regions of the cell that are affected by NCL. These findings can then be applied to therapy design efforts to determine which parts of the cell need to be targeted in patients. Unlike other model systems, researchers can also exploit the Dictyostelium life cycle to assess protein-protein interactions during both growth and multicellular development. This can then lay the foundation for future work that assesses the effects of mutations on the association of NCL proteins with each other, as well as proteins critical to their function.
However, with all that said, no model is perfect, and researchers must consider that Dictyostelium does present some limitations as a model organism for NCL research. For one, the limited number of cell types in Dictyostelium may limit the translation of findings to specific tissues or organs in mammalian systems. Dictyostelium also lacks a central nervous system, so discoveries made in the organism must be evaluated in the relevant mammalian cell type. Nonetheless, Dictyostelium presents a useful model system for exploring the functions of NCL protein homologs in a simple eukaryotic organism with measurable phenotypic outcomes.
Alternate perspectives and considerations
Our current understanding of the mechanisms underlying the NCLs is based on findings from various models ranging from organisms such as Dictyostelium, to mice, immortalized mammalian cells, and reprogrammed patient-derived cell lines. However, we must acknowledge that research in these systems may not accurately reflect the situation in patients, especially when one considers the variety of cell types affected by the disease. Multicellular organisms such as Dictyostelium may also display cell type-specific effects not observed in mammalian models or patients. With those caveats in mind, conserved findings among multiple systems were highlighted throughout this review to demonstrate their potential significance to NCL pathology. While it is important to acknowledge the caveats of previous work, the findings summarized in this review can be used as a springboard to help guide us into new viable avenues of research.
Finally, the broadly similar clinical and pathological profiles of the different NCL subtypes suggest that common disease mechanisms may be involved, which has been the focus of this review. However, a growing body of evidence also suggests that distinct mechanisms may be implicated in some forms of NCL, which is not surprising given the diversity of processes affected by mutations in NCL genes and the emerging differences in the nature and timing of pathology for the different subtypes [123]. Therefore, future work that explores both these avenues will be necessary to broaden our understanding of the cellular and molecular mechanisms underlying the NCLs.
Conclusions
The goal of this review was to integrate findings across Dictyostelium and mammalian models to shed light on the networking of NCL proteins within the eukaryotic cell. The localizations of NCL proteins suggest they play a concerted role in regulating the trafficking of material within the cell. This is supported by work showing that some NCL proteins bind to each other or have common binding partners. In addition, loss or mutation of individual NCL genes affects the expression and/or activities of other NCL genes/proteins. Finally, genetic models of NCL display similar phenotypes, which provides cellular and organismal evidence potentially linking the NCL proteins to a common cellular pathway. This review also highlighted an important observation; the links between CTSD/CLN10 and half of the NCL genes/proteins. While the precise role of CTSD/CLN10 in the NCLs is not known, that data point toward CTSD/CLN10 possibly being a common pathogenic feature of all NCL subtypes.
The continued use of mammalian and non-mammalian cellular models is vital for providing new insight into the networking of NCL proteins within cells, which in turn will facilitate to the identification of new molecular targets for therapy development. As previously mentioned, recent genetic advances show a strong overlap of some NCL subtypes with later onset forms of neurodegeneration including Alzheimer’s disease, Parkinson’s disease, and frontotemporal dementia. Therefore, the development of new therapies for the NCLs could also be highly beneficial for people affected by other forms of neurodegeneration.
Availability of data and materials
N/A
Change history
21 September 2020
An amendment to this paper has been published and can be accessed via the original article.
Abbreviations
- AprA:
-
Autocrine proliferation repressor A
- ATP13A2:
-
ATPase cation-transporting 13A2
- Bip2:
-
Luminal-binding protein 2
- CadA:
-
Calcium-dependent cell adhesion protein A
- CfaD:
-
Counting factor-associated protein D
- CLN:
-
Ceroid lipofuscinosis neuronal
- Cpr:
-
Cysteine proteinase
- CTS:
-
Cathepsin
- CV:
-
Contractile vacuole
- DBH:
-
Dopamine beta-hydroxylase
- DNAJC5:
-
DnaJ homolog subfamily C member 5
- ER:
-
Endoplasmic reticulum
- FTD:
-
Frontotemporal dementia
- GluA:
-
Beta-glucosidase
- GPR78:
-
Glucose-regulated protein 78
- GPHR:
-
Golgi pH regulator
- iPSC:
-
induced pluripotent stem cell
- KCTD7:
-
Potassium channel tetramerization domain-containing protein 7
- ManA:
-
Alpha-mannosidase
- Mcf:
-
Mitochondrial substrate carrier family protein
- MEF:
-
Mouse embryonic fibroblast
- MFSD8:
-
Major facilitator superfamily domain-containing protein 8
- NagA:
-
N-acetylglucosaminidase
- NCL:
-
Neuronal ceroid lipofuscinosis
- PGRN:
-
Progranulin
- PPT1:
-
Palmitoyl protein thioesterase 1
- TPP1:
-
Tripeptidyl peptidase 1
References
Mink JW, Augustine EF, Adams HR, Marshall FJ, Kwon JM. Classification and natural history of the neuronal ceroid lipofuscinoses. J Child Neurol. 2013;28:1101–5.
Cárcel-Trullols J, Kovács AD, Pearce DA. Cell biology of the NCL proteins: what they do and don’t do. Biochim Biophys Acta. 1852;2015:2242–55.
Butz ES, Chandrachud U, Mole SE, Cotman SL. Moving towards a new era of genomics in the neuronal ceroid lipofuscinoses. Biochim Biophys Acta Mol basis Dis. 2019;1:165571.
Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases - clinical perspectives. Biochim Biophys Acta. 1832;2013:1801–6.
Radke J, Stenzel W, Goebel HH. Human NCL neuropathology. Biochim Biophys Acta. 1852;2015:2262–6.
Lewis G, Morrill AM, Conway-Allen SL, Kim B. Review of cerliponase alfa: recombinant human enzyme replacement therapy for late-infantile neuronal ceroid lipofuscinosis type 2. J Child Neurol. 2019;1:348.
Minnis CJ, Thornton CD, FitzPatrick LM, McKay TR. Cellular models of batten disease. Biochim Biophys Acta Mol basis Dis. 2019;1:165559.
Huber RJ, Hughes SM, Liu W, Morgan A, Tuxworth RI, Russell C. The contribution of multicellular model organisms to neuronal ceroid lipofuscinosis research. Biochim Biophys Acta Mol basis Dis. 2019;1:165614.
Huber RJ. Using the social amoeba Dictyostelium to study the functions of proteins linked to neuronal ceroid lipofuscinosis. J Biomed Sci. 2016;23:83.
McLaren MD, Mathavarajah S, Huber RJ. Recent insights into NCL protein function using the model organism Dictyostelium discoideum. Cells. 2019;8:E115.
Rehberg M, Kleylein-Sohn J, Faix J, Ho TH, Schulz I, Gräf R. Dictyostelium LIS1 is a centrosomal protein required for microtubule/cell cortex interactions, nucleus/centrosome linkage, and actin dynamics. Mol Biol Cell. 2005;16:2759–71.
Myre MA, Lumsden AL, Thompson MN, Wasco W, MacDonald ME, Gusella JF. Deficiency of huntingtin has pleiotropic effects in the social amoeba Dictyostelium discoideum. PLoS Genet. 2011;7:e1002052.
Malinovska L, Alberti S. Protein misfolding in Dictyostelium: using a freak of nature to gain insight into a universal problem. Prion. 2015;9:339–46.
Chen S, Annesley SJ, Jasim RAF, Musco VJ, Sanislav O, Fisher PR. The Parkinson’s disease-associated protein DJ-1 plays a positive nonmitochondrial role in endocytosis in Dictyostelium cells. Dis Model Mech. 2017;10:1261–71.
Kelly E, Sharma D, Wilkinson CJ, Williams RSB. Diacylglycerol kinase (DGKA) regulates the effect of the epilepsy and bipolar disorder treatment valproic acid in Dictyostelium discoideum. Dis Model Mech. 2018;11:dmm035600.
Sharma D, Otto G, Warren EC, Beesley P, King JS, Williams RSB. Gamma secretase orthologs are required for lysosomal activity and autophagic degradation in Dictyostelium discoideum, independent of PSEN (presenilin) proteolytic function. Autophagy. 2019;15:1407–18.
Mathavarajah S, Flores A, Huber RJ. Dictyostelium discoideum: a model system for cell and developmental biology. Curr Prot Essen Lab Tech. 2017;15:14.1.1–19.
Bozzaro S. The past, present and future of Dictyostelium as a model system. Int J Dev Biol. 2019;63:321–31.
Persaud-Sawin DA, Mousallem T, Wang C, Zucker A, Kominami E, Boustany RM. Neuronal ceroid lipofuscinosis: a common pathway? Pediatr Res. 2007;61:146–52.
Journet A, Klein G, Brugière S, Vandenbrouck Y, Chapel A, Kieffer S, et al. Investigating the macropinocytic proteome of Dictyostelium amoebae by high-resolution mass spectrometry. Proteomics. 2012;12:241–5.
Weimer JM, Kriscenski-Perry E, Elshatory Y, Pearce DA. The neuronal ceroid lipofuscinoses: mutations in different proteins result in similar disease. NeuroMolecular Med. 2002;1:111–24.
Mukherjee AB, Appu AP, Sadhukhan T, Casey S, Mondal A, Zhang Z, et al. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol Neurodegener. 2019;14:4.
Hofmann SL, Lee LA, Lu JY, Verkruyse LA. Palmitoyl-protein thioesterase and the molecular pathogenesis of infantile neuronal ceroid lipofuscinosis. Neuropediatrics. 1997;28:27–30.
Passini MA, Dodge JC, Bu J, Yang W, Zhao Q, Sondhi D, et al. Intracranial delivery of CLN2 reduces brain pathology in a mouse model of classical late infantile neuronal ceroid lipofuscinosis. J Neurosci. 2006;26:1334–42.
Isosomppi J, Vesa J, Jalanko A, Peltonen L. Lysosomal localization of the neuronal ceroid lipofuscinosis CLN5 protein. Hum Mol Genet. 2002;11:885–91.
Huber RJ, Mathavarajah S. Cln5 is secreted and functions as a glycoside hydrolase in Dictyostelium. Cell Signal. 2018;42:236–48.
Barrett AJ. Lysosomal acid proteinase of rabbit liver. Biochem J. 1967;104:601–8.
Jerič B, Dolenc I, Mihelič M, Klarić M, Zavašnik-Bergant T, Gunčar G, et al. N-terminally truncated forms of human cathepsin F accumulate in aggresome-like inclusions. Biochim Biophys Acta. 1833;2013:2254–66.
Poole AR, Hembry RM, Dingle JT. Extracellular localization of cathepsin D in ossifying cartilage. Calcif Tissue Res. 1973;12:313–21.
Kremmidiotis G, Lensink IL, Bilton RL, Woollatt E, Chataway TK, Sutherland GR, et al. The batten disease gene product (CLN3p) is a Golgi integral membrane protein. Hum Mol Genet. 1999;8:523–31.
Golabek AA, Kaczmarski W, Kida E, Kaczmarski A, Michalewski MP, Wisniewski KE. Expression studies of CLN3 protein (battenin) in fusion with the green fluorescent protein in mammalian cells in vitro. Mol Genet Metab. 1999;66:277–82.
Steenhuis P, Herder S, Gelis S, Braulke T, Storch S. Lysosomal targeting of the CLN7 membrane glycoprotein and transport via the plasma membrane require a dileucine motif. Traffic. 2010;11:987–1000.
Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–91.
Mole SE, Michaux G, Codlin S, Wheeler RB, Sharp JD, Cutler DF. CLN6, which is associated with a lysosomal storage disease, is an endoplasmic reticulum protein. Exp Cell Res. 2004;298:399–406.
Lonka L, Kyttälä A, Ranta S, Jalanko A, Lehesjoki AE. The neuronal ceroid lipofuscinosis CLN8 membrane protein is a resident of the endoplasmic reticulum. Hum Mol Genet. 2000;9:1691–7.
Johnson JN, Ahrendt E, Braun JE. CSPalpha: the neuroprotective J protein. Biochem Cell Biol. 2010;88:157–65.
Azizieh R, Orduz D, Van Bogaert P, Bouschet T, Rodriguez W, Schiffmann SN, et al. Progressive myoclonic epilepsy-associated gene KCTD7 is a regulator of potassium conductance in neurons. Mol Neurobiol. 2011;44:111–21.
Bateman A, Belcourt D, Bennett H, Lazure C, Solomon S. Granulins, a novel class of peptide from leukocytes. Biochem Biophys Res Commun. 1990;173:1161–8.
Stumpf M, Müller R, Gaßen B, Wehrstedt R, Fey P, Karow MA, et al. A tripeptidyl peptidase 1 is a binding partner of the Golgi pH regulator (GPHR) in Dictyostelium. Dis Model Mech. 2017;10:897–907.
Domínguez-Martín E, Hernández-Elvira M, Vincent O, Coria R, Escalante R. Unfolding the endoplasmic reticulum of a social amoeba: Dictyostelium discoideum as a new model for the study of endoplasmic reticulum stress. Cells. 2018;7:E56.
Müller-Taubenberger A, Lupas AN, Li H, Ecke M, Simmeth E, Gerisch G. Calreticulin and calnexin in the endoplasmic reticulum are important for phagocytosis. EMBO J. 2001;20:6772–82.
Journet A, Chapel A, Jehan S, Adessi C, Freeze H, Klein G, et al. Characterization of Dictyostelium discoideum cathepsin D. J Cell Sci. 1999;112:3833–43.
Huber RJ, Myre MA, Cotman SL. Loss of Cln3 function in the social amoeba Dictyostelium discoideum causes pleiotropic effects that are rescued by human CLN3. PLoS One. 2014;9:e110544.
Phillips JE, Gomer RH. Partial genetic suppression of a loss-of-function mutant of the neuronal ceroid lipofuscinosis-associated protease TPP1 in Dictyostelium discoideum. Dis Model Mech. 2015;8:147–56.
Huber RJ, Myre MA, Cotman SL. Aberrant adhesion impacts early development in a Dictyostelium model for juvenile neuronal ceroid lipofuscinosis. Cell Adhes Migr. 2017;11:399–418.
Huber RJ, Mathavarajah S, Yap SQ. Mfsd8 localizes to endocytic compartments and influences the secretion of Cln5 and cathepsin D in Dictyostelium. Cell Signal. 2020;70:109572.
Huber RJ. Loss of Cln3 impacts protein secretion in the social amoeba Dictyostelium. Cell Signal. 2017;35:61–72.
Huber RJ, Mathavarajah S. Secretion and function of Cln5 during the early stages of Dictyostelium development. Biochim Biophys Acta, Mol Cell Res. 1865;2018:1437–50.
Moharir A, Peck SH, Budden T, Lee SY. The role of N-glycosylation in folding, trafficking, andfunctionality of lysosomal protein CLN5. PLoS One. 2013;8:e74299.
Hughes SM, Hope KM, Xu JB, Mitchell NL, Palmer DN. Inhibition of storage pathology in prenatal CLN5-deficient sheep neural cultures by lentiviral gene therapy. Neurobiol Dis. 2014;62:543–50.
Sriskanthadevan S, Brar SK, Manoharan K, Siu CH. Ca (2+)-calmodulin interacts with DdCAD-1 and promotes DdCAD-1 transport by contractile vacuoles in Dictyostelium cells. FEBS J. 2013;280:1795–806.
Hacker U, Albrecht R, Maniak M. Fluid-phase uptake by micropinocytosis in Dictyostelium. J Cell Sci. 1997;110:105–12.
Gabriel D, Hacker U, Kohler J, Muller-Taubenberger A, Schwartz JM, Westphal M, et al. The contractile vacuole network of Dictyostelium as a distinct organelle: its dynamics visualized by a GFP marker protein. J Cell Sci. 1999;112:3995–4005.
Mercanti V, Charette SJ, Bennett N, Ryckewaert JJ, Letourneur F, Cosson P. Selective membrane exclusion in phagocytic and macropinocytic cups. J Cell Sci. 2006;119:4079–87.
Getty A, Kovács AD, Lengyel-Nelson T, Cardillo A, Hof C, Chan CH, et al. Osmotic stress changes the expression and subcellular localization of the batten disease protein CLN3. PLoS One. 2013;8:e66203.
Bakthavatsalam D, Gomer RH. The secreted proteome profile of developing Dictyostelium discoideum cells. Proteomics. 2010;10:2556–9.
Kline RA, Wishart TM, Mills K, Heywood WE. Applying modern omic technologies to the neuronal ceroid lipofuscinoses. Biochim Biophys Acta Mol basis Dis. 2019;1:165498.
Scifo E, Szwajda A, Dębski J, Uusi-Rauva K, Kesti T, Dadlez M, et al. Drafting the CLN3 protein interactome in SH-SY5Y human neuroblastoma cells: a label-free quantitative proteomics approach. J Proteome Res. 2013;12:2101–15.
Scifo E, Szwajda A, Soliymani R, Pezzini F, Bianchi M, Dapkunas A, et al. Proteomic analysis of the palmitoyl protein thioesterase 1 interactome in SH-SY5Y human neuroblastoma cells. J Proteome. 2015;123:42–53.
Cubells JF, Zabetian CP. Human genetics of plasma dopamine beta-hydroxylase activity: applications to research in psychiatry and neurology. Psychopharmacology. 2004;174:463–76.
Ruottinen HM, Rinne JO, Haaparanta M, Solin O, Bergman J, Oikonen VJ, et al. [18F] fluorodopa PET shows striatal dopaminergic dysfunction in juvenile neuronal ceroid lipofuscinosis. J Neurol Neurosurg Psychiatry. 1997;62:622–5.
Weimer JM, Benedict JW, Elshatory YM, Short DW, Ramirez-Montealegre D, Ryan DA, et al. Alterations in striatal dopamine catabolism precede loss of substantia nigra neurons in a mouse model of juvenile neuronal ceroid lipofuscinosis. Brain Res. 2007;1162:98–112.
Le NM, Parikh S. Late infantile neuronal ceroid lipofuscinosis and dopamine deficiency. J Child Neurol. 2012;27:234–7.
Tang S, Yao B, Li N, Lin S, Huang Z. Association of dopamine beta-hydroxylase polymorphisms with Alzheimer's disease, Parkinson's disease and schizophrenia: evidence based on currently available loci. Cell Physiol Biochem. 2018;51:411–28.
Gonzalez-Lopez E, Vrana KE. Dopamine beta-hydroxylase and its genetic variants in human health and disease. J Neurochem. 2020;152:157–81.
Chaouch A, Porcelli V, Cox D, Edvardson S, Scarcia P, De Grassi A, et al. Mutations in the mitochondrial citrate carrier SLC25A1 are associated with impaired neuromuscular transmission. J Neuromuscul Dis. 2014;1:75–90.
Majd H, King MS, Smith AC, Kunji ERS. Pathogenic mutations of the human mitochondrial citrate carrier SLC25A1 lead to impaired citrate export required for lipid, dolichol, ubiquinone and sterol synthesis. Biochim Biophys Acta. 1859;2018:1–7.
Bourre JM, Haltia M, Daudu O, Monge M, Baumann N. Infantile form of so-called neuronal ceroid lipofuscinosis: lipid biochemical studies, fatty acid analysis of cerebroside sulfatides and sphingomyelin, myelin density profile and lipid composition. Eur Neurol. 1979;18:312–21.
Granier LA, Langley K, Leray C, Sarlieve LL. Phospholipid composition in late infantile neuronal ceroid lipofuscinosis. Eur J Clin Investig. 2000;30:1011–7.
Käkelä R, Somerharju P, Tyynelä J. Analysis of phospholipid molecular species in brains from patients with infantile and juvenile neuronal-ceroid lipofuscinosis using liquid chromatography-electrospray ionization mass spectrometry. J Neurochem. 2003;84:1051–65.
Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, et al. Citrin and aralar1 are ca (2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001;20:5060–9.
Thangaratnarajah C, Ruprecht JJ, Kunji ER. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nat Commun. 2014;5:5491.
Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. 1999;22:159–63.
Jolly RD, Brown S, Das AM, Walkley SU. Mitochondrial dysfunction in the neuronal ceroid-lipofuscinoses (batten disease). Neurochem Int. 2002;40:565–71.
Kang S, Seo JH, Heo TH, Kim SJ. Batten disease is linked to altered expression of mitochondria-related metabolic molecules. Neurochem Int. 2013;62:931–5.
Kim JB, Lim N, Kim SJ, Heo TH. N-acetylcysteine normalizes the urea cycle and DNA repair in cells from patients with batten disease. Cell Biochem Funct. 2012;30:677–82.
Vesa J, Chin MH, Oelgeschläger K, Isosomppi J, DellAngelica EC, Jalanko A, et al. Neuronal ceroid lipofuscinoses are connected at molecular level: interaction of CLN5 protein with CLN2 and CLN3. Mol Biol Cell. 2002;13:2410–20.
Lyly A, von Schantz C, Heine C, Schmiedt ML, Sipilä T, Jalanko A, et al. Novel interactions of CLN5 support molecular networking between neuronal ceroid lipofuscinosis proteins. BMC Cell Biol. 2009;10:83.
Zhou X, Paushter DH, Feng T, Pardon CM, Mendoza CS, Hu F. Regulation of cathepsin D activity by the FTLD protein progranulin. Acta Neuropathol. 2017;134:151–3.
Valdez C, Wong YC, Schwake M, Bu G, Wszolek ZK, Krainc D. Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet. 2017;26:4861–72.
Henderson MX, Wirak GS, Zhang YQ, Dai F, Ginsberg SD, Dolzhanskaya N, et al. Neuronal ceroid lipofuscinosis with DNAJC5/CSPα mutation has PPT1 pathology and exhibit aberrant protein palmitoylation. Acta Neuropathol. 2016;131:621–37.
Cárcel-Trullols J, Kovács AD, Pearce DA. Role of the lysosomal membrane protein, CLN3, in the regulation of cathepsin D activity. J Cell Biochem. 2017;118:3883–90.
Qureshi YH, Patel VM, Berman DE, Kothiya MJ, Neufeld JL, Vardarajan B, et al. An Alzheimer's disease-linked loss-of-function CLN5 variant impairs cathepsin D maturation, consistent with a retromer trafficking defect. Mol Cell Biol. 2018;38:e00011–8.
Mamo A, Jules F, Dumaresq-Doiron K, Costantino S, Lefrancois S. The role of ceroid lipofuscinosis neuronal protein 5 (CLN5) in endosomal sorting. Mol Cell Biol. 2012;32:1855–66.
Yasa S, Modica G, Sauvageau E, Kaleem A, Hermey G, Lefrancois S. CLN3 regulates endosomal function by modulating Rab7A-effector interactions. J Cell Sci. 2020;133:jcs234047.
Jules F, Sauvageau E, Dumaresq-Doiron K, Mazzaferri J, Haug-Kröper M, Fluhrer R, et al. CLN5 is cleaved by members of the SPP/SPPL family to produce a mature soluble protein. Exp Cell Res. 2017;357:40–50.
Chandra G, Bagh MB, Peng S, Saha A, Sarkar C, Moralle M, et al. Cln1 gene disruption in mice reveals a common pathogenic link between two of the most lethal childhood neurodegenerative lysosomal storage disorders. Hum Mol Genet. 2015;24:5416–32.
Domowicz MS, Chan WC, Claudio-Vázquez P, Henry JG, Ware CB, Andrade J, et al. Global brain transcriptome analysis of a Tpp1 neuronal ceroid lipofuscinoses mouse model. ASN Neuro. 2019;11:1759091419843393.
Appu AP, Bagh MB, Sadhukhan T, Mondal A, Casey S, Mukherjee AB. Cln3-mutations underlying juvenile neuronal ceroid lipofuscinosis cause significantly reduced levels of Palmitoyl-protein thioesterases-1 (Ppt1)-protein and Ppt1-enzyme activity in the lysosome. J Inherit Metab Dis. 2019;42:944–54.
Schmidtke C, Tiede S, Thelen M, Käkelä R, Jabs S, Makrypidi G, et al. Lysosomal proteome analysis reveals that CLN3-defective cells have multiple enzyme deficiencies associated with changes in intracellular trafficking. J Biol Chem. 2019;294:9592–604.
Danyukova T, Ariunbat K, Thelen M, Brocke-Ahmadinejad N, Mole SE, Storch S. Loss of CLN7 results in depletion of soluble lysosomal proteins and impaired mTOR reactivation. Hum Mol Genet. 2018;27:1711–22.
Ward ME, Chen R, Huang HY, Ludwig C, Telpoukhovskaia M, Taubes A, et al. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med. 2017;9:eaah5642.
Matsui H, Sato F, Sato S, Koike M, Taruno Y, Saiki S, et al. ATP13A2 deficiency induces a decrease in cathepsin D activity, fingerprint-like inclusion body formation, and selective degeneration of dopaminergic neurons. FEBS Lett. 2013;587:1316–25.
Huber RJ, Mathavarajah S. Comparative transcriptomics reveals mechanisms underlying cln3-deficiency phenotypes in Dictyostelium. Cell Signal. 2019;58:79–90.
Mathavarajah S, McLaren MD, Huber RJ. Cln3 function is linked to osmoregulation in a Dictyostelium model of batten disease. Biochim Biophys Acta Mol basis Dis. 1864;2018:3559–73.
Casas C. GRP78 at the Centre of the stage in cancer and neuroprotection. Front Neurosci. 2017;11:177.
Uusi-Rauva K, Luiro K, Tanhuanpää K, Kopra O, Martín-Vasallo P, Kyttälä A, et al. Novel interactions of CLN3 protein link batten disease to dysregulation of fodrin-Na+, K+ ATPase complex. Exp Cell Res. 2008;314:2895–905.
Dearborn JT, Harmon SK, Fowler SC, O'Malley KL, Taylor GT, Sands MS, et al. Comprehensive functional characterization of murine infantile batten disease including Parkinson-like behavior and dopaminergic markers. Sci Rep. 2015;5:12752.
Deng H, Xiu X, Jankovic J. Genetic convergence of Parkinson's disease and lysosomal storage disorders. Mol Neurobiol. 2015;51:1554–68.
Fabritius AL, Vesa J, Minye HM, Nakano I, Kornblum H, Peltonen L. Neuronal ceroid lipofuscinosis genes, CLN2, CLN3 and CLN5 are spatially and temporally co-expressed in a developing mouse brain. Exp Mol Pathol. 2014;97:484–91.
von Schantz C, Saharinen J, Kopra O, Cooper JD, Gentile M, Hovatta I, et al. Brain gene expression profiles of Cln1 and Cln5 deficient mice unravels common molecular pathways underlying neuronal degeneration in NCL diseases. BMC Genomics. 2008;9:146.
He Z, Ismail A, Kriazhev L, Sadvakassova G, Bateman A. Progranulin (PC-cell-derived growth factor/acrogranin) regulates invasion and cell survival. Cancer Res. 2002;62:5590–6.
Tsukamoto T, Iida J, Dobashi Y, Furukawa T, Konishi F. Overexpression in colorectal carcinoma of two lysosomal enzymes, CLN2 and CLN1, involved in neuronal ceroid lipofuscinosis. Cancer. 2006;106:1489–97.
Makoukji J, Raad M, Genadry K, El-Sitt S, Makhoul NJ, Saad Aldin E, et al. Association between CLN3 (neuronal ceroid Lipofuscinosis, CLN3 type) gene expression and clinical characteristics of breast cancer patients. Front Oncol. 2015;5:215.
Zhang C, Zhang M, Song S. Cathepsin D enhances breast cancer invasion and metastasis through promoting hepsin ubiquitin-proteasome degradation. Cancer Lett. 2018;438:105–15.
Vizovišek M, Fonović M, Turk B. Cysteine cathepsins in extracellular matrix remodeling: extracellular matrix degradation and beyond. Matrix Biol. 2019;75:141–59.
Parviainen L, Dihanich S, Anderson GW, Wong AM, Brooks HR, Abeti R, et al. Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathol Commun. 2017;5:74.
Sleat DE, Tannous A, Sohar I, Wiseman JA, Zheng H, Qian M, et al. Proteomic analysis of brain and cerebrospinal fluid from the three major forms of neuronal ceroid lipofuscinosis reveals potential biomarkers. J Proteome Res. 2017;16:3787–804.
Haddad SE, Khoury M, Daoud M, Kantar R, Harati H, Mousallem T, et al. CLN5 and CLN8 protein association with ceramide synthase: biochemical and proteomic approaches. Electrophoresis. 2012;33:3798–809.
Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93:1015–34.
Cao Y, Espinola JA, Fossale E, Massey AC, Cuervo AM, MacDonald ME, et al. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J Biol Chem. 2006;281:20483–93.
Cannelli N, Garavaglia B, Simonati A, Aiello C, Barzaghi C, Pezzini F, et al. Variant late infantile ceroid lipofuscinoses associated with novel mutations in CLN6. Biochem Biophys Res Commun. 2009;379:892–7.
Gusdon AM, Zhu J, Van Houten B, Chu CT. ATP13A2 regulates mitochondrial bioenergetics through macroautophagy. Neurobiol Dis. 2012;45:962–72.
Vidal-Donet JM, Cárcel-Trullols J, Casanova B, Aguado C, Knecht E. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS One. 2013;8:e55526.
Tanaka Y, Chambers JK, Matsuwaki T, Yamanouchi K, Nishihara M. Possible involvement of lysosomal dysfunction in pathological changes of the brain in aged progranulin-deficient mice. Acta Neuropathol Commun. 2014;2:78.
Brandenstein L, Schweizer M, Sedlacik J, Fiehler J, Storch S. Lysosomal dysfunction and impaired autophagy in a novel mouse model deficient for the lysosomal membrane protein Cln7. Hum Mol Genet. 2016;25:777–91.
Leinonen H, Keksa-Goldsteine V, Ragauskas S, Kohlmann P, Singh Y, Savchenko E, et al. Retinal degeneration in a mouse model of CLN5 disease is associated with compromised autophagy. Sci Rep. 2017;7:1597.
Nicastri MC, Rebecca VW, Amaravadi RK, Winkler JD. Dimeric quinacrines as chemical tools to identify PPT1, a new regulator of autophagy in cancer cells. Mol Cell Oncol. 2017;5:e1395504.
Metz KA, Teng X, Coppens I, Lamb HM, Wagner BE, Rosenfeld JA, et al. KCTD7 deficiency defines a distinct neurodegenerative disorder with a conserved autophagy-lysosome defect. Ann Neurol. 2018;84:766–80.
Marques ARA, Di Spiezio A, Thießen N, Schmidt L, Grötzinger J, Lüllmann-Rauch R, et al. Enzyme replacement therapy with recombinant pro-CTSD (cathepsin D) corrects defective proteolysis and autophagy in neuronal ceroid lipofuscinosis. Autophagy. 2019;16:1–15.
Blom T, Schmiedt ML, Wong AM, Kyttälä A, Soronen J, Jauhiainen M, et al. Exacerbated neuronal ceroid lipofuscinosis phenotype in Cln1/5 double-knockout mice. Dis Model Mech. 2013;6:342–57.
Smith PK, Sen MG, Fisher PR, Annesley SJ. Modelling of neuronal ceroid lipofuscinosis type 2 in Dictyostelium discoideum suggests that cytopathological outcomes result from altered TOR signalling. Cells. 2019;8:E469.
Nelvagal HR, Lange J, Takahashi K, Tarczyluk-Wells MA, Cooper JD. Pathomechanisms in the neuronal ceroid lipofuscinoses. Biochim Biophys Acta Mol basis Dis. 2019;1:165570.
Acknowledgements
The author would like to thank the anonymous reviewers for their helpful and constructive comments.
Funding
RJH is supported by grants from the Natural Sciences and Engineering Research Council of Canada (Discovery Grant, RGPIN-2018-04855) and the Canadian Institutes of Health Research (Project Grant, PJT165873).
Author information
Authors and Affiliations
Contributions
RJH is the sole author of this paper. The author(s) read and approved the final manuscript.
Author’s information
RJH is an Assistant Professor at Trent University. His lab uses Dictyostelium discoideum as a model system for studying the localizations and functions of NCL proteins.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
N/A
Consent for publication
N/A
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huber, R.J. Molecular networking in the neuronal ceroid lipofuscinoses: insights from mammalian models and the social amoeba Dictyostelium discoideum. J Biomed Sci 27, 64 (2020). https://doi.org/10.1186/s12929-020-00653-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-020-00653-y