
Abstract
Background
Stress-induced analgesia (SIA) is an evolutionarily conserved phenomenon during stress. Neuropeptide S (NPS), orexins, substance P, glutamate and endocannabinoids are known to be involved in stress and/or SIA, however their causal links remain unclear. Here, we reveal an unprecedented sequential cascade involving these mediators in the lateral hypothalamus (LH) and ventrolateral periaqueductal gray (vlPAG) using a restraint stress-induced SIA model.
Methods
Male C57BL/6 mice of 8–12 week-old were subjected to intra-cerebroventricular (i.c.v.) and/or intra-vlPAG (i.pag.) microinjection of NPS, orexin-A or substance P alone or in combination with selective antagonists of NPS receptors (NPSRs), OX1 receptors (OX1Rs), NK1 receptors (NK1Rs), mGlu5 receptors (mGlu5Rs) and CB1 receptors (CB1Rs), respectively. Antinociceptive effects of these mediators were evaluated via the hot-plate test. SIA in mice was induced by a 30-min restraint stress. NPS levels in the LH and substance P levels in vlPAG homogenates were compared in restrained and unrestrained mice.
Results
NPS (i.c.v., but not i.pag.) induced antinociception. This effect was prevented by i.c.v. blockade of NPSRs. Substance P (i.pag.) and orexin-A (i.pag.) also induced antinociception. Substance P (i.pag.)-induced antinociception was prevented by i.pag. Blockade of NK1Rs, mGlu5Rs or CB1Rs. Orexin-A (i.pag.)-induced antinociception has been shown previously to be prevented by i.pag. blockade of OX1Rs or CB1Rs, and here was prevented by NK1R or mGlu5R antagonist (i.pag.). NPS (i.c.v.)-induced antinociception was prevented by i.pag. blockade of OX1Rs, NK1Rs, mGlu5Rs or CB1Rs. SIA has been previously shown to be prevented by i.pag. blockade of OX1Rs or CB1Rs. Here, we found that SIA was also prevented by i.c.v. blockade of NPSRs or i.pag. blockade of NK1Rs or mGlu5Rs. Restrained mice had higher levels of NPS in the LH and substance P in the vlPAG than unrestrained mice.
Conclusions
These results suggest that, during stress, NPS is released and activates LH orexin neurons via NPSRs, releasing orexins in the vlPAG. Orexins then activate OX1Rs on substance P-containing neurons in the vlPAG to release substance P that subsequently. Activates NK1Rs on glutamatergic neurons to release glutamate. Glutamate then activates perisynaptic mGlu5Rs to initiate the endocannabinoid retrograde inhibition of GABAergic transmission in the vlPAG, leading to analgesia.
Similar content being viewed by others


Background
Stress-induced analgesia (SIA) is an evolutionarily protective system in mammals for coping with environmental stressors [1]. Several neuropeptides released during stress, such as orexins [2, 3], neuropeptide S (NPS) [4] and substance P [5], may contribute to SIA. However, how these neuropeptide-mediated signals interact to elicit SIA remains unknown.
Orexins, consisting of orexin-A and orexin-B [6], also known as hypocretin-1 and hypocretin-2 [7], are processed from preprohypocretin in hypothalamic neurons in the perifornical area (PFA), lateral hypothalamus (LH) and dorsomedial hypothalamus (DMH) [6, 7]. Orexin receptors, OX1 receptors (OX1Rs) and OX2 receptors (OX2Rs), belong to the G-protein coupled receptor (GPCR) family [8]. In addition to being involved in arousal and reward regulation [9], orexins are antinociceptive [10,11,12] and are involved in SIA [2, 3, 12, 13]. Previously, we have shown that orexins can be released during stress and contribute to SIA, at least in part, via opioid-independent and endocannabinoid (eCB)-dependent signaling [11, 12] in the ventrolateral periaqueductal gray (vlPAG), a crucial midbrain region for the initiation of descending pain inhibition [14, 15]. Specifically, orexins are released during stress [12], and orexins are known to induce antinociception by activating postsynaptic OX1Rs to generate 2-arachidonoylglycerol (2-AG) [16, 17], an eCB, through a Gq protein-coupled enzymatic cascade mediated by phospholipase C (PLC) and diacylglycerol lipase (DAGL) [18], culminating in retrograde inhibition of GABA release (disinhibition) in the vlPAG [11, 12].
NPS is an icosapeptide named due to its conserved N-terminal residue, serine, in all species [4]. Central administration of NPS (intra-cerebroventricular, i.c.v.) is antinociceptive [19,20,21]. The site of this antinociceptive action could be the PAG, where the mRNA transcript of NPS receptors (NPSRs) is abundant [22, 23], or other NPSR-rich brain regions, such as the amygdala and hypothalamus [22]. All three areas are commonly associated with emotional behaviours, and NPS is therefore implicated in stress-related behaviours. Indeed, forced swimming or restraint stress significantly activated NPS neurons in the pericoerulear region (peri-LC) and the Kölliker-Fuse nucleus of the lateral parabrachial area (KF-PBN) [24]. Intra-paraventricular nucleus (PVN) or i.c.v. administration of NPS in mice increased their locomotor and rearing activity, and plasma adrenocorticotropic hormone (ACTH) and corticosterone levels, suggesting that NPS can activate the arousal system and the hypothalamus-pituitary axis (HPA) [25].
The findings that both NPS and orexins are involved in the regulation of arousal, reward and pain suggest an interaction between the NPS [26] and orexin systems [9]. Indeed, it has been demonstrated that NPS (i.c.v.) can activate orexin neurons in the LH, PFA and DMH of rats [27, 28], where NPSRs are abundantly expressed [23]. Moreover, NPS has been reported to be an upstream activator of hypothalamic orexin neurons in feeding [27] and addiction [28, 29] behaviours. This suggests that NPS can activate orexin neurons and exert its biological functions, possibly including SIA, indirectly, by promoting the release of orexins.
Substance P is an undecapeptide belonging to the neurokinin (tachykinin) family [30] and exerts its effects mainly via NK1 receptors (NK1Rs) [31], a member of the GPCR family. Substance P is a well-known peripheral pronociceptive mediator [32] while it is antinociceptive at the supraspinal level [33]. In fact, intra-PAG microinjection (i.pag.) of substance P induces antinociception [34]. This effect may be mediated by the NK1Rs in the PAG since it is blocked by an NK1R antagonist and NK1Rs are densely distributed in pain-modulating brain regions including the PAG [35].
Using an electrophysiological approach, Drew et al. (2009) [36] have investigated how substance P modulates synaptic transmission in brain slices containing the vlPAG. They demonstrated that substance P decreased evoked GABA release in vlPAG slices. This effect was abolished by an inhibitor of DAGL, a degradation enzyme of 2-AG, and an antagonist of mGlu5 receptors (mGlu5R). Importantly, a glutamate transporter inhibitor mimicked the GABA-reducing effect of substance P, but also occluded such action of substance P [37]. However, substance P markedly increased action potential-driven spontaneous glutamate release. It is suggested that substance P induces an enormous release of glutamate that may activate perisynaptic mGlu5R to initiate the eCB-mediated retrograde disinhibition mechanism in the vlPAG. They suggested this effect may contribute to the substance P-induced analgesic effect in the vlPAG [37], however no pain behaviors were not evaluated. Substance P in the PAG may also contribute to SIA since restraint stress [38] and LH stimulation [39] increased the substance P level in the PAG and i.pag. blockade of NK1Rs abolished LH-stimulation-induced antinociception. However, there are no direct in vivo studies supporting the involvement of PAG substance P in SIA. Taking into consideration the the complexity of the aforementioned neuropeptides in SIA, a scheme depicting the possible relationships among NPS, orexins, substance P, mGlu5R and eCB (2-AG) during SIA, based on the available literature, is illustrated in Fig. 1.

A schema depicting the possible relationships among NPS, orexins, substance P, mGlu5R and endocannabinoid (2-AG) during SIA, based on the available literature. The cascades occurring in the locus coeruleus (LC)/ parabrachial nucleus (PBN), lateral hypothalamus (LH) and periaqueductal gray (PAG) during stress or exposing to NPS (purple), orexins (red) or substance P (blue) are depicted in the right box. The findings that have been reported are shown by solid lines with the numbers of referred literatures. To fulfil our hypothesis, the links that have to be proven are now established in this study, which are denoted by broken lines marked with [★]. The images of mouse brain and neurons are adapted from Illustration Toolkit Neuroscience by Motifolio. PN: projection neuron; SubP: substance P; Glu: glutamate
Substance P-induced vlPAG disinhibition, which is mediated through mGlu5R-initiated eCB signaling, highly resembles the OX1R-initiated 2-AG/CB1R signaling we reported previously [11], which contributes to SIA [12]. Moreover, mGlu5R [40] and eCBs [41, 42] are also involved in SIA. These events, all taking place in the vlPAG, prompted us to hypothesize the involvement of NK1Rs and mGlu5Rs in orexin-induced antinociception, and subsequently their involvement in SIA, possibly as downstream effectors of NPS. In this study, via behavioral, pharmacological and neurochemical approaches, we first examined the involvement of NK1R, mGlu5Rs and CB1Rs in substance P-induced antinociception. Next, we investigated whether orexins are upstream to substance P in the vlPAG in eliciting antinociceptive effects. Then, we examined whether NPS is an upstream modulator of LH orexin neurons. Lastly, we studied the involvement of the NPSR-OX1R-NK1R-mGlu5R-CB1R pathway in SIA.
Materials and methods
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee of College of Medicine, National Taiwan University following ARRIVE guidelines. Male C57BL/6 mice of 8–12 weeks were housed in groups of 10 in plastic cages and maintained in a holding room with a 12 h light-dark cycle with free access to food and water ad libitum. On the experimental day, mice were moved in their home cages to the behaviour room and acclimated there for at least 1 h before testing.
Hot-plate test
The hot-plate test in mice was performed as reported previously [12]. Briefly, the mouse was placed on a hot-plate maintained at 50 °C and paw withdrawal latency was recorded with a cut-off time of 60 s to prevent tissue damage. The antinociceptive effect in each mouse at each time point was calculated as the % of the maximal possible effect (MPE) by the equation: %MPE = 100 x (Latencyafter treatment - Latencybefore treatment) / (60s - Latencybefore treatment). The AUC of withdrawal latencies during the 60 min-recording period was calculated as the total antinociceptive effect in each mouse.
SIA
To induce SIA, mice were restrained in a 50-ml centrifugal tube with several small holes for 30 min as reported previously [12]. The control non-stress group of mice remained at their home cages for the same 30 min before being subjected to the hot-plate test.
Spontaneous locomotor activity
Locomotor activity was assessed by an open-field test in a 48 × 48 × 40 cm3 acrylic chamber with the arena floor divided into 36 squares, as described previously [43]. The mouse was placed in the center of the chamber, and the number of squares the mouse transpassed with forepaws (number of crossing) and the number of times the mouse stood up with forepaws on the floor (number of rearing) were counted for 5 min.
Intra-vlPAG (i.pag.) and intra-cerebroventricular (i.c.v.) microinjection
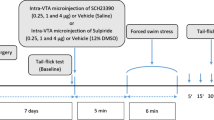
When drugs were given by i.pag. or i.c.v. microinjection, mice received the i.pag. or i.c.v. cannulation surgery 1 week before the microinjection experiment, as reported previously [12]. Briefly, under anesthesia with 50 mg/kg Zoletil® 50 (a mixture of tiletamine and zolazepam) and xylazine (10 mg/kg), the mouse was placed in a stereotaxic apparatus and a 24-gauge, 10 mm stainless-steel guide cannula was implanted into the right vlPAG (− 4.8 mm caudal, − 0.5 mm lateral, − 2.8 mm ventral from bregma, Additional file 1: Figure S1A) or the right ventricle (− 0.5 mm caudal, − 1.0 mm lateral, − 2.2 mm ventral from bregma, Additional file 1: Figure S1B), according to stereotaxic coordinates of mice [44]. On the day of experiments, i.pag. or i.c.v. microinjection was performed through a 30-gauge injection needle (10 mm) connected to a Hamilton syringe (1.0 μl) on a microinfusion pump (KDS311, KD Scientific Inc., Holliston, MA, USA). The drug solution (0.1 μl) was delivered in 60 s, followed by a 240 s-residual time to avoid back-flow of drug solution. Nociceptive responses were measured 5 mins before as well as 5, 10, 20, 30 and 40 mins after i.pag. or i.c.v. microinjections. For the mice that underwent restraint stress, i.pag or i.c.v. microinjections of the antagonists were performed 5 mins before stress, and nociceptive responses were measured immediately, 5, 10, 20, 30 and 40 mins after stress. After the final behavioural evaluation, the animals were microinjected with 0.5 μl of 0.4% trypan blue solution (Sigma-Aldrich, St. Louis, MO, USA) through the guide cannula to verify the injection tract location. Animals were then sacrificed by decapitation, coronal brain sections (300 μm) were prepared on a vibratome (DSK microslicer DTK-1000, Dosaka, Japan). The injection site was identified by the presence of trypan blue stain diffusion in the vlPAG tissue. Only animals with the cannula correctly targeting the ventricle or vlPAG were included in data analysis.
Measurements of substance P in vlPAG and NPS in LH of brain tissue homogenates
The preparation of the mouse vlPAG and LH homogenate is the same as previously reported [12]. Briefly, immediately after restraint stress, the mouse was sacrificed. Its brain was removed, placed in a pre-cooled adult mouse brain slicer matrix (Roboz Surgical Instrument, Gaithersburg, MD, USA), and sliced into 1 mm-thick coronal sections. vlPAG or LH brain tissues were bilaterally punched out with a 0.5 mm-tip according to a mouse brain atlas [44]. Each vlPAG sample was collected from one mouse brain, whereas each LH sample was from two mouse brains. After ultrasonication in lysis buffer, the lysates were homogenized and centrifuged (1900 g,14,000 rpm, 15 min) and supernatants collected. The protein concentration in the supernatant was measured by the Bradford method [45].
The substance P level in the vlPAG homogenate was measured with an EIA kit (Cat. No. 583751, Cayman Chemical. Ann Arbor, MI, USA) with a detection range of 3.9–500 pg/ml. The NPS level in the LH homogenates was measured with an ELISA kit. (Cat. No. CSB-EL016026MO, Cusabio, College Park, MD, USA) with a detection range of 4.69–300 pg/ml.
Chemicals
NPS and [tBu-D-Gly5] NPS were synthesised and purified as previously described [46]. N-(2-methyl-6-benzoxazolyl)-N-1,5-naphthyridin-4-yl-urea (SB-334867, a selective OX1R antagonist), 6-methyl-2-(phenylethynyl) pyridine hydrochloride (MPEP, a selective mGlu5R antagonist) and orexin-A were purchased from Tocris Bioscience (Bristol, UK). Substance P, 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide (AM251, a CB1R antagonist), and cis-2-(Diphenylmethyl)-N-[(2-iodophenyl)methyl]-1-azabicyclo [2.2.2] octan-3-amine oxalate salt (L-703,606, a selective NK1R antagonist) were purchased from Sigma-Aldrich. NPS and [tBu-D-Gly5] NPS were dissolved in 0.9% normal saline. Substance P was dissolved in 0.1 M acetic acid. SB-334867, L-703,606, MPEP and AM251 were dissolved in dimethyl sulfoxide (DMSO). All drugs were prepared at the working concentration for the intended i.pag. or i.c.v. injection doses.
Statistical analysis
Data are expressed as the mean ± S.E.M. and the “n” indicates the number of mice tested in each group. In the hotplate test, two-way ANOVA with post hoc Bonferroni test was used to analyse time courses of antinociceptive effects among different groups. The antinociceptive effect was also assessed by the area under the curve (AUC) of the time courses of quantification of the line graph from baseline to the last time-point of the experiment. Each AUC bar graph was calculated by one-way ANOVA followed by Tukey’s multiple comparison test. Student’s T-test was employed to analyse the results obtained in EIA and ELISA tests. Differences were considered significant if p < 0.05.
Results
NPS is antinociceptive when given by i.c.v. but not i.pag. microinjection in mice
NPS when given by i.c.v. injection at the doses 0.3 and 1.0 nmol, which did not affect the spontaneous locomotor activity of mice (Additional file 2: Figure S2), significantly prolonged the latency of nociceptive response in the hot-plate test (Fig. 2a and b) in a time- (F6,38 = 5.696, p < 0.001, two-way ANOVA, Fig. 2a) and treatment-dependent (F4,23 = 10.25, p < 0.001, two-way ANOVA, Fig. 2a) manner. However, when NPS was given by i.pag. microinjection, it did not produce significant antinociceptive effect at either 0.3 or 1.0 nmol (Fig. 2a and b). This suggests that the site of action for NPS-induced supraspinal antinociception is brain region(s) other than the vlPAG.

Antinociceptive effects induced by NPS, orexin-A and substance P in the mouse hot-plate test. a-b: Antinociceptive effects of NPS (0.3 & 1.0 nmol) by i.c.v. or i.pag. microinjection. c-d: Antinociceptive effects of i.c.v. NPS challenged by an NPSR antagonist, [tBu-D-Gly5] NPS (10 nmol, i.c.v.). e-f: A comparison of antinociceptive effects of orexin-A (1 nmol, i.pag.), substance P (5 nmol, i.pag.), and NPS (0.3 nmol, i.c.v.). a, c and e: The time course of the antinociceptive effect expressed as the percentage of the maximal possible effect (MPE) (two-way ANOVA /post hoc Bonferroni test). b, d and f: The area under the curve (AUC) of the % MPE measured within 40 min in each treatment group (one-way ANOVA /post hoc Tukey test). The number denoted in the parentheses above each bar is the n number of mice tested in each group. Data are mean ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001 vs. the vehicle control group, ###p < 0.001 vs. NPS 0.3 or 1.0 group
NPS (i.c.v.)-induced antinociception was antagonized by i.c.v. blockade of NPSRs
To investigate if the central antinociceptive effect of NPS is mediated via NPSR, we co-administered [tBu-D-Gly5] NPS (10 nmol, i.c.v.), a selective and potent NPSR antagonist [47], along with NPS (0.3 or 1.0 nmol, i.c.v.) to mice before the hot-plate test. [tBu-D-Gly5] NPS at 10 nmol (i.c.v.) did not affect the nociceptive response in naïve mice, but completely blocked the antinociceptive effect of i.c.v. NPS at the doses of 0.3 and 1.0 nmol (Fig. 2c and d) The overall comparison of the time course of the antinociceptive effect showed a significant difference between time and treatment (F30,222 = 1.872, p = 0.0057, two-way ANOVA, Fig. 2c). This suggests the central antinociceptive effect of NPS is mediated by NPSRs in the brain.
NPS (0.3 nmol, i.c.v.), substance P (5 nmol, i.pag.) and orexin-A (1 nmol, i.pag.) induced comparable antinociceptive effects in mice
To substantiate our hypothesis that a cascade mediated by NPS, orexins and substance P sequentially is involved in SIA, we evaluated equipotent doses of these three neuropeptides in a concurrent behavioural assay. As shown in Fig. 2e and f, NPS (0.3 nmol, i.c.v.) produced a antinociceptive effect in the mouse hot-plate test that was comparable to the effects induced by i.pag. microinjection of orexin-A at 1 nmol and i.pag. substance P at 5 nmol, respectively, with a significant difference between time and treatment (F18,120 = 1.924, p = 0.0198, two-way ANOVA, Fig. 2e).
Substance P (i.pag)-induced antinociception was antagonized by i.pag. blockade of NK1Rs, mGlu5Rs or CB1Rs
To ascertain if the NK1R-mGlu5R-CB1R pathway revealed by the electrophysiological study of Drew et al. (2009) [36] is involved in i.pag. substance P-induced antinociceptive effect, we challenged the antinociceptive effect of substance P with selective antagonists of NK1Rs (L-703,606), mGlu5Rs (MPEP) and CB1Rs (AM251), respectively, in a treatment-dependent manner (F3,18 = 5.316, p = 0.0084, two-way ANOVA, Fig. 3a; F3,18 = 10.97, p = 0.0003, two-way ANOVA, Fig. 3b; F3,17 = 5.929, p = 0.0059, two-way ANOVA, Fig. 3c). Indeed, i.pag. co-administration of L-703,606 (10 nmol), MPEP (30 nmol) or AM251 (30 nmol) with substance P (5 nmol) significantly antagonized the antinociceptive effect of i.pag. substance P (Fig. 3).

Substance P (i.pag.)-induced antinociception is antagonized by i.pag. blockade of NK1Rs, mGlu5Rs or CB1Rs. a-c: Time courses of antinociceptive effects (expressed as % MPE) induced by substance P (5 nmol, i.pag.) in combination with the vehicle or the antagonist of NK1Rs (L-703,606, 10 nmol, i.pag., a, mGlu5Rs (MPEP, 30 nmol, i.pag., b, and CB1Rs (AM251, 30 nmol, i.pag., c in the mouse hot-plate test. (two-way ANOVA /post hoc Bonferroni test). d: The AUC of the antinociceptive effect in each treatment group (one-way ANOVA /post hoc Tukey test). The antagonist was i.pag. co-administered with i.pag. substance P. The data presentation and statistics are the same as in Fig. 2. *p < 0.05, **p < 0.01, ***p < 0.001 vs. the vehicle control group; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. the Substance P group
Orexin-A (i.pag.)-induced antinociception was antagonized by i.pag. blockade of NK1Rs and mGlu5Rs
Our previous findings that i.pag. orexin-A induced antinociception through an OX1R-initiated eCB signalling [11], where the downstream mechanism was highly similar to i.pag. substance P induced antinociception as shown in the subsection above. In order to ascertain the interaction between orexin-A and substance P in the vlPAG, we challenged orexin-A-induced antinociception with i.pag. NK1R and mGlu5R antagonists, respectively. Co-administration of L-703,606 (10 nmol, i.pag.) or MPEP (30 nmol, i.pag.) significantly antagonized i.pag. orexin-A (1 nmol)-induced antinociception (Fig. 4). The overall comparison of time courses of the antinociceptive effect showed a significant difference between time and treatment (F18,108 = 3.841, p < 0.001, two-way ANOVA, Fig. 4a; F18,108 = 4.597, p < 0.001, two-way ANOVA, Fig. 4b). These results in combination with our previous findings suggest that orexin-A-induced antinociception is mediated by OX1Rs, NK1Rs, mGlu5Rs and CB1Rs sequentially in the vlPAG.

Orexin-A (i.pag.)-induced antinociception is antagonized by i.pag. blockade of NK1Rs or mGlu5Rs. a-b: Time courses of antinociceptive effects (expressed as % MPE) induced by orexin-A (1.0 nmol, i.pag.) in combination with the vehicle or the antagonist of NK1Rs (L-703,606, 10 nmol, i.pag., a or mGlu5Rs (MPEP, 30 nmol, i.pag., b in the mouse hot-plate test. (two-way ANOVA /post hoc Bonferroni test). c: The AUC of the antinociceptive effect in each treatment group (one-way ANOVA /post hoc Tukey test). The antagonist was i.pag. co-administered with i.pag. orexin-A. The data presentation and statistics are the same as in Fig. 2. *p < 0.05, **p < 0.01, ***p < 0.001 vs. the vehicle control group; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. the orexin-A group
NPS (i.c.v.)-induced antinociception was antagonized by i.pag. blockade of OX1Rs, NK1Rs, mGlu5Rs or CB1Rs
Next, we investigated whether the now established OX1R-NK1R-mGlu5R-CB1R cascade in the vlPAG is involved in the supraspinal antinociceptive effect of NPS. Co-administration of the respective antagonists of OX1Rs (SB-334867, 15 nmol, i.pag.), NK1Rs (L-703,606, 10 nmol, i.pag.), mGlu5Rs (MPEP, 30 nmol, i.pag.) or CB1Rs (AM251, 30 nmol, i.pag.), significantly supressed the antinociceptive effect of i.c.v. NPS (0.3 nmol) (Fig. 5), in a time- (F6,114 = 3.252, p = 0.0055, two-way ANOVA, Fig. 5a; F6,114 = 2.936, p = 0.0106, two-way ANOVA, Fig. 5b; F6,114 = 2.603, p = 0.211, two-way ANOVA, Fig. 5c; F6,114 = 2.2, p = 0.0479, two-way ANOVA, Fig. 5d) and treatment- (F3,19 = 36.96, p < 0.001, two-way ANOVA, Fig. 5a; F3,19 = 28.58, p < 0.001, two-way ANOVA, Fig. 5b; F3,19 = 67.33, p < 0.001, two-way ANOVA, Fig. 5c; F3,19 = 23.44, p < 0.001, two-way ANOVA, Fig. 5d) dependent manner. Thus, i.c.v. NPS-induced analgesia is mediated by the OX1Rs, NK1Rs, mGlu5Rs and CB1Rs in the vlPAG.

NPS (i.c.v.)-induced antinociception is antagonized by i.pag. blockade of OX1Rs, NK1Rs, mGlu5Rs or CB1Rs. a-d: Time courses of antinociceptive effects (expressed as % MPE) induced by NPS (0.3 nmol, i.c.v.) in combination with the vehicle or the antagonists of OX1Rs (SB-334867, 15 nmol, i.pag.), NK1Rs (L-703,606, 10 nmol, i.pag.), mGlu5Rs (MPEP, 30 nmol, i.pag.) or CB1Rs (AM251, 30 nmol, i.pag.) in the mouse hot-plate test. (two-way ANOVA /post hoc Bonferroni test). e: The AUC of the antinociceptive effect in each treatment group (one-way ANOVA /post hoc Tukey test). The antagonist was i.pag. Administered immediately before i.c.v. injection of NPS. The data presentation and statistics are the same as in Fig. 2. *p < 0.05, **p < 0.01, ***p < 0.001 vs. the vehicle control group; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. the NPS group
Restraint stress-induced analgesia was prevented by i.c.v. blockade of NPSR or i.pag. blockade of OX1Rs, NK1Rs or mGlu5Rs
We have previously demonstrated that the vlPAG OX1R-CB1R pathway is involved in the SIA induced by an acute restraint stress in mice [12]. We subsequently examined whether the now established NPSR-OX1R-NK1R-mGlu5R-CB1R cascade is involved in the SIA induced by the same restraint stress protocol. Mice receiving an acute restraint stress for 30 min exhibited significant reduced paw withdrawal response in the hot-plate test. This SIA diminished within 20 min (Fig. 6a) [12] and was significantly prevented in mice i.c.v. pre-treated with an NPSR antagonist ([tBu-D-Gly5] NPS, 10 nmol) (Fig. 6a and d) or with i.pag. Pretreated with NK1R (L-703,606, 10 nmol) (Fig. 6b and d) and mGlu5R (MPEP, 30 nmol) antagonists (Fig. 6c and d), respectively. The overall comparison of the time course of the antinociceptive effect showed a significant difference between time and treatment (F18,114 = 4.317, p < 0.001, two-way ANOVA, Fig. 6a; F18,108 = 3.780, p < 0.001, two-way ANOVA, Fig. 6b; F18,108 = 3.501, p < 0.001, two-way ANOVA, Fig. 6c). Take together with our previous findings that SB-334867 (15 nmol, i.pag.) and AM251 (30 nmol, i.pag.) prevented SIA [12], it is suggested that SIA is mediated via the NPSR-evoked OX1R-NK1R-mGlu5R-CB1R cascade in the vlPAG.

Restraint stress-induced antinociception (SIA) is prevented by i.c.v. blockade of NPSR or by i.pag. blockade of NK1Rs or mGlu5Rs. a-c: Time courses of antinociceptive effects (expressed as % MPE) induced by a 30 min-restraint stress (horizontal bars) in mice pre-treated with vehicle or the antagonist of NPSRs ([tBu-D-Gly5] NPS, 10 nmol, i.c.v.), NK1Rs (L-703,606, 10 nmol, i.pag.) or mGlu5Rs (MPEP, 30 nmol, i.pag.) in the hot-plate test. (two-way ANOVA /post hoc Bonferroni test). d: The AUC of the antinociceptive effect in each treatment group (one-way ANOVA /post hoc Tukey test). The antagonist was i.c.v. or i.pag. administered immediately before restraint stress. The data presentation and statistics are the same as in Fig. 2. *p < 0.05, **p < 0.01, ***p < 0.001 vs. the vehicle control group; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. the Stress group
All the tested antagonists at the administered doses that attenuated SIA had no effect on the number of crossing and rearing, including the NPSR antagonist ([tBu-D-Gly5] NPS, 10 nmol, i.c.v.) (Additional file 3: Figure S3a and b), the NK1R antagonist (L-703,606, 10 nmol, i.pag.) (Additional file 3: Figure S3, C and D) and the mGlu5R antagonist (MPEP, 30 nmol, i.pag.) (Additional file 3: Figure S3, E and F), similar to the OX1R antagonist (SB-334867, 15 nmol, i.pag.) and the CB1R antagonist (AM251, 30 nmol, i.pag.) which we have reported previously [12]. This supports that these antagonists attenuate SIA by blocking their respective endogenous ligands.
Restraint stress elevated NPS level in LH and substance P level in vlPAG
Measurement of the neuropeptide content in the brain homogenate revealed that restrain stress significantly elevated the NPS level in the LH (df = 12, t = 2.987, p < 0.05, Student’s t-test, Fig. 7a) and the substance P level in the vlPAG (df = 9, t = 2.72, p < 0.05, Student’s t-test, Fig. 7b). Similar elevations in orexin-A levels were observed in the vlPAG of restrained mice as we previously reported [12].

Restraint stress increases the NPS level in the LH (a) and the substance P level in the vlPAG. (b) Brain tissues containing the LH or vlPAG were punched and homogenized from restrained mice immediately after a 30 min-restraint stress (stress group) or from unrestrained control mice (non-stress group). NPS levels in LH homogenates were measured by an ELISA kit (Cusabio, College Park, MD, USA), whereas substance P level in vlPAG homogenates were measured by an EIA kit (Caymon Chemical. Ann Arbor, MI, USA). *p < 0.05, **p < 0.01 vs. the Non-stress control group (Student’s t-test)
Discussion
In this study, we found that the antinociceptive effect of i.c.v. NPS was blocked by i.c.v. injection of an NPSR antagonist and i.pag. injection of antagonists for OX1Rs, NK1Rs, mGlu5Rs and CB1Rs, respectively. These results suggest that orexins, substance P, glutamate and eCBs in the vlPAG are involved in supraspinal NPS-induced antinociception. In addition, blockade of either NPSRs, OX1Rs, NK1Rs, mGlu5Rs or CB1Rs suppressed the antinociception induced by a 30 min-restraint stress that increased the NPS level in the LH as well as the substance P level in the vlPAG. This suggests that NPS plays a role in SIA by activating the OX1R-NK1R-mGlu5R-CB1R-mediated sequential cascade that leads to antinociception through a disinhibition mechanism (i.e. inhibition of GABA release) mediated by GqPCR-PLC-DAGL-2-AG-CB1R signalling in the vlPAG [11, 12] (Fig. 8). Our results also suggest that restraint stress suppresses pain sensitivity in vivo by engaging the NPS-orexin-A-substance P-glutamate signalosome to initiate the eCB-mediated retrograde disinhibition mechanism in the vlPAG. Integrating with the existing literature, the findings from the present study may fill in the gaps, denoted as [★], among the signalling pathways of SIA as demonstrated by several research groups, as illustrated in Fig. 1.

A proposed schema illustrating how NPS, orexins, substance P, mGlu5R and endocannabinoid (2-AG) may be involved in SIA. Before stress, the projection neurons in the vlPAG is under GABAergic inhibitory control. During stress, hypothalamic orexin neurons (OX) are activated by NPS, which is released possibly from the NPS neurosn in peri-LC and/or the KF-PBN in mice [24], releasing orexins that activate the OX1Rs on neurokinin (SubP) neurons and release substance P in the vlPAG. Then, substance P activates the NK1R-containing glutamate (Glu) neurons, yielding massive glutamate that in turn activates perisynaptic mGlu5Rs to initiate the GqPCR signalling and generate 2-AG. This endocannabinoid then retrogradely activates presynaptic CB1Rs to inhibit GABA release in the vlPAG, ultimately leading to analgesia. The points of pharmacological intervention performed in this study are marked with blunt arrows, labelled with the respective antagonists. The images of neurons are adapted from Illustration Toolkit Neuroscience by Motifolio. PN: projection neuron. GABAAR: GABAA receptor
Substance P exerted antinociceptive effect via NK1Rs, mGlu5Rs and CB1Rs in the vlPAG
Drew et al. [36], using an electrophysiological approach, has shown that, in the vlPAG, substance P can facilitate the release of glutamate that subsequently activates postsynaptic mGlu5Rs located at perisynaptic sites, leading to the synthesis of 2-AG that retrogradely inhibits presynaptic GABA release via CB1Rs. Gregg et al. [40], using a behavioural approach, also demonstrated that activating mGlu5Rs in the PAG can induce an antinociceptive effect mediated by 2-AG and CB1Rs. Here, we further demonstrated that this substance P-initiated and mGlu5R-mediated eCB retrograde signalling contributes to the antinociceptive effect of substance P in the vlPAG since i.pag. substance P-induced antinociception was antagonized by i.pag. blockade of NK1Rs, mGlu5Rs or CB1Rs. The present study also supports that substance P is antinociceptive at the supraspinal level and the vlPAG is one of the sites of action.
The substance P-NK1R-glutamate-mGlu5R cascade acts downstream of orexin-induced antinociception in the vlPAG
The finding that the NK1R antagonist attenuated orexin-induced antinociception in the vlPAG (Fig. 4) suggests that substance P acts downstream of orexin-induced antinociception. This finding is in agreement with a recent study that reported that the level of substance P in the vlPAG was increased following i.pag. orexin-A administration in rats [48]. Previously, we have shown that orexin via OX1Rs induces analgesia through a GqPCR-PLC-DAGL-2-AG-CB1R retrograde disinhibition mechanism in the vlPAG [11]. Given that mGlu5R, a GPCR, is also coupled to Gq proteins and mediates the antinociceptive effect via the same 2-AG-dependent disinhibition mechanism in the PAG [40] as the OX1R [11], it is reasonable to suggest that the mGlu5R is a downstream target after OX1R-NK1R activation. That is, orexin may induce analgesia via a cascade mediated by the OX1R-substance P-NK1R-glutamate-mGlu5R-PLC-DAGL-2-AG-CB1R signalling sequentially in the PAG (Fig. 8).
This sequential cascade may be able to explain the previous finding that i.pag. blockade of NK1Rs attenuated LH stimulation-induced antinociception [49]. It is likely that orexin is the mediator released from the LH to induce antinociception indirectly via the NK1R in the PAG. Additionally, involvement of substance P in the antinociceptive effect of orexin may also explain our previous electrophysiological finding that, in certain recorded vlPAG neurons, orexin-A did not induce postsynaptic depolarization but attenuated GABA release via presynaptic CB1Rs [11]. In addition to the 2-AG spill-over hypothesis, orexin-A may activate neurokinin neurons to release substance P that indirectly inhibits GABA release via mGlu5R-eCB signalling in those neurons that were not depolarized by orexin-A.
The PAG is not the site of action for NPS-induced supraspinal antinociception
In agreement with previous studies that i.c.v. NPS was antinociceptive in swiss mice [19,20,21], we also found i.c.v. NPS reduced the hot-plate nociceptive response in C57BL/6JNarl mice. Peng et al. [20] suggested that the PAG is likely the site of action of NPS since i.c.v. NPS increased the c-Fos expression in the PAG where the NPSR mRNA is abundant [23]. However, our findings that direct i.pag. microinjection of NPS failed to induce antinociception and that i.c.v., but not i.pag., blockade of NPSRs antagonized i.c.v. NPS-induced antinociception indicate that i.c.v. NPS may act in brain regions other than the PAG to exert its antinociceptive effect.
NPS-induced antinociception is mediated via OX1R-NK1R-mGlu5R-CB1R sequentially in the vlPAG
The findings that i.pag. blockade of OX1Rs, NK1Rs, mGlu5Rs and CB1Rs prevented i.c.v. NPS-induced antinociception suggest the involvement of the OX1R-NK1R-mGlu5R-CB1R signalling in the vlPAG in the supraspinal antinociceptive action of NPS. The site of action is likely in the hypothalamic areas where orexin neurons are located, especially the LH that is involved in pain regulation. Ideally, it would be more precise to study the action of NPS and its antagonist on orexin neurons in the LH via intra-LH microinjection. However, due to the difficulty of performing both intra-LH and i.pag. cannulations in mice, i.c.v. and i.pag. microinjections were employed (Figs. 5 and 6). Nevertheless, several studies have suggested an interaction between NPS and orexin systems. Anatomical and functional studies suggest that NPS can activate orexin neurons and may modulate biological functions indirectly via released orexins. First, the hypothalamic regions where orexin neurons are located, including the LH, PFA, and DMH, are enriched with NPSRs [23]. Second, after i.c.v. injection of NPS in rats, fos-immunoreactive cells in the hypothalamus, especially in the LH, were orexin-A-positive [27, 28]. Third, NPS has been reported to be an upstream activator of the orexin system in feeding [27] and addiction [28] behaviours acting in the hypothalamus. Therefore, it is likely that NPS activates the orexin neurons at the LH, releasing orexins in the vlPAG to induce antinociception.
SIA is mediated by endogenous NPS-initiated hypothalamic orexins via the OX1R-NK1R-mGlu5R-CB1R-mediated sequential cascade in the vlPAG
Previously, we have demonstrated that SIA is mediated by orexins released from the LH, an important region for SIA [13], via an OX1R-initiated 2-AG-dependent disinhibition mechanism in the vlPAG [12]. Here, we extend the findings in this study to suggest that NPS activates hypothalamic orexin neurons and add substance P as downstream of vlPAG OX1R activation in this SIA mechanism. That is, during stress, hypothalamic orexin neurons are activated by NPS, which is released possibly from the peri-LC and/or the KF-PBN in mice [24], releasing orexins that activate the OX1Rs on neurokinin neurons in the vlPAG. Then, substance P is released and activates the NK1R-containing glutamate neurons, yielding massive glutamate release that in turn activates perisynaptic mGlu5Rs to initiate GqPCR signalling and generation of 2-AG. This eCB then retrogradely activates presynaptic CB1Rs to inhibit GABA release in the vlPAG, ultimately leading to analgesia (Figs. 1 and 8). This conclusion is based on the following findings, which may fill the gaps [★] in the schema depicted in Fig. 1, that (1) stress increased NPS levels in the LH (Fig. 7a) and SIA was reduced by blocking NPSRs (Fig. 6a); (2) stress increased orexin levels in the vlPAG and SIA was reduced by blocking OX1Rs in the vlPAG [12]; (3) stress increased substance P levels (Fig. 7b) and SIA was reduced by blocking NK1Rs in the vlPAG (Fig. 6b); (4) SIA was reduced by blocking either mGlu5Rs (Fig. 6c), CB1Rs or DAGL in the vlPAG [12]. The antagonist/inhibitor of NPSRs (BuG-NPS, Fig. 6a), OX1Rs (SB-334867) [12], NK1Rs (L-703,606, Fig. 6b), mGlu5Rs (MPEP, Fig. 6c), CB1Rs (AM251) or DAGL (tetrahydrolipstatin) [12] employed at the dose blocking SIA, per se, did not affect nociceptive threshold in unrestrained normal mice, suggesting no non-specific effects of these antagonists employed at the concentrations used in this study.
Since 1990s, substance P has been reported to play a role in SIA while the site(s) of action remain unidentified. Rosen et al. [38] reported that substance P was released from the PAG of animals in response to a behavioural stress, suggesting that endogenous substance P contributes to SIA originated from the PAG-mediated descending pain inhibition. The finding that the antinociceptive effect induced by stimulating the LH was abolished by i.pag. L-703,606 [39], suggesting that stimulating the LH can release substance P to induce antinociception via the NK1Rs in the PAG. Here, we provided direct evidence supporting that SIA is mediated by elevated substance P in the PAG.
Several lines of evidence have indicated the involvement of NPS in stress-induced responses. NPSRs are enriched in the amygdala and hypothalamus [22], stress-related brain regions. The number of c-fos-containing NPS neurons in the peri-LC and KF-PBN was increased after a short-term forced swim stress or restraint stress [24]. The current finding that the acute restraint stress that induces analgesia can increase the NPS level in the LH directly supports that NPS is released during stress and contributes to SIA.
Several reports have indicated a cross-modulatory relationship between NPS and the corticotrophin releasing factor (CRF) system in stress-related responses. Paneda et al. [50] reported that CRF1 receptor may mediate NPS-induced cocaine reinstatement in mice. Conversely, Jungling et al. [4]. demonstrated that CRF can modulate NPS neurons in the LC of mice following acute stress. It remains to be elucidated if interactions between the CRF system and the NPSR-OX1R-NK1R-mGlu5R-CB1R-mediated sequential cascade in SIA occur.
Limitations of the current study
In the current study, we found that NPS (i.c.v.) at 0.3 and 1 nmol in C57BL/6 did not induce significant hyperlocomotion (Additional file 2: Figure S2). This is different with previous studies, where i.c.v. NPS at doses of 0.1 and 1.0 nmol induced hyperlocomotion in C57BL/6 [50] and Swiss mice [4]. However, Rizzi et al. [51], Castro et al. [52] and Boeck et al. [53] consistently demonstrated that i.c.v. NPS, only at the dose of 0.1 nmol, but not 0.01 and 1.0 nmol, exhibited significant hyperlocomotion in CF-1 mice. Furthemore, Holanda et al. [21] reported that i.c.v. NPS at 0.1 nmol did not increase locomotor activity in CF-1 mice. The discrepancy among studies is unclear. It may be that the i.c.v. NPS doses employed under the conditions (mouse strain and the motor activity assessment) in the present study fall outside the optimal dose for inducing hyperlocomotion.
Conclusions
During stress, NPS is released to activate hypothalamic orexin neurons, releasing orexins that activate OX1Rs on neurokinin neurons in the vlPAG, releasing substance P that activates NK1Rs on glutamate neurons, yielding massive glutamate that in turn activates perisynaptic mGlu5Rs to initiate the GqPCR signalling and then generate 2-AG, which then. Retrogradely activates presynaptic CB1Rs to inhibit GABA release in the vlPAG, ultimately leading to analgesia (Fig. 8).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- 2-AG:
-
2-arachidonoylglycerol
- AM251:
-
1-(2,4-Dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide,CB1R, CB1 receptor
- CRF:
-
Corticotrophin releasing factor
- DAGL:
-
Diacylglycerol lipase
- DMH:
-
Dorsomedial hypothalamus
- eCB:
-
endocannabinoid
- EIA:
-
Enzyme immunoassay
- ELISA:
-
Enzyme-linked immunosorbent assay
- GqPCR:
-
Gq-protein coupled receptor
- HPA:
-
Hypothalamus-pituitary axis
- i.c.v. :
-
intra-cerebroventricular
- i.pag :
-
intra-ventrolateral periaqueductal gray
- L-703,606:
-
cis-2-(Diphenylmethyl)-N-[(2-iodophenyl)methyl]-1-azabicyclo [2.2.2] octan-3-amine oxalate salt
- LC:
-
Locus coeruleus
- LH:
-
Lateral hypothalamus
- mGlu5R:
-
mGlu5 receptor
- MPE:
-
Maximal possible effect
- MPEP:
-
2-methyl-6-(phenylethynyl) pyridine hydrochloride
- NK1R:
-
NK1 receptor
- NPS:
-
Neuropeptide S
- NPSR:
-
Neuropeptide S receptor
- OX1R:
-
OX1 receptor
- OX2R:
-
OX2 receptor
- PBN:
-
Parabrachial nucleus
- PFA:
-
Perifornical area
- PLC:
-
Phospholipase C
- PVN:
-
Paraventricular nucleus
- SB-334867:
-
N-(2-Methyl-6-benzoxazolyl)-N′-1,5-naphthyridin-4-yl urea
- SIA:
-
Stress-induced analgesia
- vlPAG:
-
ventrolateral periaqueductal gray
References
Lewis JW, Cannon JT, Stapleton JM, Liebeskind JC. Stress activates endogenous pain-inhibitory systems: opioid and non-opioid mechanisms. Proc West Pharmacol Soc. 1980;23:85–8.
Watanabe S, Kuwaki T, Yanagisawa M, Fukuda Y, Shimoyama M. Persistent pain and stress activate pain-inhibitory orexin pathways. Neuroreport. 2005;16:5–8.
Xie X, Wisor JP, Hara J, Crowder TL, LeWinter R, Khroyan TV, Yamanaka A, Diano S, Horvath TL, Sakurai T, et al. Hypocretin/orexin and nociceptin/orphanin FQ coordinately regulate analgesia in a mouse model of stress-induced analgesia. J Clin Invest. 2008;118:2471–81.
Jungling K, Liu X, Lesting J, Coulon P, Sosulina L, Reinscheid RK, Pape HC. Activation of neuropeptide S-expressing neurons in the locus coeruleus by corticotropin-releasing factor. J Physiol. 2012;590:3701–17.
Ebner K, Muigg P, Singewald G, Singewald N. Substance P in stress and anxiety: NK-1 receptor antagonism interacts with key brain areas of the stress circuitry. Ann N Y Acad Sci. 2008;1144:61–73.
Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–85.
de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS 2nd, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–7.
Tsujino N, Sakurai T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol Rev. 2009;61:162–76.
Harris GC, Aston-Jones G. Arousal and reward: a dichotomy in orexin function. Trends Neurosci. 2006;29:571–7.
Chiou LC, Lee HJ, Ho YC, Chen SP, Liao YY, Ma CH, Fan PC, Fuh JL, Wang SJ. Orexins/hypocretins: pain regulation and cellular actions. Curr Pharm Des. 2010;16:3089–100.
Ho YC, Lee HJ, Tung LW, Liao YY, Fu SY, Teng SF, Liao HT, Mackie K, Chiou LC. Activation of orexin 1 receptors in the periaqueductal gray of male rats leads to antinociception via retrograde endocannabinoid (2-arachidonoylglycerol)-induced disinhibition. J Neurosci. 2011;31:14600–10.
Lee HJ, Chang LY, Ho YC, Teng SF, Hwang LL, Mackie K, Chiou LC. Stress induces analgesia via orexin 1 receptor-initiated endocannabinoid/CB1 signaling in the mouse periaqueductal gray. Neuropharmacology. 2016;105:577–86.
Gerashchenko D, Horvath TL, Xie XS. Direct inhibition of hypocretin/orexin neurons in the lateral hypothalamus by nociceptin/orphanin FQ blocks stress-induced analgesia in rats. Neuropharmacology. 2011;60:543–9.
Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–25.
Bannister K, Dickenson AH. The plasticity of descending controls in pain: translational probing. J Physiol. 2017;595:4159–66.
Turunen PM, Jantti MH, Kukkonen JP. OX1 orexin/hypocretin receptor signaling through arachidonic acid and endocannabinoid release. Mol Pharmacol. 2012;82:156–67.
Cristino L, Luongo L, Imperatore R, Boccella S, Becker T, Morello G, Piscitelli F, Busetto G, Maione S, Di Marzo V. Orexin-A and Endocannabinoid activation of the descending Antinociceptive pathway underlies altered pain perception in Leptin signaling deficiency. Neuropsychopharmacology. 2016;41:508–20.
Kukkonen JP. G-protein-dependency of orexin/hypocretin receptor signalling in recombinant Chinese hamster ovary cells. Biochem Biophys Res Commun. 2016;476:379–85.
Li W, Chang M, Peng YL, Gao YH, Zhang JN, Han RW, Wang R. Neuropeptide S produces antinociceptive effects at the supraspinal level in mice. Regul Pept. 2009;156:90–5.
Peng YL, Zhang JN, Chang M, Li W, Han RW, Wang R. Effects of central neuropeptide S in the mouse formalin test. Peptides. 2010;31:1878–83.
Holanda AD, Asth L, Santos AR, Guerrini R, de PS-RV CG, Andre E, Gavioli EC. Central adenosine A1 and A2A receptors mediate the antinociceptive effects of neuropeptide S in the mouse formalin test. Life Sci. 2015;120:8–12.
Xu YL, Gall CM, Jackson VR, Civelli O, Reinscheid RK. Distribution of neuropeptide S receptor mRNA and neurochemical characteristics of neuropeptide S-expressing neurons in the rat brain. J Comp Neurol. 2007;500:84–102.
Clark SD, Duangdao DM, Schulz S, Zhang L, Liu X, Xu YL, Reinscheid RK. Anatomical characterization of the neuropeptide S system in the mouse brain by in situ hybridization and immunohistochemistry. J Comp Neurol. 2011;519:1867–93.
Liu X, Zeng J, Zhou A, Theodorsson E, Fahrenkrug J, Reinscheid RK. Molecular fingerprint of neuropeptide S-producing neurons in the mouse brain. J Comp Neurol. 2011;519:1847–66.
Smith KL, Patterson M, Dhillo WS, Patel SR, Semjonous NM, Gardiner JV, Ghatei MA, Bloom SR. Neuropeptide S stimulates the hypothalamo-pituitary-adrenal axis and inhibits food intake. Endocrinology. 2006;147:3510–8.
Okamura N, Reinscheid RK. Neuropeptide S: a novel modulator of stress and arousal. Stress. 2007;10:221–6.
Niimi M. Centrally administered neuropeptide S activates orexin-containing neurons in the hypothalamus and stimulates feeding in rats. Endocrine. 2006;30:75–9.
Kallupi M, Cannella N, Economidou D, Ubaldi M, Ruggeri B, Weiss F, Massi M, Marugan J, Heilig M, Bonnavion P, et al. Neuropeptide S facilitates cue-induced relapse to cocaine seeking through activation of the hypothalamic hypocretin system. Proc Natl Acad Sci U S A. 2010;107:19567–72.
Ubaldi M, Giordano A, Severi I, Li H, Kallupi M, de Guglielmo G, Ruggeri B, Stopponi S, Ciccocioppo R, Cannella N. Activation of Hypocretin-1/Orexin-A neurons projecting to the bed nucleus of the Stria Terminalis and Paraventricular nucleus is critical for reinstatement of alcohol seeking by neuropeptide S. Biol Psychiatry. 2016;79:452–62.
Hokfelt T, Pernow B, Wahren J. Substance P: a pioneer amongst neuropeptides. J Intern Med. 2001;249:27–40.
Mantyh PW. Neurobiology of substance P and the NK1 receptor. J Clin Psychiatry. 2002;63(Suppl 11):6–10.
Minami M, Kuraishi Y, Kawamura M, Yamaguchi T, Masu Y, Nakanishi S, Satoh M. Enhancement of preprotachykinin a gene expression by adjuvant-induced inflammation in the rat spinal cord: possible involvement of substance P-containing spinal neurons in nociception. Neurosci Lett. 1989;98:105–10.
Stewart JM, Getto CJ, Neldner K, Reeve EB, Krivoy WA, Zimmermann E. Substance P and analgesia. Nature. 1976;262:784–5.
Rosen A, Zhang YX, Lund I, Lundeberg T, Yu LC. Substance P microinjected into the periaqueductal gray matter induces antinociception and is released following morphine administration. Brain Res. 2004;1001:87–94.
Quirion R, Shults CW, Moody TW, Pert CB, Chase TN, O'Donohue TL. Autoradiographic distribution of substance P receptors in rat central nervous system. Nature. 1983;303:714–6.
Drew GM, Lau BK, Vaughan CW. Substance P drives endocannabinoid-mediated disinhibition in a midbrain descending analgesic pathway. J Neurosci. 2009;29:7220–9.
Drew GM, Mitchell VA, Vaughan CW. Glutamate spillover modulates GABAergic synaptic transmission in the rat midbrain periaqueductal grey via metabotropic glutamate receptors and endocannabinoid signaling. J Neurosci. 2008;28:808–15.
Rosen A, Brodin K, Eneroth P, Brodin E. Short-term restraint stress and s.c. saline injection alter the tissue levels of substance P and cholecystokinin in the peri-aqueductal grey and limbic regions of rat brain. Acta Physiol Scand. 1992;146:341–8.
Holden JE, Pizzi JA, Jeong Y. An NK1 receptor antagonist microinjected into the periaqueductal gray blocks lateral hypothalamic-induced antinociception in rats. Neurosci Lett. 2009;453:115–9.
Gregg LC, Jung KM, Spradley JM, Nyilas R, Suplita RL 2nd, Zimmer A, Watanabe M, Mackie K, Katona I, Piomelli D, Hohmann AG. Activation of type 5 metabotropic glutamate receptors and diacylglycerol lipase-alpha initiates 2-arachidonoylglycerol formation and endocannabinoid-mediated analgesia. J Neurosci. 2012;32:9457–68.
Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–12.
Suplita RL 2nd, Farthing JN, Gutierrez T, Hohmann AG. Inhibition of fatty-acid amide hydrolase enhances cannabinoid stress-induced analgesia: sites of action in the dorsolateral periaqueductal gray and rostral ventromedial medulla. Neuropharmacology. 2005;49:1201–9.
Hwang LL, Wang CH, Li TL, Chang SD, Lin LC, Chen CP, Chen CT, Liang KC, Ho IK, Yang WS, Chiou LC. Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity (Silver Spring). 2010;18:463–9.
Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates: Academic. San Diego; 1997.
Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54.
Guerrini R, Camarda V, Trapella C, Calo G, Rizzi A, Ruzza C, Fiorini S, Marzola E, Reinscheid RK, Regoli D, Salvadori S. Synthesis and biological activity of human neuropeptide S analogues modified in position 5: identification of potent and pure neuropeptide S receptor antagonists. J Med Chem. 2009;52:524–9.
Ruzza C, Rizzi A, Camarda V, Pulga A, Marzola G, Filaferro M, Novi C, Ruggieri V, Marzola E, Vitale G, et al. [tBu-D-Gly5] NPS, a pure and potent antagonist of the neuropeptide S receptor: in vitro and in vivo studies. Peptides. 2012;34:404–11.
Raoof M, Soofiabadi S, Abbasnejad M, Kooshki R, Esmaeili-Mahani S, Mansoori M. Activation of orexin-1 receptors in the ventrolateral periaqueductal grey matter (vlPAG) modulates pulpal nociception and the induction of substance P in vlPAG and trigeminal nucleus caudalis. Int Endod J. 2019;52:318–28.
Holden JE, Pizzi JA. Lateral hypothalamic-induced antinociception may be mediated by a substance P connection with the rostral ventromedial medulla. Brain Res. 2008;1214:40–9.
Paneda C, Huitron-Resendiz S, Frago LM, Chowen JA, Picetti R, de Lecea L, Roberts AJ. Neuropeptide S reinstates cocaine-seeking behavior and increases locomotor activity through corticotropin-releasing factor receptor 1 in mice. J Neurosci. 2009;29:4155–61.
Rizzi A, Vergura R, Marzola G, Ruzza C, Guerrini R, Salvadori S, Regoli D, Calo G. Neuropeptide S is a stimulatory anxiolytic agent: a behavioural study in mice. Br J Pharmacol. 2008;154:471–9.
Castro AA, Moretti M, Casagrande TS, Martinello C, Petronilho F, Steckert AV, Guerrini R, Calo G, Dal Pizzol F, Quevedo J, Gavioli EC. Neuropeptide S produces hyperlocomotion and prevents oxidative stress damage in the mouse brain: a comparative study with amphetamine and diazepam. Pharmacol Biochem Behav. 2009;91:636–42.
Boeck CR, Martinello C, de Castro AA, Moretti M, Dos Santos CT, Guerrini R, Calo G, Gavioli EC. Blockade of adenosine A2A receptor counteracts neuropeptide-S-induced hyperlocomotion in mice. Naunyn Schmiedeberg's Arch Pharmacol. 2010;381:153–60.
Acknowledgements
We appreciate the contribution from Prof. Ken Mackie (Indiana University Bloomington, USA) for providing critical comments and suggestions in improving this manuscript.
Funding
This study was supported by grants from the Ministry of Science and Technology, Taiwan (MOST104–2314-B-002-053-MY3, MOST106–2321-B-002-019, MOST106–2811-B-002-124, MOST107–2321-B-002-010, MOST107–2811-B-002-008, MOST 108–2321-B-002-005 and MOST 108–2320-B-002-029-MY3), National Health Research Institutes, Taiwan (NHRI-EX102-10251NI, NHRI-EX107-10733NI) and Ministry of Education, Taiwan (107 M4022–3).
Author information
Authors and Affiliations
Contributions
MTL contributed to data interpretation, data analysis, results discussion and paper writing; YTC and YCC conducted experiments, analyzed data and paper writing; CCH and HJL conducted the experiments; RG and GC synthesized and provided compounds as well as contributed to results discussion; LCC designed experiments, analyzed data and wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal experiments were approved by the Institutional Animal Care and Use Committee of College of Medicine, National Taiwan University following ARRIVE guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Figure S1. The representative diagram of i.pag. (A) and i.c.v. (B) microinjections in mice. The diagrams were adapted from mouse brain atlas [44]. The black dots (A) and white dots (B) represent the microinjection sites.
Additional file 2:
Figure S2. Effects of NPS on locomotor activity. Locomotor activity in the open field test was measured before and 10 min after i.c.v. administration of 0.3 nmol (A-B) or 1 nmol (C-D) of NPS. Locomotor activity was assessed by the number of crossing (A & C) and rearing (B & D) in the open field test for 5 min. Data are expressed as the mean ± S.E.M. (Unpaired t-test)
Additional file 3:
Figure S3. Effects of [tBu-D-Gly5] NPS, L-703,606 or MPEP on locomotor activity. Locomotor activity in the open field test was measured before and 10 min after administration of [tBu-D-Gly5] NPS (10 nmol, i.c.v.) (A-B), L-703,606 (10 nmol, i.pag.) (C-D), or MPEP (30 nmol, i.pag.) (E-F). Locomotor activity was assessed by the number of crossing (A, C & E) and rearing (B, D & F) in the open field test for 5 min. Data are expressed as the mean ± S.E.M. (Unpaired t-test)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lee, M.T., Chiu, YT., Chiu, YC. et al. Neuropeptide S-initiated sequential cascade mediated by OX1, NK1, mGlu5 and CB1 receptors: a pivotal role in stress-induced analgesia. J Biomed Sci 27, 7 (2020). https://doi.org/10.1186/s12929-019-0590-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-019-0590-1