
Abstract
Enterovirus A71 (EV-A71) is an important emerging virus posing a threat to children under five years old. EV-A71 infection in infants or young children can cause hand-foot-and-mouth disease, herpangina, or severe neurological complications. However, there are still no effective antivirals for treatment of these infections. In this review, we summarize the antiviral compounds developed to date based on various targets of the EV-A71 life cycle. Moreover, development of a vaccine would be the most effective approach to prevent EV-A71 infection. Therefore, we also summarize the development and clinical progress of various candidate EV-A71 vaccines, including inactivated whole virus, recombinant VP1 protein, synthetic peptides, viral-like particles, and live attenuated vaccines.
Similar content being viewed by others

Background
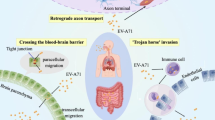
Enterovirus A71 (EV-A71), a positive-strand RNA virus of the family Picornaviridae, represents a persistent global health problem and has caused large outbreaks in the Asia-Pacific region in recent years [1]. Infection by EV-A71 can result in hand-foot-and-mouth disease (HFMD) and herpangina. Children under five years old are particularly susceptible to the most severe forms of EV-A71-associated neurological complications, including aseptic meningitis, brainstem and/or cerebellar encephalitis, myocarditis, acute flaccid paralysis, and rapid fatal pulmonary edema and hemorrhage [2]. Owing to the lack of effective drugs for inhibiting EV-A71 infection, supportive therapy remains the primary means of managing severe cases. Nevertheless, there has been substantial progress in identifying candidate targets for anti-viral drugs and vaccines.
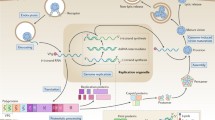
The enterovirus genome encodes four structural capsid proteins (VP1, VP2, VP3, and VP4) that facilitate the cellular entry and delivery of the viral genome into the cytosol of the host cell, and seven non-structural proteins (2Apro, 2B, 2C, 3A, 3B, 3Cpro, and 3Dpol) that mediate viral RNA replication [3]. Antiviral therapy and vaccines can have a variety of mechanisms of action and each step of the virus replication cycle can be targeted. Here, we summarize these recent advances and some of the key compounds showing potential for new therapeutic strategies in the development of vaccine and antiviral compounds that target the life cycle of EV-A71, and discuss the prospects and challenges in this field.
Inhibitors of the EV-A71 life cycle
Capsid inhibitors
The first step in successful viral infection is receptor binding, and the capsid protein VP1 is involved in the recognition of EV-A71 receptors on the surface of host cells. Numerous viral receptors that are responsible for the entry of EV-A71 into host cells have been characterized, including human scavenger receptor class B member 2 (hSCARB2) [4], human P-selectin glycoprotein ligand 1 (PSGL-1) [5], annexin A2 (Anx2) [6], heparan sulfate [7], sialylated glycan [8], and dendritic cell-specific intercellular adhesion molecule-3 grabbing nonintegrin (DC-SIGN) [9]. Various approaches have been proposed for the discovery of antivirals targeting EV-A71 host receptor binding.
Antibodies against SCARB2, PSGL-1, and DC-SIGN effectively inhibited EV-A71 infection in a dose-dependent manner [4, 5, 10, 11]. EV-A71 VP1 inhibitors were the first candidates proposed for developing antivirals against viral infection. To date, pleconaril and vapendavir have been identified to bind to the viral capsid and thus inhibit EV-A71 infection [12]. Moreover, the capsid binder pyridyl imidazolidinone showed notable potency against EV-A71 infection in several consecutive studies [13,14,15]. Pyridyl imidazolidinone fits into the viral hydrophobic pocket of VP1 to inhibit viral infection [16, 17]. In addition, an imidazolidinone derivative (PR66) was found to inhibit EV-A71 infection by impeding the uncoating process via its interaction with the capsid protein VP1. PR66 could also protect against EV-A71-induced neurological symptoms in vivo by suppressing EV-A71 replication [18].
One compound (14) of aminopyridyl 1,2,5-thiadiazolidine 1,1-dioxides, which was capsid inhibitor, showed anti-EV-A71 infection effects (EC50 = 4 nM) and exhibited good in vivo efficiency in an EV-A71-infected mouse model [19]. A sulfonated food azo dye, Brilliant Black BN (E151), was reported to inhibit EV-A71 infection by competing with EV71 attachment factors for viral binding, thereby blocking viral attachment/entry to host cells in vitro; moreover, in vivo studies demonstrated that daily administration of E151 at 200 mg/kg given in the initial four days of challenge protected AG129 mice challenged with a 10 of 50% lethal dose (LD50) of EV-A71 [20].
2Apro inhibitors
2Apro is enterovirus-encoded protease ad is important for viral polyprotein processing [21, 22]. 2Apro could also cleave the host factor eIF4GI to inhibit the cap-dependent translation of cellular mRNA. Falah and coworkers showed that a six-amino acid peptide (LVLQTM) exhibited antiviral potencies against EV-A71 in HeLa cells. This peptide bound to the 2Apro active site and inhibited eIF4GI cleavage by 2Apro [23].
2B inhibitor
Viral protein 2B and its precursor 2 BC have been suggested to be responsible for membranous alteration and inhibition of cellular protein secretion in infected cells [24, 25]. EV-A71 2B protein also induced cell apoptosis by modulating Bax protein activation [26]. Xie and colleagues reported that 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS), which is a chloride-dependent current inhibitor, could prevent EV-A71 2B activity and lead to the inhibition of virus production in RD cells [27].
2C inhibitor
The 2C protein plays a role in viral replication complex formation and is involved in processing nucleoside triphosphatase activity and in the synthesis of RNA-negative strands [28, 29]. Two adenosine analogs, metrifudil and N6-benzyladenosine, have been demonstrated to interact with 2C protein to inhibit EV-A71 infection [30].
3A inhibitor
The 3A protein also plays a role in viral replication complex formation and inhibits cellular protein secretion. An enviroxime mimetic compound, AN-12-H5, was found to inhibit EV-A71 infection. Assays with resistant mutants have suggested that AN-12-H5 blocked replication by targeting 3A and also inhibited an early stage of infection by targeting VP1 and VP3 [31]. Another compound, GW5074, 3-(3,5-dibromo-4-hydroxybenzylidine-5-iodo-1,3-dihydro-indol-2-one), which is a Raf-1 inhibitor, has been demonstrated to target 3A to inhibit EV-A71 infection [30, 32].
3Cpro inhibitor
The 3C protein acts as a protease to cleave viral polypeptides toward their conversion to mature viral proteins during viral infection, and is thus another useful target for antiviral therapy. The compound rupintrivir (also known as AG7088) inhibited EV-A71 3C protein by mimicking the substrate of the 3C protein in vitro and protected suckling mice from EV-A71-caused limb paralysis in vivo [33, 34]. On the other hand, a series of rupintrivir analogues have also been synthesized and more inhibited EV-A71 3C protease activity and viral growth than rupintrivir [35]. Another 3C protease inhibitor (SG85) also inhibited the replication of 21 EV-A71 strains [12].
Cyanohyfdrin (R)-1 is another potent inhibitor of EV-A71 3Cpro but was unstable and showed potential toxicity. Modifying the labile cyanohydrin moiety led to the discovery of the 4-iminooxazolidin-2-one-based inhibitors 4e and 4 g with potent inhibitory activity and significantly improved stability [36]. One small-molecule inhibitor, DC07090, inhibited EV-A71 replication with an EC50 value of 22.09 ± 1.07 μM by targeting 3C protease [37]. Luteoloside is a member of the flavonoids family that exhibits several bioactivities, including anti-microbial and anti-cancer activities, and was also shown to act as a 3C protease inhibitor of EV-A71 in vitro [38].
3Dpol inhibitor
3Dpol of EV-A71 is an RNA-dependent RNA polymerase that plays a role in viral RNA synthesis. DTriP-22 is a non-nucleoside analogue that was shown to inhibit EV-A71 infection by reducing the accumulation of viral RNA [39]. Aurintricarboxylic acid, a compound of a group of polyanionic compounds, could also prevent EV-A71 infection through interference with 3Dpol in vitro [40]. As another antiviral strategy, monoclonal antibodies against EV-A71 3Dpol were generated to inhibit polymerase activity and viral replication [41].
Apolipoprotein B messenger RNA-editing enzyme catalytic polypeptide-like 3G (APOBEC3G or A3G) can interact with viral 3Dpol and viral RNA and can be packaged into progeny virions to reduce the infectivity. APOBEC3G is also a mediator of the antiviral activity of IMB-Z, an N-phenylbenzamide derivative [42].
Viral release inhibitor
Retro-2cycl and Retro-2.1 are inhibitors of several pathogens specifically targeting intracellular vesicle transport, and also participate in EV-A71 life cycle processes, including progeny virus release in vitro. Administration of Retro-2cycl at 10 mg/kg significantly protected 90% of newborn mice from lethal EV-A71 challenge [43].
Internal ribosome entry site (IRES) inhibitor
The 5′ untranslated region (UTR) of the EV-A71 genome is about 745 nucleotides long and highly structured, containing a cloverleaf-like structure that is critical for viral RNA synthesis and an IRES that is important for viral translation. Idarubicin (IDR) is an anthracycline compound and a USA Food and Drug Administration-approved anticancer drug. IDR inhibits EV-A71 through impaired binding between the EV-A71 IRES RNA and hnRNP A1, a known host IRES trans-acting factor [44].
Other strategies targeting EV-A71
Ribavirin
Ribavirin is a nucleotide analogue that can serve as a base analogue of either ATP or GTP, and was reported to reduce the EV-A71 titer in vitro. Ribavirin also significantly reduced the mortality, morbidity, and subsequent paralysis sequelae in EV-A71-infected mice [45, 46].
RNA interference
RNA interference, a native and specific post-transcriptional gene silencing mechanism, has also been exploited as another antiviral tool against EV-A71 infection in vitro and in vivo. Short hairpin RNA (shRNA) expression plasmids or small interfering RNAs (siRNAs) that specifically targeted viral genome to inhibit viral protein expression and viral infection [47,48,49,50,51,52].
MicroRNA (miRNA)
MiRNAs are approximately 19–24-nucleotide-long non-coding RNAs that post-transcriptionally repress gene expression by targeting mRNAs, and play a pivotal role in the complicated interaction networks between viruses and their hosts. MiRNAs regulate viral replication through multiple mechanisms. For example, miR-9-5p was shown to exert an anti-EV-A71 effect in cells and in a mouse model via mediating the nuclear factor-kappa B (NF-κB) activity of the RIG-I signaling pathway [53]. In addition, miR-2911 inhibited EV-A71 replication via targeting the VP1 gene [54]. MiR-23b could also inhibit EV-A71 replication through downregulation of EV-A71 VPl protein [55]. Overexpression of miR-16-5p enhanced EV-A71-induced apoptosis and inhibited viral replication [56]. MiR-134 inhibited both EV-A71 and poliovirus infection [57], and miR-27a suppressed EV-A71 replication by directly targeting the epidermal growth factor receptor gene [58]. The human miRNA hsa-miR-296-5p suppressed EV-A71 replication by targeting the viral genome located in the regions of nt 2115 to 2135 and nt 2896 to 2920 (strain BrCr) [59]. These studies provide novel mechanisms for the miRNA-mediated regulation of EV-A71 in host cells, suggesting a novel approach in combating infection and in the development of antiviral strategies.
Heparan sulfate (HS) mimetics
HS is present in the extracellular matrix, on cell surfaces, and in the intracellular granule secretions of all types of animal tissues. HS mimetics are a group of soluble synthetic or semi-synthetic compounds that are structurally related to cellular HS, and can stimulate the functions of cell-surface HS. HS is also a receptor of EV-A71. HS mimetics exhibited anti-EV-A71 activity at less than 250 mg/ml in Vero cells [60].
Signal pathway targets
GS-9620, a potent and selective agonist of Toll-like receptor 7, could inhibit EV-A71 replication mainly through the NF-κB and PI3K-AKT signaling pathways [61]. Berberine inhibited EV-A71 replication by downregulating autophagy and the MEK/ERK signaling pathway [62]. Isochlorogenic acid C showed antioxidant activity and prevented EV-A71 infection by modulating the redox homeostasis of glutathione [63].
Development of an EV-A71 vaccine
Inactivated whole EV-A71 vaccine
Vaccination is considered to be one of the most effective ways to protect against virus infection. Although there are many different approaches available for developing EV-A71 vaccines, including inactivating the whole virus, a live attenuated virus, virus-like particles (VLPs), recombinant subunits, and synthetic peptides, currently, only an inactivated whole virus vaccine for EV-A71 is the only candidate that has proceeded to a completed human clinical trial. To date, inactivated whole EV-A71 vaccines have been established in Taiwan, China, and Singapore. Three vaccine organizations, including Beijing Vigoo Biological Co., Ltd. (Vigoo), Sinovac Biotech Co., Ltd. (Sinovac), and the Chinese Academy of Medical Sciences (CAMS) in China completed EV-A71 vaccine phase III clinical trials in 2013 and received a license for their administration that was approved by China’s Food and Drug Administration in 2015 [64, 65].
These three vaccine organizations in China used different technologies to develop an EV-A71 vaccine. CAMS used KMB-17 human diploid cells as a cell bank that were cultured using a cell factory, whereas Vigoo and Sinovac used Vero cells to amplify EV-A71 with a microcarrier bioreactor and a cell factory, respectively. All organizations selected the EV-A71 C4 subgenotype as a virus seed for vaccine development, which is the most prevalent genotype circulating in China, although they each used a different virus strain: CAMS chose the EV-A71 FY-23 strain, Vigoo chose the FY7VP5 strain, and Sinovac chose the H07 strain. The three organizations began their phase I clinical trials in 2010 to 2011, and completed their phase III clinical trials in 2013. In the Vigoo phase III clinical trial, a total of 10,245 participants aged 6–35 months randomly received a 320 U (EV-A71 antigen unit) alum-adjuvant vaccine (5120 participants) or a placebo control (5125 participants) at days 0 and 28, and were then followed-up for 1 [66] and 2 years [67] (ClinicalTrials.gov, number NCT01508247). The efficacy of the Vigoo EV-A71 vaccine against EV-A71-associated HFMD was 90%, and that against other EV-A71-associated diseases was 80.4% during the 1-year surveillance period. In addition, the vaccine efficacy against EV-A71-associated HFMD was 100% during the second year, and no serious adverse events were reported. Thus, Vigoo claimed that their EV-A71 vaccine is safe and had good efficacy for protecting against EV-A71-associated HFMD in children. Sinovac also conducted a follow-up study for 1 and 2 years [68, 69] in which a total of 10,077 participants aged 6–35 months were assigned to two groups receiving 400 U of the alum-adjuvant Sinovac EV-A71 vaccine or a placebo control at days 0 and 28 (ClinicalTrials.gov, number NCT01507857). During the 1-year surveillance period, the vaccine efficacy was 94.8% against EV-A71-associated HFMD or herpangina, and was 100% against EV-A71-associated HFMD with neurological complications. Given this success, they extended their study to follow-up the vaccine efficacy for another 12 months [69], and reported a vaccine efficacy of 95.1% for the second year; the overall efficacy of the Sinovac EV-A71 vaccine against EV-A71-associated HFMD was 94.7% [68]. Recently, a five-year follow-up study also indicated that the Sinovac EV-A71 vaccine showed long-term immunity persistence [70]. In the phase III clinical trial of the CAMS EV-A71 vaccine, 12,000 children of 6–71 months of age were assigned (at a 1:1 ratio) to receive 100 U of the alum-adjuvant vaccine or placebo control (ClinicalTrials.gov number, NCT01569581). The vaccine efficacy against EV-A71-associated HFMD was 97.4% [71]. However, both the Sinovac and CAMS EV-A71 vaccines showed no efficacy against HFMD caused by coxsackievirus A16 (CV-A16), demonstrating their specificity [68, 71].
In contrast to these three organizations in China, the National Health Research Institutes (NHRI) in Taiwan used the EV-A71 clinical isolate E59 strain (B4 subgenotype) as a virus seed, which was grown in Vero cells cultured with roller-bottle technology. This strain was chosen for producing the EV-A71 inactivated vaccine because of its confirmed genetic stability over several passages and its ability to grow well in Vero cells [72]. The phase I clinical trial of the NHRI was completed in 2012 (ClinicalTrials.gov number, NCT01268787). Sixty heathy adults aged 20–60 years randomly received two intramuscular doses of either 5 μg of EV71 antigen with 150 μg of aluminum adjuvant or 10 μg of EV71 antigen with 300 μg of aluminum adjuvant, 21 days apart. The immunogenicity results indicated that the EV-A71 vaccine produced from the NHRI was safe and immunogenic in healthy adults [73]. Moreover, over 85% of the participants developed a strong cross-neutralizing antibody response against subgenotypes B1, B5, and C4a; however, only 20% of the participants developed a weak cross-neutralizing antibody response against subgenotype C4b and CV-A16 [74]. Two organizations of Taiwan, Enimmune Corp. and Medigen Vaccinology Corp., continue to evaluate the safety and immunogenicity of the E59 strain EV-A71 vaccine in phase II clinical trials (ClinicalTrials.gov number, NCT02777411, NCT03268083 and NCT02200237). In the clinical trial of Medigen Vaccinology Corp., a total of 365 infants or children aged 2 months to 11 years received different doses (low, mid, or high) of alum-adjuvant EV-A71 vaccine or the placebo control in a double-blind and randomized design (ClinicalTrials.gov number, NCT02200237). No vaccine-related serious adverse events were reported in this trial. In addition, the EV-A71 vaccine could elicit an immune response against not only subgenotype B4 but also B5, C4a, C4b, and C5. The EV-A71 vaccine also showed persistence for 2 years [75]. Based on these findings, Medigen Vaccinology Corp. is initiating a phase III clinical trial (ClinicalTrials.gov number, NCT03865238) in 2019, which is expected to be completed in 2022.
In Singapore, Inviragen Inc. (Takeda Pharmaceuticals International, Inc.) completed a phase I clinical trial of an EV-A71 vaccine in April 2012 (ClinicalTrials.gov number, NCT01376479). In contrast to the organizations of China and Taiwan, Inviragen used the B3 subgenotype as the virus seed for EV-A71 vaccine production, which was named INV21. A total of 36 adults aged 21–45 years received two doses (low or high) of INV21 or placebo control 28 days apart. Inviragen claimed that INV21 induced a high immune response against HFMD caused by EV-A71. However, there has been no further clinical trial conducted in Singapore recently.
Recombinant VP1 vaccine
VP1 is not only a structural protein of EV-A71 but also exhibits strong antigenicity. Accordingly, several research groups have adopted various strategies to express EV-A71 VP1. Wu et al. [76] produced recombinant VP1 proteins of EV-A71 expressed by Escherichia coli (E. coli). The purified VP1 proteins were then injected into adult female mice through an intraperitoneal route. Although the VP1 subunit vaccine could protect suckling mice against a lower challenge dose of EV-A71 (230 LD50 virus/mouse), the inactivated EV-A71 vaccine still elicited a greater immune response than the VP1 subunit vaccine and protected suckling mice against a lethal dose (2300 LD50 virus/mouse) of EV-A71. Zhou et al. [77] also expressed recombinant VP1 protein in E. coli, and then vaccinated rabbits with the purified VP1 protein or heat-inactivated EV-A71 virus, which elicited comparable humoral and cellular immune responses. Moreover, maternal antibodies protect newborn mice against EV-A71 challenge. EV-A71-specific antibodies of immunized mice were elicited by purified recombinant baculovirus expressing VP1. In addition, the antisera exhibited cross-neutralization activities against different subgenotypes of EV-A71 [78]. Wang et al. [79] generated an HIV-gag-based VLP as a carrier to express EV-A71 VP1 protein, which provided passive protection of newborn mice against EV-A71 infection.
VP1 protein has also been developed as an antigen for oral vaccine development. Adult female BALB/c mice were orally immunized with transgenic tomato fruit expressing VP1 protein [80], attenuated Salmonella enterica serovar Typhimurium expressing VP1 [81], VP1-expressing Bifidobacterium longum [82], surface-displayed VP1 Saccharomyces cerevisiae [83], or recombinant Lactococcus lactis expressing secretory VP1 [84]. All of these VP1-expressing vaccines elicited immune responses by oral immunization and could protect newborn mice against EV-A71 infection. Chen et al. [85] generated a transgenic mouse that can express VP1 and secrete into their milk, which could protect suckling mice against EV-A71 challenge. However, the recombinant VP1 proteins generally exhibited lower protective efficacy in mice compared to the inactivated EV-A71 virus.
Synthetic peptide vaccines
Synthetic peptides have also been tested as an alternative strategy to develop EV-A71 vaccines, which are considered to be safe and efficacious for multivalent vaccines development. The majority of research related to antigen peptides has focused on mapping EV-A71 structural proteins (VP1, VP2, VP3, and VP4). Initially, Foo et al. [86] found that two peptides, SP55 (amino acids 163–177 of VP1) and SP70 (amino acids 208–222 of VP1), could elicit neutralizing antibodies against EV-A71. SP70 elicited a higher titer of neutralizing antibody (1:32) than the neutralizing antibody of SP55 (1:8); however, antisera from heat-inactivated EV-A71-immunized mice elicited the highest neutralization titer of 1:128 [86]. Moreover, Foo et al. [87] found that anti- SP70 antisera passively protected suckling mice against both homologous and heterologous EV-A71 strains. In another strategy, six synthetic peptides (P70–159 in VP2, P140–249 in VP2, P324–443 in VP2, and P746–876 in VP1) were combined, which induced the antisera and passively protected newborn mice against EV-A71 infection [88]. The synthetic peptide VP2–28 (amino acids 136–150 of VP2) showed cross-neutralizing activity against EV-A71 and can bind to the anti-EV-A71 monoclonal antibody MAB979 [89]. Xu et al. [90] generated a fusion protein with hepatitis B virus core protein (HBc) and VP2 epitope corresponding to amino acids 141–155 of VP2, named HBc-VP2 (aa141–155), which induced cross-neutralizing EV-A71 antibodies, and the anti-sera from HBc-VP2 (aa141–155) immunized mice protected newborn mice from EV-A71 infection. Huo et al. [91] used the same strategy to display EV-A71 epitopes (SP70, amino acids 208–222 of VP1) and CV-A16 (PEP91, amino acids 271–285 of VP1) using HBc as a carrier protein. The chimeric VLP expressing SP70 and PEP91 epitopes induced an immune response and protected suckling mice against both EV-A71 and CV-A16 infection. Aw-Yong et al. [92] sought to comprehensively map the potential synthetic peptides within the structural and non-structural proteins of EV-A71. A total of 63 synthetic peptides were synthesized and used for characterization of EV-A71 B-cell linear epitopes. Among these, synthetic peptide PEP27 (VP1 residues 142–156) was recognized as an EV-A71 IgM-specific immunodominant epitope; moreover, synthetic peptide PEP23 (VP1 residues 41–55) was identified as an EV-A71 IgG cross-reactive immunodominant epitope. Jiang et al. [93] utilized the norovirus P protein as a carrier for delivery of the EV-A71 epitope, which is the 71–6 epitope spanning amino acids 176–190 of VP3. Sera from mice immunized with chimeric P protein displaying the 71–6 epitope protected suckling mice against a lethal dose of EV-A71 challenge.
VLP-based vaccines
VLPs have been applied in the production of other viral vaccines such as hepatitis B virus and human papillomavirus, and could also be a suitable choice for EV-A71 vaccine development. The morphological characteristic and antigenicity of VLPs are similar to those of the naïve virus. Moreover, VLPs are associated with greater safety because they lack the viral genome and thus cannot replicate in the host. However, VLPs can still effectively elicit innate and adaptive immunity.
The baculovirus expression system has been widely used for VLP production. A recombinant baculovirus co-expressing the P1 region and the viral protease 3CD of EV-A71 with different promoters was infected to insect cells for VLPs production (subgenotype C2, neu strain) [94, 95]. The viral protease 3CD can cleave the P1 region of structural proteins [VP0 (VP4 and VP2), VP3, and VP1], which is important to constitute the virus capsid. However, this VLP production method suffers from low yields and excessive VLP degradation. Several factors influencing the expression yields of VLP, including the control of various promoters, insect cell types, and incubation time. After researchers serially modified the system for the construction of recombinant baculoviruses, the yield of EV-A71 VLPs improved [96]. EV-A71 VLPs elicited humoral and cellular immune responses in immunized mice, and vaccination of female mice with VLPs, protected the neonatal mice from a lethal dose of EV-A71 challenge [97]. In another study, Macaque monkeys were vaccinated with EV-A71 VLPs produced from baculovirus, which elicited immune responses [98]. In addition to the baculovirus expression system, EV-A71 VLP also can be generated in yeast such as Saccharomyces cerevisiae or Pichia pastoris, which showed protective efficacy against EV-A71 challenge in mice. In addition, maternal immunization with VLPs could also protect neonate mice against lethal EV-A71 challenge [96, 99].
Chimeric VLPs, including adenovirus or varicella-zoster virus-based VLPs, have also been applied to co-express the P1 and 3CD regions of EV-A71, which could both induce an EV-A71-specific immune response and neutralization antibodies in vaccinated mice, and exhibited protective efficacy against EV-A71 infection [100, 101].
Live-attenuated vaccines
According to the experience in developing the poliovirus Sabin vaccine, and the numerous advantages of live-attenuated vaccines, including elicitation of long-lasting immunity and cost-effective production, researchers have continued to investigate potential candidates for an EV-A71 live-attenuated vaccine. EV-A71(S1–3′) was derived from the prototype EV-A71(BrCr) strain. Five cynomolgus monkeys were inoculated with EV-A71 (S1–3′) via an intravenous route, followed by challenge with a lethal dose of EV-A71(BrCr-TR), demonstrating induction of an efficient immune response, and the sera showed neutralization activity against EV-A71(BrCr-TR) (subgenotype A) and other subgenotypes, including B1, B4, C2, and C4. However, EV71(S1–3′) caused tremor in the inoculated monkeys, and the virus was isolated from the lumbar spinal cord of inoculated monkeys on days 4 or 10 post-inoculation [102]. Therefore, the safety issue of live-attenuated vaccine remains a concern.
Because the detailed molecular pathogenic mechanism of EV-A71 infection remains unexplored, the virulence determinants of EV-A71 are still being investigated. The amino acid residue 145 in VP1 is considered to be an important factor for EV-A71 virulence and receptor attachment [103, 104]. Mutation of a single amino acid, glutamine (Q) to glutamic acid (E), at residue 145 of VP1 in the subgenotype C4 of EV-A71 was used to generate a mouse-virulent EV-A71 strain [105]. Viruses harboring the VP1–145E mutation could also induce neurological symptoms in cynomolgus monkeys; therefore, VP1–145E viruses are more virulent than VP1–145G viruses in cynomolgus monkeys [103]. The nucleotide 158 in the stem loop II region of the EV-A71 5′ UTR play a pivotal role in EV-A71 virulence. The nucleotide substitution of C158U reduced the translation activity of EV-A71, and attenuated EV-A71 virulence in a mouse model [106].
Moreover, the nucleotide substitutions of G64R, G64 T, and S264 L in EV-A71 3D polymerase were shown to contribute to EV-A71 replication fidelity. Enhancement of the fidelity of 3D polymerase can improve the stability and safety of live-attenuated vaccines [107]. Another study also indicated that EV-A71 with the RdRp-G64R and RdRp-L123F mutations attenuated the virulence of the virus in an AG129 mouse model [108]. Yee et al. [109] constructed an miRNA-based EV-A71 vaccine strain, pIY, which carried let-7a and miR-124a target genes. They found that the viral yield of the pIY strain was much lower than that of the EV-A71 wild-type B4 strain 41 in SHY-5Y cells. Moreover, the pIY strain could still protect mice against EV-A71 in a mouse-adapted strain challenge.
In recent years, a new combination strategy of codon deoptimization and synthetic virus production has emerged for vaccine development. Tsai et al. [110] found that rgEV-A71-CD-HF, a virus with a deoptimized VP1 codon, and a high-fidelity virus with nucleotide substitutions of G64R and L123F in 3D polymerase showed less virulence in a mouse model.
Mucosal vaccines
The mucosal immune response, which is effectively induced by the administration of a vaccine onto the mucosal surface, is the first line of defense against pathogen invasion. Several mucosal vaccines have been licensed for use in humans, such as oral vaccines against poliovirus, rotavirus, Vibrio cholera, and Salmonella Typhi, and an intranasal vaccine against influenza virus. The advantages of mucosal vaccines are that they are good inducers of mucosal and systematic immunity, and the needle-free administration is more acceptable for infants and young children [111, 112]. Although the poliovirus Sabin vaccine is a successful example of a mucosal vaccine, the safety issue of live-attenuated vaccine remains a concern. To date, there have been few studies focused on development of a mucosal vaccine for EV-A71. As summarized above, several studies involved immunizing mice with recombinant VP1 protein by an oral route [80,81,82,83,84]; however, these vaccines are still at the preclinical stage of research and validation. Recently, Lin et al. [113] found that the titers of EV-A71-specific IgG and IgA, T-cell proliferative response, and interleukin-17 secretion were increased in a group of BALB/c mice immunized with a CpG-adjuvanted inactivated EV-A71 vaccine via an intranasal route. In addition, this vaccine could also protect human scavenger receptor class B, member 2 transgenic (hSCARB2-Tg) mice against lethal EV-A71 challenge. Although a mucosal vaccine seems to be another good choice for EV-A71 vaccine development, there are still some challenges in mucosal vaccine design to overcome, including how to effectively breach the epithelial barrier, and the relatively large amounts of vaccine needed for mucosal immunization.
Conclusions and prospects
EV-A71 is one of the most pathogenic enteroviruses infecting humans, with many outbreaks occurring worldwide causes a wide range of human diseases. However, there is still no clinically approved antiviral drug available for the prevention and treatment of the EV-A71 infection. Although the development of antiviral therapy and vaccine represents a major challenge, the progress made so far in understanding the viral replication mechanism has provided novel targets for antiviral therapy and the characterization of compounds with antiviral activity. The development of pan-enteroviruses vaccine and anti-viral drugs is an important and achievable goal in the future.
Availability of data and materials
Not applicable.
Abbreviations
- 5′ UTR:
-
5′ untranslated region
- Anx2:
-
Annexin A2
- APOBEC3G or A3G:
-
Apolipoprotein B messenger RNA-editing enzyme catalytic polypeptide-like 3G
- CAMS:
-
Chinese Academy of Medical Sciences
- CV-A16:
-
Coxsackievirus A16
- DC-SIGN:
-
Dendritic cell-specific intercellular adhesion molecule-3 grabbing nonintegrin
- DIDS:
-
4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid
- E. coli :
-
Escherichia coli
- EV-A71:
-
Enterovirus A71
- HFMD:
-
Hand-foot-and-mouth disease
- HS:
-
Heparan sulfate
- IDR:
-
Idarubicin
- IRES:
-
Internal ribosome entry site
- NF-κB:
-
Nuclear factor-kappa B
- NHRI:
-
National Health Research Institutes
- PSGL-1:
-
P-selectin glycoprotein ligand 1
- SCARB2:
-
Scavenger receptor class B member 2
- Sinovac:
-
Sinovac Biotech Co., Ltd
- Vigoo:
-
Beijing Vigoo Biological Co., Ltd
- VLP:
-
Virus-like particle
References
Huang PN, Shih SR. Update on enterovirus 71 infection. Curr Opin Virol. 2014;5:98–104.
McMinn PC. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol Rev. 2002;26(1):91–107.
Bedard KM, Semler BL. Regulation of picornavirus gene expression. Microbes Infect. 2004;6(7):702–13.
Yamayoshi S, Yamashita Y, Li J, Hanagata N, Minowa T, Takemura T, Koike S. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat Med. 2009;15(7):798–801.
Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat Med. 2009;15(7):794–7.
Yang SL, Chou YT, Wu CN, Ho MS. Annexin II binds to capsid protein VP1 of enterovirus 71 and enhances viral infectivity. J Virol. 2011;85(22):11809–20.
Tan CW, Poh CL, Sam IC, Chan YF. Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J Virol. 2013;87(1):611–20.
Yang B, Chuang H, Yang KD. Sialylated glycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol J. 2009;6:141.
Ren XX, Ma L, Liu QW, Li C, Huang Z, Wu L, Xiong SD, Wang JH, Wang HB. The molecule of DC-SIGN captures enterovirus 71 and confers dendritic cell-mediated viral trans-infection. Virol J. 2014;11:47.
He QQ, Ren S, Xia ZC, Cheng ZK, Peng NF, Zhu Y. Fibronectin Facilitates Enterovirus 71 Infection by Mediating Viral Entry. J Virol. 2018;92(9).
Lin JY, Chen TC, Weng KF, Chang SC, Chen LL, Shih SR. Viral and host proteins involved in picornavirus life cycle. J Biomed Sci. 2009;16:103.
Tijsma A, Franco D, Tucker S, Hilgenfeld R, Froeyen M, Leyssen P, Neyts J. The capsid binder Vapendavir and the novel protease inhibitor SG85 inhibit enterovirus 71 replication. Antimicrob Agents Chemother. 2014;58(11):6990–2.
Chang CS, Lin YT, Shih SR, Lee CC, Lee YC, Tai CL, Tseng SN, Chern JH. Design, synthesis, and antipicornavirus activity of 1-[5-(4-arylphenoxy)alkyl]-3-pyridin-4-ylimidazolidin-2-one derivatives. J Med Chem. 2005;48(10):3522–35.
Chern JH, Lee CC, Chang CS, Lee YC, Tai CL, Lin YT, Shia KS, Lee CY, Shih SR. Synthesis and antienteroviral activity of a series of novel, oxime ether-containing pyridyl imidazolidinones. Bioorg Med Chem Lett. 2004;14(20):5051–6.
Shia KS, Li WT, Chang CM, Hsu MC, Chern JH, Leong MK, Tseng SN, Lee CC, Lee YC, Chen SJ, Peng KC, Tseng HY, Chang YL, Tai CL, Shih SR. Design, synthesis, and structure-activity relationship of pyridyl imidazolidinones: a novel class of potent and selective human enterovirus 71 inhibitors. J Med Chem. 2002;45(8):1644–55.
Chen TC, Liu SC, Huang PN, Chang HY, Chern JH, Shih SR. Antiviral activity of pyridyl imidazolidinones against enterovirus 71 variants. J Biomed Sci. 2008;15(3):291–300.
Shih SR, Tsai MC, Tseng SN, Won KF, Shia KS, Li WT, Chern JH, Chen GW, Lee CC, Lee YC, Peng KC, Chao YS. Mutation in enterovirus 71 capsid protein VP1 confers resistance to the inhibitory effects of pyridyl imidazolidinone. Antimicrob Agents Chemother. 2004;48(9):3523–9.
Ho JY, Chern JH, Hsieh CF, Liu ST, Liu CJ, Wang YS, Kuo TW, Hsu SJ, Yeh TK, Shih SR, Hsieh PW, Chiu CH, Horng JT. In vitro and in vivo studies of a potent capsid-binding inhibitor of enterovirus 71. J Antimicrob Chemother. 2016;71(7):1922–32.
Li P, Yu J, Hao F, He H, Shi X, Hu J, Wang L, Du C, Zhang X, Sun Y, Lin F, Gu Z, Xu D, Chen X, Shen L, Hu G, Li J, Chen S, Xiao W, Wang Z, Guo Q, Chang X, Tian X, Lin T. Discovery of potent EV71 capsid inhibitors for treatment of HFMD. ACS Med Chem Lett. 2017;8(8):841–6.
Meng T, Jia Q, Wong SM, Chua KB. In vitro and in vivo inhibition of infectivity of human Enterovirus 71 by a sulfonated food azo dye, Brilliant Black BN. J Virol. 2019.
Krausslich HG, Nicklin MJ, Lee CK, Wimmer E. Polyprotein processing in picornavirus replication. Biochimie. 1988;70(1):119–30.
Laitinen OH, Svedin E, Kapell S, Nurminen A, Hytonen VP, Flodstrom-Tullberg M. Enteroviral proteases: structure, host interactions and pathogenicity. Rev Med Virol. 2016;26(4):251–67.
Falah N, Montserret R, Lelogeais V, Schuffenecker I, Lina B, Cortay JC, Violot S. Blocking human enterovirus 71 replication by targeting viral 2A protease. J Antimicrob Chemother. 2012;67(12):2865–9.
Aldabe R, Barco A, Carrasco L. Membrane permeabilization by poliovirus proteins 2B and 2BC. J Biol Chem. 1996;271(38):23134–7.
Barco A, Carrasco L. A human virus protein, poliovirus protein 2BC, induces membrane proliferation and blocks the exocytic pathway in the yeast Saccharomyces cerevisiae. EMBO J. 1995;14(14):3349–64.
Cong H, Du N, Yang Y, Song L, Zhang W, Tien P. Enterovirus 71 2B induces cell apoptosis by directly inducing the conformational activation of the Proapoptotic protein Bax. J Virol. 2016;90(21):9862–77.
Xie S, Wang K, Yu W, Lu W, Xu K, Wang J, Ye B, Schwarz W, Jin Q, Sun B. DIDS blocks a chloride-dependent current that is mediated by the 2B protein of enterovirus 71. Cell Res. 2011;21(8):1271–5.
Banerjee R, Echeverri A, Dasgupta A. Poliovirus-encoded 2C polypeptide specifically binds to the 3′-terminal sequences of viral negative-strand RNA. J Virol. 1997;71(12):9570–8.
Rodriguez PL, Carrasco L. Poliovirus protein 2C has ATPase and GTPase activities. J Biol Chem. 1993;268(11):8105–10.
Arita M, Wakita T, Shimizu H. Characterization of pharmacologically active compounds that inhibit poliovirus and enterovirus 71 infectivity. J Gen Virol. 2008;89(Pt 10):2518–30.
Arita M, Takebe Y, Wakita T, Shimizu H. A bifunctional anti-enterovirus compound that inhibits replication and the early stage of enterovirus 71 infection. J Gen Virol. 2010;91(Pt 11):2734–44.
Arita M, Wakita T, Shimizu H. Cellular kinase inhibitors that suppress enterovirus replication have a conserved target in viral protein 3A similar to that of enviroxime. J Gen Virol. 2009;90(Pt 8):1869–79.
Kuo CJ, Shie JJ, Fang JM, Yen GR, Hsu JT, Liu HG, Tseng SN, Chang SC, Lee CY, Shih SR, Liang PH. Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg Med Chem. 2008;16(15):7388–98.
Zhang X, Song Z, Qin B, Zhang X, Chen L, Hu Y, Yuan Z. Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection: evaluation of antiviral efficacy in a murine infection model. Antivir Res. 2013;97(3):264–9.
Tan YW, Ang MJ, Lau QY, Poulsen A, Ng FM, Then SW, Peng J, Hill J, Hong WJ, Chia CS, Chu JJ. Antiviral activities of peptide-based covalent inhibitors of the Enterovirus 71 3C protease. Sci Rep. 2016;6:33663.
Ma Y, Shang C, Yang P, Li L, Zhai Y, Yin Z, Wang B, Shang L. 4-Iminooxazolidin-2-one as a Bioisostere of the cyanohydrin moiety: inhibitors of Enterovirus 71 3C protease. J Med Chem. 2018;61(22):10333–9.
Ma GH, Ye Y, Zhang D, Xu X, Si P, Peng JL, Xiao YL, Cao RY, Yin YL, Chen J, Zhao LX, Zhou Y, Zhong W, Liu H, Luo XM, Chen LL, Shen X. Identification and biochemical characterization of DC07090 as a novel potent small molecule inhibitor against human enterovirus 71 3C protease by structure-based virtual screening. Eur J Med Chem. 2016;124:981–91.
Cao Z, Ding Y, Ke Z, Cao L, Li N, Ding G, Wang Z, Xiao W. Luteoloside acts as 3C protease inhibitor of Enterovirus 71 in vitro. PLoS One. 2016;11(2):e0148693.
Chen TC, Chang HY, Lin PF, Chern JH, Hsu JT, Chang CY, Shih SR. Novel antiviral agent DTriP-22 targets RNA-dependent RNA polymerase of enterovirus 71. Antimicrob Agents Chemother. 2009;53(7):2740–7.
Hung HC, Chen TC, Fang MY, Yen KJ, Shih SR, Hsu JT, Tseng CP. Inhibition of enterovirus 71 replication and the viral 3D polymerase by aurintricarboxylic acid. J Antimicrob Chemother. 2010;65(4):676–83.
Li Y, Yu J, Qi X, Yan H. Monoclonal antibody against EV71 3D(pol) inhibits the polymerase activity of RdRp and virus replication. BMC Immunol. 2019;20(1):6.
Wang H, Zhong M, Li Y, Li K, Wu S, Guo T, Cen S, Jiang J, Li Z, Li Y. APOBEC3G is a restriction factor of EV71 and mediator of IMB-Z antiviral activity. Antivir Res. 2019;165:23–33.
Dai W, Wu Y, Bi J, Lu X, Hou A, Zhou Y, Sun B, Kong W, Barbier J, Cintrat JC, Gao F, Gillet D, Su W. And Jiang C. antiviral effects of Retro-2(cycl) and Retro-2.1 against Enterovirus 71 in vitro and in vivo. Antivir Res. 2017;144:311–21.
Hou HY, Lu WW, Wu KY, Lin CW, Kung SH. Idarubicin is a broad-spectrum enterovirus replication inhibitor that selectively targets the virus internal ribosomal entry site. J Gen Virol. 2016;97(5):1122–33.
Li ZH, Li CM, Ling P, Shen FH, Chen SH, Liu CC, Yu CK, Chen SH. Ribavirin reduces mortality in enterovirus 71-infected mice by decreasing viral replication. J Infect Dis. 2008;197(6):854–7.
Zhang G, Zhou F, Gu B, Ding C, Feng D, Xie F, Wang J, Zhang C, Cao Q, Deng Y, Hu W, Yao K. In vitro and in vivo evaluation of ribavirin and pleconaril antiviral activity against enterovirus 71 infection. Arch Virol. 2012;157(4):669–79.
Deng JX, Nie XJ, Lei YF, Ma CF, Xu DL, Li B, Xu ZK, Zhang GC. The highly conserved 5′ untranslated region as an effective target towards the inhibition of Enterovirus 71 replication by unmodified and appropriate 2′-modified siRNAs. J Biomed Sci. 2012;19:73.
Liu H, Qin Y, Kong Z, Shao Q, Su Z, Wang S, Chen J. siRNA Targeting the 2Apro Genomic Region Prevents Enterovirus 71 Replication In Vitro. PLoS One. 2016;11(2):e0149470.
Lu WW, Hsu YY, Yang JY, Kung SH. Selective inhibition of enterovirus 71 replication by short hairpin RNAs. Biochem Biophys Res Commun. 2004;325(2):494–9.
Sim AC, Luhur A, Tan TM, Chow VT, Poh CL. RNA interference against enterovirus 71 infection. Virology. 2005;341(1):72–9.
Tan EL, Tan TM, Chow VT, Poh CL. Enhanced potency and efficacy of 29-mer shRNAs in inhibition of Enterovirus 71. Antivir Res. 2007;74(1):9–15.
Tan EL, Tan TM, Tak Kwong Chow V, Poh CL. Inhibition of enterovirus 71 in virus-infected mice by RNA interference. Mol Ther. 2007;15(11):1931–8.
Li B, Zheng J. MicroR-9-5p suppresses EV71 replication through targeting NFkappaB of the RIG-I-mediated innate immune response. FEBS Open Bio. 2018;8(9):1457–70.
Li X, Huang Y, Sun M, Ji H, Dou H, Hu J, Yan Y, Wang X, Chen L. Honeysuckle-encoded microRNA2911 inhibits Enterovirus 71 replication via targeting VP1 gene. Antivir Res. 2018;152:117–23.
Wen BP, Dai HJ, Yang YH, Zhuang Y, Sheng R. MicroRNA-23b inhibits enterovirus 71 replication through downregulation of EV71 VPl protein. Intervirology. 2013;56(3):195–200.
Zheng C, Zheng Z, Sun J, Zhang Y, Wei C, Ke X, Liu Y, Deng L, Wang H. MiR-16-5p mediates a positive feedback loop in EV71-induced apoptosis and suppresses virus replication. Sci Rep. 2017;7(1):16422.
Orr-Burks NL, Shim BS, Wu W, Bakre AA, Karpilow J, Tripp RA. MicroRNA screening identifies miR-134 as a regulator of poliovirus and enterovirus 71 infection. Sci Data. 2017;4:170023.
Zhang L, Chen X, Shi Y, Zhou B, Du C, Liu Y, Han S, Yin J, Peng B, He X, Liu W. miR-27a suppresses EV71 replication by directly targeting EGFR. Virus Genes. 2014;49(3):373–82.
Zheng Z, Ke X, Wang M, He S, Li Q, Zheng C, Zhang Z, Liu Y, Wang H. Human microRNA hsa-miR-296-5p suppresses enterovirus 71 replication by targeting the viral genome. J Virol. 2013;87(10):5645–56.
Pourianfar HR, Poh CL, Fecondo J, Grollo L. In vitro evaluation of the antiviral activity of heparan sulfate mimetic compounds against Enterovirus 71. Virus Res. 2012;169(1):22–9.
Chen J, Zhang C, Zhou Y, Zhang X, Shen C, Ye X, Jiang W, Huang Z, Cong Y. A 3.0-Angstrom Resolution Cryo-Electron Microscopy Structure and Antigenic Sites of Coxsackievirus A6-Like Particles. J Virol. 2018;92(2).
Wang H, Li K, Ma L, Wu S, Hu J, Yan H, Jiang J, Li Y. Berberine inhibits enterovirus 71 replication by downregulating the MEK/ERK signaling pathway and autophagy. Virol J. 2017;14(1):2.
Cao Z, Ding Y, Cao L, Ding G, Wang Z, Xiao W. Isochlorogenic acid C prevents enterovirus 71 infection via modulating redox homeostasis of glutathione. Sci Rep. 2017;7(1):16278.
Mao Q, Wang Y, Bian L, Xu M, Liang Z. EV-A71 vaccine licensure: a first step for multivalent enterovirus vaccine to control HFMD and other severe diseases. Emerg Microbes Infect. 2016;5(7):e75.
Mao QY, Wang Y, Bian L, Xu M, Liang Z. EV71 vaccine, a new tool to control outbreaks of hand, foot and mouth disease (HFMD). Expert Rev Vaccines. 2016;15(5):599–606.
Zhu FC, Meng FY, Li JX, Li XL, Mao QY, Tao H, Zhang YT, Yao X, Chu K, Chen QH, Hu YM, Wu X, Liu P, Zhu LY, Gao F, Jin H, Chen YJ, Dong YY, Liang YC, Shi NM, Ge HM, Liu L, Chen SG, Ai X, Zhang ZY, Ji YG, Luo FJ, Chen XQ, Zhang Y, Zhu LW, Liang ZL, Shen XL. Efficacy, safety, and immunology of an inactivated alum-adjuvant enterovirus 71 vaccine in children in China: a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2013;381(9882):2024–32.
Wei M, Meng F, Wang S, Li J, Zhang Y, Mao Q, Hu Y, Liu P, Shi N, Tao H, Chu K, Wang Y, Liang Z, Li X, Zhu F. 2-year efficacy, immunogenicity, and safety of Vigoo Enterovirus 71 vaccine in healthy Chinese children: a randomized open-label study. J Infect Dis. 2017;215(1):56–63.
Li JX, Song YF, Wang L, Zhang XF, Hu YS, Hu YM, Xia JL, Li J, Zhu FC. Two-year efficacy and immunogenicity of Sinovac Enterovirus 71 vaccine against hand, foot and mouth disease in children. Expert Rev Vaccines. 2016;15(1):129–37.
Zhu F, Xu W, Xia J, Liang Z, Liu Y, Zhang X, Tan X, Wang L, Mao Q, Wu J, Hu Y, Ji T, Song L, Liang Q, Zhang B, Gao Q, Li J, Wang S, Hu Y, Gu S, Zhang J, Yao G, Gu J, Wang X, Zhou Y, Chen C, Zhang M, Cao M, Wang J, Wang H, Wang N. Efficacy, safety, and immunogenicity of an enterovirus 71 vaccine in China. N Engl J Med. 2014;370(9):818–28.
Hu Y, Zeng G, Chu K, Zhang J, Han W, Zhang Y, Li J, Zhu F. Five-year immunity persistence following immunization with inactivated enterovirus 71 type (EV71) vaccine in healthy children: a further observation. Hum Vaccin Immunother. 2018;14(6):1517–23.
Li R, Liu L, Mo Z, Wang X, Xia J, Liang Z, Zhang Y, Li Y, Mao Q, Wang J, Jiang L, Dong C, Che Y, Huang T, Jiang Z, Xie Z, Wang L, Liao Y, Liang Y, Nong Y, Liu J, Zhao H, Na R, Guo L, Pu J, Yang E, Sun L, Cui P, Shi H, Wang J, Li Q. An inactivated enterovirus 71 vaccine in healthy children. N Engl J Med. 2014;370(9):829–37.
Chang JY, Chang CP, Tsai HH, Lee CD, Lian WC, Ih JS, Sai IH, Liu CC, Chou AH, Lu YJ, Chen CY, Lee PH, Chiang JR, Chong PC. Selection and characterization of vaccine strain for Enterovirus 71 vaccine development. Vaccine. 2012;30(4):703–11.
Cheng A, Fung CP, Liu CC, Lin YT, Tsai HY, Chang SC, Chou AH, Chang JY, Jiang RH, Hsieh YC, Su IJ, Chong PC, Hsieh SM. A phase I, randomized, open-label study to evaluate the safety and immunogenicity of an enterovirus 71 vaccine. Vaccine. 2013;31(20):2471–6.
Chou AH, Liu CC, Chang JY, Jiang R, Hsieh YC, Tsao A, Wu CL, Huang JL, Fung CP, Hsieh SM, Wang YF, Wang JR, Hu MH, Chiang JR, Su IJ, Chong PC. Formalin-inactivated EV71 vaccine candidate induced cross-neutralizing antibody against subgenotypes B1, B4, B5 and C4A in adult volunteers. PLoS One. 2013;8(11):e79783.
Huang LM, Chiu CH, Chiu NC, Lin CY, Li MT, Kuo TY, Weng YJ, Hsieh EF, Tai IC. Immunogenicity, safety, cross-reaction, and immune persistence of an inactivated enterovirus A71 vaccine in children aged from two months to 11 years in Taiwan. Vaccine. 2019;37(13):1827–35.
Wu CN, Lin YC, Fann C, Liao NS, Shih SR, Ho MS. Protection against lethal enterovirus 71 infection in newborn mice by passive immunization with subunit VP1 vaccines and inactivated virus. Vaccine. 2001;20(5–6):895–904.
Zhou SL, Ying XL, Han X, Sun XX, Jin Q, Yang F. Characterization of the enterovirus 71 VP1 protein as a vaccine candidate. J Med Virol. 2015;87(2):256–62.
Meng T, Kolpe AB, Kiener TK, Chow VT, Kwang J. Display of VP1 on the surface of baculovirus and its immunogenicity against heterologous human enterovirus 71 strains in mice. PLoS One. 2011;6(7):e21757.
Wang X, Dong K, Long M, Lin F, Gao Z, Wang L, Zhang Z, Chen X, Dai Y, Wang H, Zhang H. Induction of a high-titered antibody response using HIV gag-EV71 VP1-based virus-like particles with the capacity to protect newborn mice challenged with a lethal dose of enterovirus 71. Arch Virol. 2018;163(7):1851–61.
Chen HF, Chang MH, Chiang BL, Jeng ST. Oral immunization of mice using transgenic tomato fruit expressing VP1 protein from enterovirus 71. Vaccine. 2006;24(15):2944–51.
Chiu CH, Chu C, He CC, Lin TY. Protection of neonatal mice from lethal enterovirus 71 infection by maternal immunization with attenuated Salmonella enterica serovar typhimurium expressing VP1 of enterovirus 71. Microbes Infect. 2006;8(7):1671–8.
Yu Z, Huang Z, Sao C, Huang Y, Zhang F, Ma G, Chen Z, Zeng Z, Qiwen D, Zeng W. Oral immunization of mice using Bifidobacterium longum expressing VP1 protein from enterovirus 71. Arch Virol. 2013;158(5):1071–7.
Zhang C, Wang Y, Ma S, Li L, Chen L, Yan H, Peng T. Human Enterovirus 71 protein displayed on the surface of Saccharomyces cerevisiae as an Oral vaccine. Viral Immunol. 2016;29(5):288–95.
Xu P, Wang Y, Tao L, Wu X, Wu W. Recombinant lactococcus lactis secreting viral protein 1 of enterovirus 71 and its immunogenicity in mice. Biotechnol Lett. 2019;41(6–7):867–72.
Chen HL, Huang JY, Chu TW, Tsai TC, Hung CM, Lin CC, Liu FC, Wang LC, Chen YJ, Lin MF, Chen CM. Expression of VP1 protein in the milk of transgenic mice: a potential oral vaccine protects against enterovirus 71 infection. Vaccine. 2008;26(23):2882–9.
Foo DG, Alonso S, Phoon MC, Ramachandran NP, Chow VT, Poh CL. Identification of neutralizing linear epitopes from the VP1 capsid protein of Enterovirus 71 using synthetic peptides. Virus Res. 2007;125(1):61–8.
Foo DG, Alonso S, Chow VT, Poh CL. Passive protection against lethal enterovirus 71 infection in newborn mice by neutralizing antibodies elicited by a synthetic peptide. Microbes Infect. 2007;9(11):1299–306.
Liu JN, Wang W, Duo JY, Hao Y, Ma CM, Li WB, Lin SZ, Gao XZ, Liu XL, Xu YF, Xu WB, Qin C, Zhang LF. Combined peptides of human enterovirus 71 protect against virus infection in mice. Vaccine. 2010;28(46):7444–51.
Liu CC, Chou AH, Lien SP, Lin HY, Liu SJ, Chang JY, Guo MS, Chow YH, Yang WS, Chang KH, Sia C, Chong P. Identification and characterization of a cross-neutralization epitope of Enterovirus 71. Vaccine. 2011;29(26):4362–72.
Xu L, He D, Li Z, Zheng J, Yang L, Yu M, Yu H, Chen Y, Que Y, Shih JW, Liu G, Zhang J, Zhao Q, Cheng T, Xia N. Protection against lethal enterovirus 71 challenge in mice by a recombinant vaccine candidate containing a broadly cross-neutralizing epitope within the VP2 EF loop. Theranostics. 2014;4(5):498–513.
Huo C, Yang J, Lei L, Qiao L, Xin J, Pan Z. Hepatitis B virus core particles containing multiple epitopes confer protection against enterovirus 71 and coxsackievirus A16 infection in mice. Vaccine. 2017;35(52):7322–30.
Aw-Yong KL, Sam IC, Koh MT, Chan YF. Immunodominant IgM and IgG epitopes recognized by antibodies induced in Enterovirus A71-associated hand, Foot and Mouth Disease Patients. PLoS One. 2016;11(11):e0165659.
Jiang L, Fan R, Sun S, Fan P, Su W, Zhou Y, Gao F, Xu F, Kong W, Jiang C. A new EV71 VP3 epitope in norovirus P particle vector displays neutralizing activity and protection in vivo in mice. Vaccine. 2015;33(48):6596–603.
Chung YC, Huang JH, Lai CW, Sheng HC, Shih SR, Ho MS, Hu YC. Expression, purification and characterization of enterovirus-71 virus-like particles. World J Gastroenterol. 2006;12(6):921–7.
Hu YC, Hsu JT, Huang JH, Ho MS, Ho YC. Formation of enterovirus-like particle aggregates by recombinant baculoviruses co-expressing P1 and 3CD in insect cells. Biotechnol Lett. 2003;25(12):919–25.
Zhang C, Ku Z, Liu Q, Wang X, Chen T, Ye X, Li D, Jin X, Huang Z. High-yield production of recombinant virus-like particles of enterovirus 71 in Pichia pastoris and their protective efficacy against oral viral challenge in mice. Vaccine. 2015;33(20):2335–41.
Chung YC, Ho MS, Wu JC, Chen WJ, Huang JH, Chou ST, Hu YC. Immunization with virus-like particles of enterovirus 71 elicits potent immune responses and protects mice against lethal challenge. Vaccine. 2008;26(15):1855–62.
Lin YL, Yu CI, Hu YC, Tsai TJ, Kuo YC, Chi WK, Lin AN, Chiang BL. Enterovirus type 71 neutralizing antibodies in the serum of macaque monkeys immunized with EV71 virus-like particles. Vaccine. 2012;30(7):1305–12.
Li HY, Han JF, Qin CF, Chen R. Virus-like particles for enterovirus 71 produced from Saccharomyces cerevisiae potently elicits protective immune responses in mice. Vaccine. 2013;31(32):3281–7.
Tsou YL, Lin YW, Shao HY, Yu SL, Wu SR, Lin HY, Liu CC, Huang C, Chong P, Chow YH. Recombinant adeno-vaccine expressing enterovirus 71-like particles against hand, foot, and mouth disease. PLoS Negl Trop Dis. 2015;9(4):e0003692.
Yan Q, Wu L, Chen L, Qin Y, Pan Z, Chen M. Vesicular stomatitis virus-based vaccines expressing EV71 virus-like particles elicit strong immune responses and protect newborn mice from lethal challenges. Vaccine. 2016;34(35):4196–204.
Arita M, Nagata N, Iwata N, Ami Y, Suzaki Y, Mizuta K, Iwasaki T, Sata T, Wakita T, Shimizu H. An attenuated strain of enterovirus 71 belonging to genotype a showed a broad spectrum of antigenicity with attenuated neurovirulence in cynomolgus monkeys. J Virol. 2007;81(17):9386–95.
Fujii K, Sudaka Y, Takashino A, Kobayashi K, Kataoka C, Suzuki T, Iwata-Yoshikawa N, Kotani O, Ami Y, Shimizu H, Nagata N, Mizuta K, Matsuzaki Y, Koike S. VP1 Amino Acid Residue 145 of Enterovirus 71 Is a Key Residue for Its Receptor Attachment and Resistance to Neutralizing Antibody during Cynomolgus Monkey Infection. J Virol. 2018;92(15).
Nishimura Y, Lee H, Hafenstein S, Kataoka C, Wakita T, Bergelson JM, Shimizu H. Enterovirus 71 binding to PSGL-1 on leukocytes: VP1-145 acts as a molecular switch to control receptor interaction. PLoS Pathog. 2013;9(7):e1003511.
Zaini Z, McMinn P. A single mutation in capsid protein VP1 (Q145E) of a genogroup C4 strain of human enterovirus 71 generates a mouse-virulent phenotype. J Gen Virol. 2012;93(Pt 9):1935–40.
Yeh MT, Wang SW, Yu CK, Lin KH, Lei HY, Su IJ, Wang JR. A single nucleotide in stem loop II of 5′-untranslated region contributes to virulence of enterovirus 71 in mice. PLoS One. 2011;6(11):e27082.
Sadeghipour S, Bek EJ, McMinn PC. Ribavirin-resistant mutants of human enterovirus 71 express a high replication fidelity phenotype during growth in cell culture. J Virol. 2013;87(3):1759–69.
Meng T, Kwang J. Attenuation of human enterovirus 71 high-replication-fidelity variants in AG129 mice. J Virol. 2014;88(10):5803–15.
Yee PTI, Tan SH, Ong KC, Tan KO, Wong KT, Hassan SS, Poh CL. Development of live attenuated Enterovirus 71 vaccine strains that confer protection against lethal challenge in mice. Sci Rep. 2019;9(1):4805.
Tsai YH, Huang SW, Hsieh WS, Cheng CK, Chang CF, Wang YF, Wang JR. Enterovirus A71 Containing Codon-Deoptimized VP1 and High-Fidelity Polymerase as Next-Generation Vaccine Candidate. J Virol. 2019;93(13).
Kim SH, Jang YS. The development of mucosal vaccines for both mucosal and systemic immune induction and the roles played by adjuvants. Clin Exp Vaccine Res. 2017;6(1):15–21.
Miquel-Clopes A, Bentley EG, Stewart JP, Carding SR. Mucosal vaccines and technology. Clin Exp Immunol. 2019;196(2):205–14.
Lin YL, Chow YH, Huang LM, Hsieh SM, Cheng PY, Hu KC, Chiang BL. A CpG-adjuvanted intranasal enterovirus 71 vaccine elicits mucosal and systemic immune responses and protects human SCARB2-transgenic mice against lethal challenge. Sci Rep. 2018;8(1):10713.
Acknowledgements
Not applicable.
Funding
This work was financially supported by the Research Center for Emerging Viral Infections from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan and the Ministry of Science and Technology (MOST), Taiwan (MOST 108–3017-F-182-001).
Author information
Authors and Affiliations
Contributions
J.-Y.L. and Y.-A.K. conducted literature research, and contributed in writing the manuscript. S.-R.S. designed, organized and edited the manuscript for submission. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lin, JY., Kung, YA. & Shih, SR. Antivirals and vaccines for Enterovirus A71. J Biomed Sci 26, 65 (2019). https://doi.org/10.1186/s12929-019-0560-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-019-0560-7