
Abstract
Dengue virus (DENV) infection is the most common mosquito-transmitted viral infection. DENV infection can cause mild dengue fever or severe dengue hemorrhagic fever (DHF)/dengue shock syndrome (DSS). Hemorrhage and vascular leakage are two characteristic symptoms of DHF/DSS. However, due to the limited understanding of dengue pathogenesis, no satisfactory therapies to treat nor vaccine to prevent dengue infection are available, and the mortality of DHF/DSS is still high. DENV nonstructural protein 1 (NS1), which can be secreted in patients’ sera, has been used as an early diagnostic marker for dengue infection for many years. However, the roles of NS1 in dengue-induced vascular leakage were described only recently. In this article, the pathogenic roles of DENV NS1 in hemorrhage and vascular leakage are reviewed, and the possibility of using NS1 as a therapeutic target and vaccine candidate is discussed.
Similar content being viewed by others
Background
General information about dengue
Dengue virus (DENV) is the most common mosquito-borne flavivirus and threatens people in tropic and subtropical areas. The World Health Organization estimates that more than 2.5 billion people representing over 40% of the world’s population are at risk of dengue infection [1]. Dengue virus infections are often asymptomatic or cause a flu-like syndrome with fever and rash. However, a small proportion of cases develop into severe illness, which is termed dengue hemorrhagic fever (DHF). DHF is characterized by vascular leakage, thrombocytopenia, and coagulopathy [2]. Vascular leakage results in hemoconcentration and serous effusions, leading to circulatory collapse, which further develops into life-threatening dengue shock syndrome (DSS) [2]. An estimated 390 million infections occur each year globally, and approximately 960,000 people with severe dengue require hospitalization [1]. Children contribute to a large proportion of the severe disease cases. In 1958, DHF was reported to carry a case fatality rate (CFR) of 13.9% in Bangkok [3]. Even with standardized diagnosis and management, the CFR remained in the range of 0.5–1.7% from 2000–2011 in the Philippines [4]. Despite the high mortality of DHF/DSS, no promising viral-specific drugs or vaccines are available due to the limited understanding of the complicated pathogenic mechanism.
Several hypotheses have been proposed to explain the pathogenesis of DHF/DSS [5]. Among them, antibody-dependent enhancement (ADE) has been proposed to explain why most DHF/DSS cases occur in children who are secondarily infected with a different serotype of DENV from the previous one [6]. Based on ADE, antibodies that are generated by a single DENV infection contribute to lasting homotypic immunity but may permit heterotypic DENV infection. Furthermore, these serotype non-specific antibodies may augment heterotypic virus entry and replication in Fcγ receptor-bearing macrophages, leading to enhanced viremia, antigenemia and cytokine storm [7]. This scenario may also explain why infants who passively acquire maternal anti-dengue antibodies are more likely to develop DHF/DSS following primary infection [8]. However, ADE dose not explain why vascular leakage and hemorrhage occur in DHF/DSS patients. Only when we better understand the molecular mechanisms of DENV pathogenesis can a more effective and specific therapy or vaccine against DHF/DSS be developed. In this review, we focus on the pathogenic roles of DENV non-structural protein 1 (NS1) in the pathogenesis of DHF/DSS. The potential of NS1 as a drug target or vaccine candidate to treat or prevent dengue will be discussed.
DENV structure
The DENV particle is approximately 500 Å in diameter and includes a positive-sense RNA genome with ~10,700 nucleotides and 3 structural proteins: capsid (C, 100 amino acids), precursor membrane (prM, 75 amino acids), and envelope (E, 495 amino acids) [9]. The capsid protein and the viral RNA genome form a nucleocapsid that buds at the endoplasmic reticulum (ER) in association with 180 copies of prM and E and carries host-derived lipids to form the immature virion [10]. Initially, the immature virion is covered by 60 spikes, each of which is composed of E trimers with associated prM proteins. The maturation process requires the host protease furin, which cleaves prM into the pr and M proteins in the Golgi after the noninfectious virion passes through the cell’s secretory system, which is an acidic environment. This cleavage results in a rearrangement of E to the immature dimer structure, in which E maintains interactions with pr and M [11]. After budding from the cell via exocytosis, the neutral pH of the extracellular environment dissociates E and pr to form mature virions, which are available to infect new cells [11]. In addition to the structural proteins, the RNA genome of dengue virus encodes 7 nonstructural proteins that are essential for viral replication (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5).
NS1 structure, expression and secretion
DENV NS1 is a 48-kDa glycoprotein that is highly conserved among all flaviviruses [12]. NS1 is essential for viral replication with an unknown mechanism that possibly involves interactions with NS4A and NS4B [13, 14]. Initially, NS1 is expressed as a monomer in infected cells. After post-translational modification in the ER lumen, it forms homodimers associated with organelle membranes and the cell membrane [12]. Despite the lack of a transmembrane region, NS1 anchors to the cell membrane through several pathways. The mechanisms are unclear, but anchorage of NS1 to glycosyl-phosphatidylinositol and lipid rafts has been shown [15, 16]. In addition, NS1 is the only protein that is continuously secreted by infected host cells. NS1 is secreted in a hexamer form, which is composed of three dimers with a detergent-sensitive hydrophobic central cavity that carries a cargo of ~70 lipid molecules; the composition is similar to a high-density lipoprotein [17, 18]. This lipid-rich structure may help secreted NS1 attach to the cell membrane by associating with glycosaminoglycans (GAGs) [19]. Due to the similarity between NS1 and high-density lipoprotein, NS1 has been proposed to disrupt the coagulation cascade possibly through interfering with the interaction or biogenesis of endogenous lipoprotein particles [18]. Accumulation of secreted NS1 in DHF/DSS patient sera has been observed during the critical phase [20]. The serum concentration of NS1 in DHF/DSS patients can reach as high as 50 μg/ml, and the concentration is positively correlated with the disease severity [21,22,23]. During the recovery phase, NS1 is cleared from the circulation by antibody-mediated effects. Because secreted NS1 can interact with complement protein, it was first described as a soluble complement-fixing (SCF) antigen that could promote C4 degradation and in turn possibly protect DENV from complement-dependent lysis [24,25,26]. Recently, pathogenic roles for secreted NS1 in DHF/DSS have been demonstrated due to its involvement in systemic immunity and endothelial cell activation. In this review, we focus on the molecular mechanisms underlying how NS1 may contribute to vascular leakage, coagulopathy and thrombocytopenia during dengue infection. The possibility of targeting NS1 as a drug and vaccine development target against dengue infection will also be discussed.
The pathogenic roles of NS1 in vascular leakage
Pathogenic factors of vascular leakage in dengue pathogenesis
Based upon in vitro data or mouse models, it was once concluded that endothelial cell apoptosis led to vascular permeability during DENV infections and that this was because direct infection of endothelial cells by DENV or damage by antibodies (Abs) against NS1 which can cross-react with endothelial cells [27,28,29,30,31,32,33,34]. However, plasma leakage improves within 1 to 2 days in DHF/DSS patients who receive appropriate fluid resuscitation, and tissue samples from these patients show little structural damage in their vessels. Therefore, apoptosis of endothelial cells induced by DENV infection or anti-NS1 antibodies is not sufficient to support the clinical outcome. As a result, endothelial dysfunction but not apoptosis induced by a dengue-specific factor is currently considered to play a more important role in causing vascular leakage in DHF/DSS [35,36,37].
Contribution of the NS1 protein to vascular leakage
DENV NS1-induced vascular leakage has been widely discussed since 2015. A previous study demonstrated that NS1 proteins induced vascular leakage, and applying anti-NS1 antibodies attenuated NS1-induced vascular leakage as well as the mortality rate in mice [38]. However, the mediating receptor of NS1 remains controversial. One study suggested that blocking TLR2 or TLR6 attenuated DENV NS1-induced secretion of TNF-α and IL-6 by peripheral blood mononuclear cells (PBMCs) [39]. TLR6 deficiency also reduced DENV NS1-induced mortality in mice [39]. However, another study demonstrated that NS1-activated TNF-α and IL-1β mRNA expression and IL-6 secretion were attenuated by blocking TLR4 in PBMCs [40]. In contrast, TLR2 inhibition did not alter the effects induced by NS1 in PBMCs [40]. These authors also showed that blocking TLR4 rescued NS1-induced endothelial hyperpermeability, indicating that NS1-induced vascular leakage was mediated by TLR4 [40]. Later, the same group published a short communication to explain that the different results might be caused by contamination of Escherichia coli-derived recombinant NS1 with multiple TLR ligands and that TLR4 should be regarded as the real NS1 receptor [41]. However, it has also been shown that DENV NS1 induces similar levels of vascular leakage in TLR4-receptor-deficient mice and wild-type animals, which indicates that NS1-induced vascular leakage can be independent of TLR4 [42]. Taken together, these results suggest that NS1 may contribute to vascular leakage through both TLR4-dependent and independent mechanisms.
In our previous study, we demonstrated that autophagy-mediated junction disruption was involved in DENV NS1-induced vascular leakage, which may explain why vascular leakage in dengue patients is a quick and reversible pathogenic change [43]. NS1-induced macrophage migration inhibitory factor (MIF) secretion is involved in NS1-induced autophagy of endothelial cells [43]. An in vitro study also showed that DENV-infected cells induced MIF secretion, which can cause endothelial hyperpermeability. Furthermore, Mif−/− mice exhibited reduced pathogenesis in a model of severe dengue [44], indicating the importance of MIF in dengue pathogenesis [45]. In fact, several clinical studies have shown that the MIF concentration is elevated in dengue patients [46, 47] and that the MIF concentration is higher in DHF patients who die than in DHF survivors and DF patients [48].
In addition to disrupting endothelial junctions, NS1 also causes vascular leakage by inducing endothelial glycocalyx degradation mediated by heparanase-1 (HPA-1) [42, 49]. The glycocalyx is a thin, negatively charged network consisting of glycoproteins, proteoglycans, and glycosaminoglycans at the luminal side of endothelial cells lining blood vessels throughout the body [50]. To maintain homeostasis, the glycocalyx acts as a barrier that controls numerous physiological processes, such as regulating vascular permeability, preventing the adhesion of leukocytes and blood platelets to the vessel walls [51, 52], mediating shear stress [53, 54], and modulating inflammatory and hemostatic processes. Damage of the endothelial glycocalyx correlates to several vascular pathologies, including ischemia/reperfusion, hypoxia, sepsis, volume overload, diabetes and atherosclerosis [50, 55].
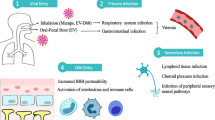
Shedding of the endothelial glycocalyx is related to activation of the heparan sulfate-specific glucuronidase HPA-1 [52, 56]. HPA-1 is synthesized as a 65-kDa non-active precursor that subsequently undergoes proteolytic cleavage to yield 8-kDa and 50-kDa subunits that heterodimerize to form an active enzyme. Activated HPA-1 enhances shedding of the transmembrane heparan sulfate proteoglycan syndecan-1 (CD138) and elevates the CD138 level in the bloodstream [57, 58]. In addition to HPA-1, metalloproteinase (MMP) family proteins are also important proteases that are capable of digesting the endothelial glycocalyx [59, 60] and increased MMP levels correlate with vascular leakage in DHF/DSS [61,62,63,64]. In 2017, Glasner suggested that DENV NS1-induced vascular leakage was independent of inflammatory cytokines, including TNF-α, IL-6 and IL-8, but was dependent on endothelial glycocalyx components, including cathepsin L and HPA-1 [42]. Recently, we further demonstrated that MIF is involved in DENV NS1-induced HPA-1 and MMP-9 secretion and degradation of the endothelial glycocalyx [65]. Taken together, the mechanisms of the vascular leakage that occurs during DENV infection may be very complex and may involve both the virus and the host immune response. The possible mechanisms by which DENV NS1 contributes to vascular leakage are shown in Fig. 1.

The possible mechanisms by which DENV NS1 causes vascular leakage. (1a) NS1 binding to TLR4 of PBMCs induces the expression and secretion of TNF-α, IL-1β and IL-6 cytokines, which may disrupt the tight junction, leading to vascular leakage [40]. (1b) NS1 binding to TLR4 or (2a) other molecules on endothelial cells induces the secretion of MIF [43]. (2b) MIF binding to its receptor on endothelial cells induces junction disruption through autophagic degradation of junction proteins such as ZO-1 and VE-cadherin [43]. (3) Binding of NS1 to endothelial cells also induces HPA-1 activation through cathepsin L, leading to endothelial glycocalyx degradation and vascular leakage [42, 49]. (4a) Additionally, NS1-induced MIF secretion is also involved in HPA-1 secretion of endothelial cells, and (4b) MMP-9 secretion of WBCs which can also contribute to endothelial glycocalyx degradation [65]
The pathogenic roles of NS1 in coagulopathy and thrombocytopenia
In addition to vascular leakage, DENV NS1 may also contribute to severe dengue by disrupting coagulation. The NS1/thrombin complex was found in the sera of dengue patients, and binding of NS1 to prothrombin inhibited its activation, leading to a prolonged activated partial thromboplastin time [66]. However, whether NS1 is involved in thrombocytopenia is still unclear. It is known that LPS can induce platelet activation and potentiate platelet aggregation via TLR4/MyD88 signal transduction [67]. Since both NS1 and LPS can activate immune cells through TLR4, NS1 may induce platelet activation and enhance aggregation, possibly leading to over-destruction of platelets during dengue infection. Collectively, increasing evidence suggest that NS1 plays a crucial role in dengue pathogenesis by contributing to both vascular leakage and hemorrhage in dengue disease.
DENV NS1 as a therapeutic target
Current status of DENV treatment
Although many dengue patients only experience asymptomatic or mild signs of a flu-like illness followed by self-recovery within one week, some patients develop worse dengue symptoms that become life-threatening. To date, the treatment of dengue disease has been mostly supportive, and no licensed therapeutic drug is available. An effective drug against DENV infection is in great demand until a satisfactory vaccine becomes available. Because earlier observational studies stated that disease severity positively correlated with the viremia level and febrile phase during infections [68, 69], dengue researchers have put great effort into anti-viral approaches targeting different structural or nonstructural proteins. The inhibitory mechanisms of viral entry [70], pH-dependent viral fusion [71, 72], enzymes required for transcription/replication [73,74,75,76,77], and viral protein modifications [78] have been widely investigated. From this perspective, some off-patent drugs and antibiotics have also been tested for repurposing [79, 80], which is beneficial for shortening development time and costs. Although some drugs lead to significant viral reduction and provide effective anti-viral activity both in vitro and in animal models, in real world situations, these types of antiviral drugs face other limitations and challenges, probably due to untimely treatment in the clinic. People often enroll in clinical studies until their viremia declines during the later phase of illness. As a result, most cases, unfortunately, fail to meet the therapeutic endpoint measurements in clinical trials [71].
From another perspective, other studies have provided different insights into host modulation and immune regulation as therapeutic targets, which include targeting host dependency factors, host restriction factors, and host-mediated pathogenesis pathways [81]. In contrast to viral targets, host-targeting antiviral approaches are believed to avoid the rapid drug resistance or mutations that arise during viral evolution. Strategies targeting host factors have been widely reviewed using advanced mass screening approaches [82,83,84]. For instance, turn-on targets of antiviral responses are often reported to act as defensive therapies [85, 86]. Interferons exert diverse antiviral functions against viral replication, which can activate interferon-stimulated genes and related mechanisms. In addition, regulation of host metabolic pathways required for viral replication, such as glycolysis and autophagy, has been studied recently [87, 88]. However, these approaches can nonspecifically regulate the functions of different cells, and most of these approaches fail in clinical trials [89]. Recently, we found that minocycline, a semi-synthetic tetracycline-derivative antibiotic, attenuates DENV replication through inhibition of MIF secretion and autophagy formation both in vitro and in vivo [90]. In addition, minocycline treatment can prolong the survival of ICR suckling mice after DENV infection. Therefore, minocycline may modulate both virus replication and the host immune response. Further clinical trials of minocycline in dengue patients may help to verify its therapeutic protection against DHF/DSS.
Antibody against NS1 as a therapeutic drug for dengue disease
In addition to small molecule drugs, antibody therapies are appreciated for their specificity against diseases. To date, many murine- or human-derived monoclonal antibodies have been developed to test their therapeutic effects against DENV infection in different studies. Antibodies targeting structural proteins, such as envelope and prM/M, have been characterized; these antibodies are termed “neutralizing antibodies” due to their blockage of viral entry and inhibition of viral attachment to host cells. However, safety issues and insufficient efficacy against all four DENV serotypes are often challenged due to the risk of ADE. The enhancement of viral infection by antibodies against DENV has been not only evaluated both in vitro and in vivo but also emphasized in a clinical cohort study [91]. Nevertheless, many researchers have identified mAbs that can neutralize DENV infection without ADE as a side effect. One study indicated that single-dose administration of human Ab513, which recognizes a linear epitope of envelope domain III, prevented DENV-induced thrombocytopenia in humanized mice with ADE [92]. Another study suggested that antibodies that recognized the envelope dimer epitope (EDE) were highly potent and broadly neutralizing antibodies [93, 94]. In addition, the human-derived mAb SIgN-3C with a LALA mutant abrogated the ADE effect and protected mice from lethal DENV-2 infection [95].
In contrast to Abs against structural proteins on the virion, such as E and PrM/M, which are effective only in the viremic phase, anti-NS1 Abs can provide different therapeutic mechanisms, by not only reducing viral propagation from infected cells in the early viremic phase but also attenuating NS1-induced disease development during the critical phase. In addition, because NS1 is not a viral structural protein, anti-NS1 Abs will not induce ADE. Indeed, anti-NS1 Abs can reduce viral replication by complement-dependent cytotoxicity (CDC) of infected cells and have been demonstrated in several flaviviruses, including DENV, in vitro and in vivo [96,97,98,99]. In addition, anti-NS1 Abs can block NS1-elicited pathogenic effects both in vitro and in vivo [38] and reduce DENV-induced mortality and morbidity in different mouse models (Table 1).
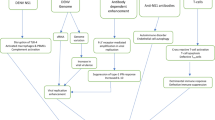
However, as previously described, many anti-NS1 Abs can cross-react with host proteins; thus, antibodies generated from NS1 immune sera may contain cross-reactive Abs with undesired side effects [32, 100]. Therefore, we need to identify a subclass of protective anti-NS1 Abs that can block the activity of all four different dengue NS1 serotypes without cross-reactivity to host proteins. In our previous study, we analyzed a mAb against NS1 and identified Abs against a region of the NS1 wing domain that were protective against DENV infection in mice. Furthermore, the amount of these anti-NS1 Abs was inversely correlated with the severity of disease in dengue patients, indicating that the Abs were protective in patients [97]. Therefore, a mAb against this region of NS1 may represent an alternative choice for a therapeutic drug that is specific against DENV infection while avoiding the risk of ADE (Fig. 2).

The possible pathogenic roles of DENV NS1 and its potential as a therapeutic target against DENV infection
DENV NS1 as a vaccine candidate
Current status of DENV vaccine development
The only licensed dengue vaccine currently available was developed by Sanofi Pasteur and has been approved in many countries. This live-attenuated tetravalent dengue vaccine (CYD-TDV; Dengvaxia) contains DENV E and prM proteins from the four serotypes in the yellow fever 17D backbone and has been used to induce preventive humoral and cell-mediated immune responses [101, 102]. Although CYD-TDV induced neutralizing Abs against all four DENV serotypes, this vaccine rendered only partial protection against serotype 2 DENV infection. In addition to the unequal efficacy of CYD-TDV against all four DENV serotypes, vaccination of this vaccine may induce non-protective Abs that enhance disease severity in persons who had not been exposed to dengue before. Indeed, it is reported that among children younger than nine years of age, the vaccine is associated with an increased incidence of hospitalization for severe dengue disease [103]. Therefore, the vaccination of seronegative individuals with Dengvaxia may enhance dengue disease severity but not protection due to ADE [104]. Conversely, some studies have suggested that the dengue vaccine fails to provide full protection, possibly due to the lack of T cell immunity elicited against nonstructural proteins or an NS1-induced protective immune response [105]. Collectively, these concerns make nonstructural proteins, including NS1, alternative options for dengue vaccine development.
NS1 as a vaccine candidate against DENV infection
Both humoral and T cell-mediated cellular immune responses are critical for protection against DENV infection. Viral structural proteins have been regarded as potent targets to induce neutralizing Abs but are associated with the intractable issue of ADE. As a nonstructural protein, NS1 does not induce Abs against the virion. NS1 is the only nonstructural protein of DENV that can be both anchored on the surface of infected cells as a membrane-associated homodimer and released from infected cells into circulation as a hexamer. These properties of NS1 make it able to trigger both cellular and humoral immunity. Because NS1 is exposed on infected cells, the complement cascade can be triggered by NS1-bound anti-NS1 Abs [106]. Complement activation can lyse infected cells via CDC and eventually reduce viral titers through suppression of viral propagation. Studies have shown that immunization with DENV NS1 via different approaches can induce protective immune responses against DENV infection in mice, as shown in Table 2.
However, immunization of mice with full length of DENV NS1 can induce Abs cross-react with host proteins have also been demonstrated by different groups and these cross-reactive Abs can cause pathological effects both in vitro and in mice [107,108,109]. Although the contributions of these cross-reactive Abs in dengue pathogenesis are still under debate, the potential side effects induced by vaccination with full-length NS1 should be avoided in dengue vaccine design. Since most of these cross-reactive Abs recognize the C-terminal region of NS1. [110], different approaches have been applied to prevent the induction of cross-reactive Abs by NS1 immunization. For instance, NS1 lacking its C-terminus (ΔC NS1) was used to immunize mice; the results showed that ΔC NS1-elicited Abs provided better protection against DENV infection than immunization with full-length NS1 [96]. However, in addition to the C-terminus, other regions of NS1 also show molecular mimicry to host proteins and can elicit auto-reactive Abs against endothelial cells and coagulation factors [111, 112]. To avoid the risk of the induction of cross-reactive antibodies by NS1 immunization, a short modified NS1 peptide containing an 11-a.a. conserved wing domain region of NS1 was designed; this peptide modified the critical pathogenic amino acids to reduce cross-reactivity but maintain immunogenicity. Importantly, both active immunization with the modified NS1 peptide and passive transfer of polyclonal Abs against the modified NS1 peptide provided protection against DENV in a hemorrhagic mouse model and a lethal infection mouse model [97].
Conclusions
Collectively, this review discusses the critical pathogenic roles of NS1 in dengue pathogenesis. NS1 is considered a unique “viral toxin” in dengue disease. Therapeutic approaches and vaccine development targeting NS1 may provide different opportunities to combat dengue disease.
Abbreviations
- CFR:
-
Case-fatality rate
- CXCR:
-
CXC chemokine receptors
- DENV:
-
Dengue virus
- DF:
-
Dengue fever
- DHF:
-
Dengue hemorrhagic fever
- DSS:
-
Dengue shock syndrome
- E:
-
Envelope protein
- ER:
-
Endoplasmic reticulum
- GAG:
-
Glycosaminoglycan
- IL:
-
Interleukin
- iNOS:
-
Inducible NO synthase
- MIF:
-
Macrophage migration inhibitory factor
- MMP:
-
Matrix metalloproteinase
- NO:
-
nitric oxide
- NS1:
-
Nonstructural protein 1
- PBMCs:
-
Peripheral blood mononuclear cells
- prM:
-
Pre-membrane protein
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
References
Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, et al. The global distribution and burden of dengue. Nature. 2013;496(7446):504–7.
Dengue haemorrhagic fever. diagnosis, treatment, prevention and control. 2nd ed. Geneva: World Health Organization; 1997.
Kalayanarooj S. Standardized Clinical Management: Evidence of Reduction of Dengue Haemorrhagic Fever Case-Fatality Rate in Thailand. Dengue Bulletin. 1999;23:10–7.
Bravo L, Roque VG, Brett J, Dizon R, L'Azou M. Epidemiology of dengue disease in the Philippines (2000-2011): a systematic literature review. PLoS Negl Trop Dis. 2014;8(11):e3027.
Lei HY, Yeh TM, Liu HS, Lin YS, Chen SH, Liu CC. Immunopathogenesis of dengue virus infection. J Biomed Sci. 2001;8(5):377–88.
Kliks SC, Nisalak A, Brandt WE, Wahl L, Burke DS. Antibody-dependent enhancement of dengue virus growth in human monocytes as a risk factor for dengue hemorrhagic fever. Am J Trop Med Hyg. 1989;40(41):444–51.
Halstead SB, Venkateshan CN, Gentry MK, Larsen LK. Heterogeneity of infection enhancement of dengue 2 strains by monoclonal antibodies. J Immunol. 1984;132(31):1529–32.
Kliks SC, Nimmanitya S, Nisalak A, Burke DS. Evidence that maternal dengue antibodies are important in the development of dengue hemorrhagic fever in infants. Am J Trop Med Hyg. 1988;38(21):411–9.
Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, et al. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 2002;108(5):717–25.
Byk LA, Gamarnik AV. Properties and Functions of the Dengue Virus Capsid Protein. Annu Rev Virol. 2016;3(1):263–81.
Christian EA, Kahle KM, Mattia K, Puffer BA, Pfaff JM, Miller A, Paes C, Davidson E, Doranz BJ. Atomic-level functional model of dengue virus Envelope protein infectivity. Proc Natl Acad Sci USA. 2013;110(46):18662–7.
Muller DA, Young PR. The flavivirus NS1 protein: Molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antiviral Res. 2013;98(2):192–208.
Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J Virol. 2012;86(13):7360–71.
Lindenbach BD, Rice CM. Genetic interaction of flavivirus nonstructural proteins NS1 and NS4A as a determinant of replicase function. J Virol. 1999;73(6):4611–21.
Jacobs MG, Robinson PJ, Bletchly C, Mackenzie JM, Young PR. Dengue virus nonstructural protein 1 is expressed in a glycosyl-phosphatidylinositol-linked form that is capable of signal transduction. FASEB J. 2000;14(11):1603–10.
Noisakran S, Dechtawewat T, Avirutnan P, Kinoshita T, Siripanyaphinyo U, Puttikhunt C, Kasinrerk W, Malasit P, Sittisombut N. Association of dengue virus NS1 protein with lipid rafts. J Gen Virol. 2008;89(Pt 10):2492–500.
Edeling MA, Diamond MS, Fremont DH. Structural basis of Flavivirus NS1 assembly and antibody recognition. Proc Natl Acad Sci U S A. 2014;111(11):4285–90.
Gutsche I, Coulibaly F, Voss JE, Salmon J, d'Alayer J, Ermonval M, Larquet E, Charneau P, Krey T, Megret F, et al. Secreted dengue virus nonstructural protein NS1 is an atypical barrel-shaped high-density lipoprotein. Proc Natl Acad Sci U S A. 2011;108(19):8003–8.
Avirutnan P, Zhang L, Punyadee N, Manuyakorn A, Puttikhunt C, Kasinrerk W, Malasit P, Atkinson JP, Diamond MS. Secreted NS1 of dengue virus attaches to the surface of cells via interactions with heparan sulfate and chondroitin sulfate E. PLoS Pathog. 2007;3(11):e183.
Chuang YC, Wang SY, Lin YS, Chen HR, Yeh TM. Re-evaluation of the pathogenic roles of nonstructural protein 1 and its antibodies during dengue virus infection. J Biomed Sci. 2013;20:42.
Alcon S, Talarmin A, Debruyne M, Falconar A, Deubel V, Flamand M. Enzyme-linked immunosorbent assay specific to Dengue virus type 1 nonstructural protein NS1 reveals circulation of the antigen in the blood during the acute phase of disease in patients experiencing primary or secondary infections. J Clin Microbiol. 2002;40(2):376–81.
Libraty DH, Young PR, Pickering D, Endy TP, Kalayanarooj S, Green S, Vaughn DW, Nisalak A, Ennis FA, Rothman AL. High circulating levels of the dengue virus nonstructural protein NS1 early in dengue illness correlate with the development of dengue hemorrhagic fever. J Infect Dis. 2002;186(8):1165–8.
Young PR, Hilditch PA, Bletchly C, Halloran W. An antigen capture enzyme-linked immunosorbent assay reveals high levels of the dengue virus protein NS1 in the sera of infected patients. J Clin Microbiol. 2000;38(3):1053–7.
Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, Atkinson JP. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J Exp Med. 2010;207(4):793–806.
Avirutnan P, Hauhart RE, Somnuke P, Blom AM, Diamond MS, Atkinson JP. Binding of flavivirus nonstructural protein NS1 to C4b binding protein modulates complement activation. J Immunol. 2011;187(1):424–33.
Smith TJ, Brandt WE, Swanson JL, McCown JM, Buescher EL. Physical and biological properties of dengue-2 virus and associated antigens. J Virol. 1970;5(41):524–32.
Liao H, Xu J, Huang J. FasL/Fas pathway is involved in dengue virus induced apoptosis of the vascular endothelial cells. J Med Virol. 2010;82(8):1392–9.
Long X, Li Y, Qi Y, Xu J, Wang Z, Zhang X, Zhang D, Zhang L, Huang J. XAF1 contributes to dengue virus-induced apoptosis in vascular endothelial cells. FASEB J. 2013;27(3):1062–73.
Chen HC, Hofman FM, Kung JT, Lin YD, Wu-Hsieh BA. Both virus and tumor necrosis factor alpha are critical for endothelium damage in a mouse model of dengue virus-induced hemorrhage. J Virol. 2007;81(11):5518–26.
Cheng HJ, Luo YH, Wan SW, Lin CF, Wang ST, Hung NT, Liu CC, Ho TS, Liu HS, Yeh TM, et al. Correlation between serum levels of anti-endothelial cell autoantigen and anti-dengue virus nonstructural protein 1 antibodies in dengue patients. Am J Trop Med Hyg. 2015;92(5):989–95.
Lin CF, Lei HY, Shiau AL, Liu HS, Yeh TM, Chen SH, Liu CC, Chiu SC, Lin YS. Endothelial cell apoptosis induced by antibodies against dengue virus nonstructural protein 1 via production of nitric oxide. J Immunol. 2002;169(2):657–64.
Chen CL, Lin CF, Wan SW, Wei LS, Chen MC, Yeh TM, Liu HS, Anderson R, Lin YS. Anti-dengue virus nonstructural protein 1 antibodies cause NO-mediated endothelial cell apoptosis via ceramide-regulated glycogen synthase kinase-3beta and NF-kappaB activation. J Immunol. 2013;191(4):1744–52.
Lin CF, Chiu SC, Hsiao YL, Wan SW, Lei HY, Shiau AL, Liu HS, Yeh TM, Chen SH, Liu CC, et al. Expression of cytokine, chemokine, and adhesion molecules during endothelial cell activation induced by antibodies against dengue virus nonstructural protein 1. J Immunol. 2005;174(1):395–403.
Cheng HJ, Lin CF, Lei HY, Liu HS, Yeh TM, Luo YH, Lin YS. Proteomic analysis of endothelial cell autoantigens recognized by anti-dengue virus nonstructural protein 1 antibodies. Exp Biol Med (Maywood). 2009;234(1):63–73.
Avirutnan P, Punyadee N, Noisakran S, Komoltri C, Thiemmeca S, Auethavornanan K, Jairungsri A, Kanlaya R, Tangthawornchaikul N, Puttikhunt C, et al. Vascular leakage in severe dengue virus infections: a potential role for the nonstructural viral protein NS1 and complement. J Infect Dis. 2006;193(8):1078–88.
Thomas SJ. NS1: A corner piece in the dengue pathogenesis puzzle? Sci Transl Med. 2015;7(304):304fs337.
Halstead SB. Pathogenesis of Dengue: Dawn of a New Era. F1000Res. 2015;4:1353–60.
Beatty PR, Puerta-Guardo H, Killingbeck SS, Glasner DR, Hopkins K, Harris E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci Transl Med. 2015;7(304):304ra141.
Chen J, Ng MM, Chu JJ. Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection. PLoS Pathog. 2015;11(7):e1005053.
Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, Hume DA, Stacey KJ, Young PR. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci Transl Med. 2015;7(304):304ra142.
Modhiran N, Watterson D, Blumenthal A, Baxter AG, Young PR, Stacey KJ. Dengue virus NS1 protein activates immune cells via TLR4 but not TLR2 or TLR6. Immunol Cell Biol. 2017;95(5):491–5.
Glasner DR, Ratnasiri K, Puerta-Guardo H, Espinosa DA, Beatty PR, Harris E. Dengue virus NS1 cytokine-independent vascular leak is dependent on endothelial glycocalyx components. PLoS Pathog. 2017;13(11):e1006673.
Chen HR, Chuang YC, Lin YS, Liu HS, Liu CC, Perng GC, Yeh TM. Dengue Virus Nonstructural Protein 1 Induces Vascular Leakage through Macrophage Migration Inhibitory Factor and Autophagy. PLoS Negl Trop Dis. 2016;10(7):e0004828.
Assuncao-Miranda I, Amaral FA, Bozza FA, Fagundes CT, Sousa LP, Souza DG, Pacheco P, Barbosa-Lima G, Gomes RN, Bozza PT, et al. Contribution of macrophage migration inhibitory factor to the pathogenesis of dengue virus infection. FASEB J. 2010;24(1):218–28.
Chuang YC, Lei HY, Liu HS, Lin YS, Fu TF, Yeh TM. Macrophage migration inhibitory factor induced by dengue virus infection increases vascular permeability. Cytokine. 2011;54(2):222–31.
Ferreira RA, de Oliveira SA, Gandini M, Ferreira Lda C, Correa G, Abiraude FM, Reid MM, Cruz OG, Kubelka CF. Circulating cytokines and chemokines associated with plasma leakage and hepatic dysfunction in Brazilian children with dengue fever. Acta Trop. 2015;149:138–47.
Yong YK, Tan HY, Jen SH, Shankar EM, Natkunam SK, Sathar J, Manikam R, Sekaran SD. Aberrant monocyte responses predict and characterize dengue virus infection in individuals with severe disease. J Transl Med. 2017;15(1):121.
Chen LC, Lei HY, Liu CC, Shiesh SC, Chen SH, Liu HS, Lin YS, Wang ST, Shyu HW, Yeh TM. Correlation of serum levels of macrophage migration inhibitory factor with disease severity and clinical outcome in dengue patients. Am J Trop Med Hyg. 2006;74(1):142–7.
Puerta-Guardo H, Glasner DR, Harris E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016;12(7):e1005738.
Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454(3):345–59.
Lipowsky HH. The endothelial glycocalyx as a barrier to leukocyte adhesion and its mediation by extracellular proteases. Ann Biomed Eng. 2012;40(4):840–8.
Schmidt EP, Yang Y, Janssen WJ, Gandjeva A, Perez MJ, Barthel L, Zemans RL, Bowman JC, Koyanagi DE, Yunt ZX, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18(8):1217–23.
Bai K, Wang W. Spatio-temporal development of the endothelial glycocalyx layer and its mechanical property in vitro. J R Soc Interface. 2012;9(74):2290–8.
Dull RO, Cluff M, Kingston J, Hill D, Chen H, Hoehne S, Malleske DT, Kaur R. Lung heparan sulfates modulate K(fc) during increased vascular pressure: evidence for glycocalyx-mediated mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2012;302(9):L816–28.
Becker BF, Chappell D, Jacob M. Endothelial glycocalyx and coronary vascular permeability: the fringe benefit. Basic Res Cardiol. 2010;105(6):687–701.
Chappell D, Jacob M, Rehm M, Stoeckelhuber M, Welsch U, Conzen P, Becker BF. Heparinase selectively sheds heparan sulphate from the endothelial glycocalyx. Biol Chem. 2008;389(1):79–82.
Yang Y, Macleod V, Miao HQ, Theus A, Zhan F, Shaughnessy JD Jr, Sawyer J, Li JP, Zcharia E, Vlodavsky I, et al. Heparanase enhances syndecan-1 shedding: a novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem. 2007;282(18):13326–33.
Purushothaman A, Uyama T, Kobayashi F, Yamada S, Sugahara K, Rapraeger AC, Sanderson RD. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood. 2010;115(12):2449–57.
Mulivor AW, Lipowsky HH. Inhibition of glycan shedding and leukocyte-endothelial adhesion in postcapillary venules by suppression of matrixmetalloprotease activity with doxycycline. Microcirculation (New York, NY : 1994). 2009;16(8):657–66.
Lipowsky HH. Protease Activity and the Role of the Endothelial Glycocalyx in Inflammation. Drug Discov Today Dis Models. 2011;8(1):57–62.
Voraphani N, Khongphatthanayothin A, Srikaew K, Tontulawat P, Poovorawan Y. Matrix metalloproteinase-9 (mmp-9) in children with dengue virus infection. Jpn J Infect Dis. 2010;63(5):346–8.
Her Z, Kam YW, Gan VC, Lee B, Thein TL, Tan JJ, Lee LK, Fink K, Lye DC, Renia L, et al. Severity of Plasma Leakage Is Associated With High Levels of Interferon gamma-Inducible Protein 10, Hepatocyte Growth Factor, Matrix Metalloproteinase 2 (MMP-2), and MMP-9 During Dengue Virus Infection. J Infect Dis. 2017;215(1):42–51.
Luplertlop N, Misse D. MMP cellular responses to dengue virus infection-induced vascular leakage. Jpn J Infect Dis. 2008;61(4):298–301.
Suwarto S, Sasmono RT, Sinto R, Ibrahim E, Suryamin M. Association of Endothelial Glycocalyx and Tight and Adherens Junctions With Severity of Plasma Leakage in Dengue Infection. J Infect Dis. 2017;215(6):992–9.
Chen HR, Chao CH, Liu CC, Ho TS, Tsai HP, Perng GC, Lin YS, Wang JR, Yeh TM. Macrophage migration inhibitory factor is critical for dengue NS1-induced endothelial glycocalyx degradation and hyperpermeability. PLoS Pathog. 2018;14(4):e1007033.
Lin SW, Chuang YC, Lin YS, Lei HY, Liu HS, Yeh TM. Dengue virus nonstructural protein NS1 binds to prothrombin/thrombin and inhibits prothrombin activation. J Infect. 2012;64(3):325–34.
Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, Du X, Li Z. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol. 2009;182(12):7997–8004.
Vaughn DW, Green S, Kalayanarooj S, Innis BL, Nimmannitya S, Suntayakorn S, Endy TP, Raengsakulrach B, Rothman AL, Ennis FA, et al. Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J Infect Dis. 2000;181(1):2–9.
Wang WK, Chao DY, Kao CL, Wu HC, Liu YC, Li CM, Lin SC, Ho ST, Huang JH, King CC. High levels of plasma dengue viral load during defervescence in patients with dengue hemorrhagic fever: implications for pathogenesis. Virology. 2003;305(2):330–8.
Altmeyer R. Virus attachment and entry offer numerous targets for antiviral therapy. Curr Pharm Des. 2004;10(30):3701–12.
Tricou V, Minh NN, Van TP, Lee SJ, Farrar J, Wills B, Tran HT, Simmons CP. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Negl Trop Dis. 2010;4(8):e785.
De La Guardia C, Lleonart R. Progress in the identification of dengue virus entry/fusion inhibitors. Biomed Res Int. 2014;2014:825039.
Nguyen NM, Tran CN, Phung LK, Duong KT, Huynh Hle A, Farrar J, Nguyen QT, Tran HT, Nguyen CV, Merson L, et al. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J Infect Dis. 2013;207(9):1442–50.
Rothan HA, Han HC, Ramasamy TS, Othman S, Rahman NA, Yusof R. Inhibition of dengue NS2B-NS3 protease and viral replication in Vero cells by recombinant retrocyclin-1. BMC Infect Dis. 2012;12:314.
van Cleef KWR, Overheul GJ, Thomassen MC, Kaptein SJF, Davidson AD, Jacobs M, Neyts J, van Kuppeveld FJM, van Rij RP. Identification of a new dengue virus inhibitor that targets the viral NS4B protein and restricts genomic RNA replication. Antivir Res. 2013;99(2):165–71.
Wang QY, Kondreddi RR, Xie XP, Rao R, Nilar S, Xu HY, Qing M, Chang D, Dong HP, Yokokawa F, et al. A Translation Inhibitor That Suppresses Dengue Virus In Vitro and In Vivo. Antimicrob Agents Ch. 2011;55(9):4072–80.
Mastrangelo E, Pezzullo M, De Burghgraeve T, Kaptein S, Pastorino B, Dallmeier K, de Lamballerie X, Neyts J, Hanson AM, Frick DN, et al. Ivermectin is a potent inhibitor of flavivirus replication specifically targeting NS3 helicase activity: new prospects for an old drug. J Antimicrob Chemoth. 2012;67(8):1884–94.
Rathore APS, Paradkar PN, Watanabe S, Tan KH, Sung C, Connolly JE, Low J, Ooi EE, Vasudevan SG. Celgosivir treatment misfolds dengue virus NS1 protein, induces cellular pro-survival genes and protects against lethal challenge mouse model. Antivir Res. 2011;92(3):453–60.
Simanjuntak Y, Liang JJ, Lee YL, Lin YL. Repurposing of prochlorperazine for use against dengue virus infection. J Infect Dis. 2015;211(3):394–404.
Leela SL, Srisawat C, Sreekanth GP, Noisakran S, Yenchitsomanus PT, Limjindaporn T. Drug repurposing of minocycline against dengue virus infection. Biochem Bioph Res Co. 2016;478(1):410–6.
Krishnan MN, Garcia-Blanco MA. Targeting host factors to treat West Nile and dengue viral infections. Viruses. 2014;6(2):683–708.
de Chassey B, Meyniel-Schicklin L, Vonderscher J, Andre P, Lotteau V. Virus-host interactomics: new insights and opportunities for antiviral drug discovery. Genome Med. 2014;6(11):115.
Savidis G, McDougall WM, Meraner P, Perreira JM, Portmann JM, Trincucci G, John SP, Aker AM, Renzette N, Robbins DR, et al. Identification of zika virus and dengue virus dependency factors using functional genomics. Cell Rep. 2016;16(1):232–46.
Wang Y, Zhang P. Recent advances in the identification of the host factors involved in dengue virus replication. Virol Sin. 2017;32(1):23–31.
Yu JS, Wu YH, Tseng CK, Lin CK, Hsu YC, Chen YH, Lee JC. Schisandrin A inhibits dengue viral replication via upregulating antiviral interferon responses through STAT signaling pathway. Sci Rep. 2017;7:45171.
Zainal N, Chang CP, Cheng YL, Wu YW, Anderson R, Wan SW, Chen CL, Ho TS, AbuBakar S, Lin YS. Resveratrol treatment reveals a novel role for HMGB1 in regulation of the type 1 interferon response in dengue virus infection. Sci Rep. 2017;7:42998.
Datan E, Roy SG, Germain G, Zali N, McLean JE, Golshan G, Harbajan S, Lockshin RA, Zakeri Z. Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis. 2016;7:e2127.
Fontaine KA, Sanchez EL, Camarda R, Lagunoff M. Dengue virus induces and requires glycolysis for optimal replication. J Virol. 2015;89(4):2358–66.
Low JG, Ooi EE, Vasudevan SG. Current status of dengue therapeutics research and development. J Infect Dis. 2017;215(suppl_2):S96–S102.
Lai YC, Chuang YC, Chang CP, Lin YS, Perng GC, Wu HC, Hsieh SL, Yeh TM. Minocycline suppresses dengue virus replication by down-regulation of macrophage migration inhibitory factor-induced autophagy. Antiviral Res. 2018;155:28–38.
Katzelnick LC, Gresh L, Halloran ME, Mercado JC, Kuan G, Gordon A, Balmaseda A, Harris E. Antibody-dependent enhancement of severe dengue disease in humans. Science. 2017;358(6365):929–32.
Robinson LN, Tharakaraman K, Rowley KJ, Costa VV, Chan KR, Wong YH, Ong LC, Tan HC, Koch T, Cain D, et al. Structure-guided design of an anti-dengue antibody directed to a non-immunodominant epitope. Cell. 2015;162(3):493–504.
Rouvinski A, Guardado-Calvo P, Barba-Spaeth G, Duquerroy S, Vaney MC, Kikuti CM, Navarro Sanchez ME, Dejnirattisai W, Wongwiwat W, Haouz A, et al. Recognition determinants of broadly neutralizing human antibodies against dengue viruses. Nature. 2015;520(7545):109–13.
Dejnirattisai W, Wongwiwat W, Supasa S, Zhang X, Dai X, Rouvinski A, Jumnainsong A, Edwards C, Quyen NTH, Duangchinda T, et al. A new class of highly potent, broadly neutralizing antibodies isolated from viremic patients infected with dengue virus. Nat Immunol. 2015;16(2):170–7.
Xu M, Zuest R, Velumani S, Tukijan F, Toh YX, Appanna R, Tan EY, Cerny D, MacAry P, Wang CI, et al. A potent neutralizing antibody with therapeutic potential against all four serotypes of dengue virus. NPJ Vaccines. 2017;2:2.
Wan SW, Lu YT, Huang CH, Lin CF, Anderson R, Liu HS, Yeh TM, Yen YT, Wu-Hsieh BA, Lin YS. Protection against dengue virus infection in mice by administration of antibodies against modified nonstructural protein 1. PLoS One. 2014;9(3):e92495.
Lai YC, Chuang YC, Liu CC, Ho TS, Lin YS, Anderson R, Yeh TM. Antibodies against modified NS1 wing domain peptide protect against dengue virus infection. Sci Rep. 2017;7(1):6975.
Schlesinger JJ, Brandriss MW, Walsh EE. Protection of mice against dengue 2 virus encephalitis by immunization with the dengue 2 virus non-structural glycoprotein NS1. J Gen Virol. 1987;68(Pt 3):853–7.
Krishna VD, Rangappa M, Satchidanandam V. Virus-specific cytolytic antibodies to nonstructural protein 1 of Japanese encephalitis virus effect reduction of virus output from infected cells. J Virol. 2009;83(10):4766–77.
Lin CF, Wan SW, Chen MC, Lin SC, Cheng CC, Chiu SC, Hsiao YL, Lei HY, Liu HS, Yeh TM, et al. Liver injury caused by antibodies against dengue virus nonstructural protein 1 in a murine model. Lab Invest. 2008;88(10):1079–89.
Vigne C, Dupuy M, Richetin A, Guy B, Jackson N, Bonaparte M, Hu B, Saville M, Chansinghakul D, Noriega F, et al. Integrated immunogenicity analysis of a tetravalent dengue vaccine up to 4 y after vaccination. Hum Vaccin Immunother. 2017;13(9):2004–16.
Guy B, Jackson N. Dengue vaccine: hypotheses to understand CYD-TDV-induced protection. Nat Rev Microbiol. 2016;14(1):45–54.
Hadinegoro SR, Arredondo-Garcia JL, Capeding MR, Deseda C, Chotpitayasunondh T, Dietze R, Muhammad Ismail HI, Reynales H, Limkittikul K, Rivera-Medina DM, et al. Efficacy and Long-Term Safety of a Dengue Vaccine in Regions of Endemic Disease. N Engl J Med. 2015;373(13):1195–206.
Halstead SB. Dengvaxia sensitizes seronegatives to vaccine enhanced disease regardless of age. Vaccine. 2017;35(47):6355–8.
Halstead SB. Licensed dengue vaccine: Public health conundrum and scientific challenge. Am J Trop Med Hyg. 2016;95(4):741–5.
Avirutnan P, Hauhart RE, Marovich MA, Garred P, Atkinson JP, Diamond MS. Complement-mediated neutralization of dengue virus requires mannose-binding lectin. MBio. 2011;2(6):e00276–11.
Lin YS, Yeh TM, Lin CF, Wan SW, Chuang YC, Hsu TK, Liu HS, Liu CC, Anderson R, Lei HY. Molecular mimicry between virus and host and its implications for dengue disease pathogenesis. Exp Biol Med. 2011;236(5):515–23.
Lin CF, Wan SW, Cheng HJ, Lei HY, Lin YS. Autoimmune pathogenesis in dengue virus infection. Viral Immunology. 2006;19(2):127–32.
Sun DS, King CC, Huang HS, Shih YL, Lee CC, Tsai WJ, Yu CC, Chang HH. Antiplatelet autoantibodies elicited by dengue virus non-structural protein 1 cause thrombocytopenia and mortality in mice. J Thromb Haemost. 2007;5(11):2291–9.
Chen MC, Lin CF, Lei HY, Lin SC, Liu HS, Yeh TM, Anderson R, Lin YS. Deletion of the C-terminal region of dengue virus nonstructural protein 1 (NS1) abolishes anti-NS1-mediated platelet dysfunction and bleeding tendency. J Immunol. 2009;183(3):1797–803.
Liu IJ, Chiu CY, Chen YC, Wu HC. Molecular mimicry of human endothelial cell antigen by autoantibodies to nonstructural protein 1 of dengue virus. J Biol Chem. 2011;286(11):9726–36.
Chuang YC, Lin YS, Liu HS, Yeh TM. Molecular mimicry between dengue virus and coagulation factors induces antibodies to inhibit thrombin activity and enhance fibrinolysis. J Virol. 2014;88(23):13759–68.
Henchal EA, Henchal LS, Schlesinger JJ. Synergistic interactions of anti-NS1 monoclonal antibodies protect passively immunized mice from lethal challenge with dengue 2 virus. J Gen Virol. 1988;69(Pt 8):2101–7.
Wan SW, Chen PW, Chen CY, Lai YC, Chu YT, Hung CY, Lee H, Wu HF, Chuang YC, Lin J, et al. Therapeutic effects of monoclonal antibody against dengue virus NS1 in a STAT1 knockout mouse model of dengue infection. J Immunol. 2017;199(8):2834–44.
Chu YT, Wan SW, Chang YC, Lee CK, Wu-Hsieh BA, Anderson R, Lin YS. Antibodies against nonstructural protein 1 protect mice from dengue virus-induced mast cell activation. Lab Invest. 2017;97(5):602.
Falgout B, Bray M, Schlesinger JJ, Lai CJ. Immunization of mice with recombinant vaccinia virus expressing authentic dengue virus nonstructural protein NS1 protects against lethal dengue virus encephalitis. J Virol. 1990;64(9):4356–63.
Srivastava AK, Putnak JR, Warren RL, Hoke CH. Mice immunized with a dengue type-2 virus-E and NS1 fusion protein made in Escherichia-Coli are protected against lethal dengue virus-infection. Vaccine. 1995;13(13):1251–8.
Amorim JH, Diniz MO, Cariri FAMO, Rodrigues JF, Bizerra RSP, Goncalves AJS, Alves AMD, Ferreira LCD. Protective immunity to DENV2 after immunization with a recombinant NS1 protein using a genetically detoxified heat-labile toxin as an adjuvant. Vaccine. 2012;30(5):837–45.
Wu SF, Liao CL, Lin YL, Yeh CT, Chen LK, Huang YF, Chou HY, Huang JL, Shaio MF, Sytwu HK. Evaluation of protective efficacy and immune mechanisms of using a non-structural protein NS1 in DNA vaccine against dengue 2 virus in mice. Vaccine. 2003;21(25-26):3919–29.
Costa SM, Freire MS, Alves AMB. DNA vaccine against the non-structural 1 protein (NS1) of dengue 2 virus. Vaccine. 2006;24(21):4562–4.
Goncalves AJ, Oliveira ER, Costa SM, Paes MV, Silva JF, Azevedo AS, Mantuano-Barradas M, Nogueira AC, Almeida CJ, Alves AM. Cooperation between CD4+ T Cells and humoral immunity is critical for protection against dengue using a DNA vaccine based on the NS1 antigen. PLoS Negl Trop Dis. 2015;9(12):e0004277.
Costa SM, Azevedo AS, Paes MV, Sarges FS, Freire MS, Alves AMB. DNA vaccines against dengue virus based on the ns1 gene: The influence of different signal sequences on the protein expression and its correlation to the immune response elicited in mice. Virology. 2007;358(2):413–23.
Acknowledgements
We thank the members of the Center of Infectious Disease and Signaling Research of NCKU for their invaluable inputs and insights throughout the course of this study.
Funding
This study was supported by grants from the Ministry of Science and Technology of Taiwan (102-2320-B-006-025-MY3) (105-2321-B-006-023) and (106-2321-B-006 -011).
Author information
Authors and Affiliations
Contributions
T-M.Y. provided the main ideas and organized and directed this article. H-R.C. contributed to “The role of NS1 in the pathogenesis of dengue” section and Y-C.L. wrote the “NS1 as a target protein for treatment and vaccination” section. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, HR., Lai, YC. & Yeh, TM. Dengue virus non-structural protein 1: a pathogenic factor, therapeutic target, and vaccine candidate. J Biomed Sci 25, 58 (2018). https://doi.org/10.1186/s12929-018-0462-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-018-0462-0