
Abstract
Nasopharyngeal carcinoma (NPC) is a head and neck cancer with poor clinical outcomes and insufficient treatments in Southeast Asian populations. Although concurrent chemoradiotherapy has improved recovery rates of patients, poor overall survival and low efficacy are still critical problems. To improve the therapeutic efficacy, we focused on a tumor-associated protein called Annexin A2 (ANXA2). This review summarizes the mechanisms by which ANXA2 promotes cancer progression (e.g., proliferation, migration, the epithelial-mesenchymal transition, invasion, and cancer stem cell formation) and therapeutic resistance (e.g., radiotherapy, chemotherapy, and immunotherapy). These mechanisms gave us a deeper understanding of the molecular aspects of cancer progression, and further provided us with a great opportunity to overcome therapeutic resistance of NPC and other cancers with high ANXA2 expression by developing this prospective ANXA2-targeted therapy.
Similar content being viewed by others

Background
Nasopharyngeal carcinoma (NPC) is a low-frequency disease in western countries; however, it is a high-risk head and neck cancer in Southeast Asia and China [1, 2]. In the clinic, concurrent chemoradiotherapy improved overall survival (OS, 94.5% at 5 years) in stage I/II patients, but the same treatment did not work effectively in stage III/IV patients (OS, 72.3% at 5 years). With monotherapy, stage III/IV patients who received radiotherapy alone had a relatively poor survival rate (an OS of 54.2% at 5 years) [3,4,5,6]. To improve the therapeutic efficacy, we tried to understand the mechanism of therapeutic resistance. A meta-analysis of 2321 cancer patients with several cancer types including head and neck cancers (esophageal, sinonasal, and oral), showed that a high expression level of Annexin A2 (ANXA2) was related to poor overall survival and disease-free survival [7]. Consistent results of high expression levels of ANXA2 being associated with advanced patients were also found for NPC [8].
ANXA2, as a tumor-associated protein, promotes cancer progression including proliferation, invasion, and metastasis in various cancer types (NPC, ovarian cancer, gliomas, hepatomas, pancreatic cancer, and breast cancer) [8,9,10,11,12,13]. In addition to cancer progression, ANX family proteins (ANXA1 and ANXA2) suppress the efficacy of both chemotherapy and radiotherapy [8, 14, 15]. In 2015, we further uncovered the interaction of ANXA2 with dendritic cell (DC)-specific intracellular adhesion molecule (ICAM)-3 grabbing non-integrin (DC-SIGN, CD209), which resulted in immunosuppression. This suppression might influence outcomes of anticancer therapies [16].
In recent years, increased attention has focused on ANXA2 and its role in regulating cancer development [17,18,19]. In this review, cellular and molecular mechanisms of ANXA2-mediated cancer progression and therapeutic resistance are addressed in the first two sections. Then, we further discuss the prospective effect of ANXA2-targeted therapy in the final section.
Cancer progression
Aberrant expression in cancers
Over the period of 2014~ 2017, researchers pointed out that high expression of ANXA2 in biopsies of epithelial ovarian cancer (56.42%) [20], urothelial carcinoma (53.02%) [21], hepatocellular carcinoma (HCC; 73.81%) [22], NPC (33%) [8], and serous ovarian cancer (57.79%) [23] was associated with poor OS (Table 1). Weihua Qiu and his colleagues performed a meta-analysis of 2321 cancer patients to confirm that high expression of ANXA2 was correlated with both OS (hazard ratio [HR] 1.56; p < 0.001) and disease-free survival (HR 1.47; p < 0.001) [7]. High ANXA2 expression was also related to a high risk of metastases (n = 48, NPC) and recurrence (n = 93, endometrioid endometrial carcinoma) [8, 24]. On the other hand, ANXA2 serves as a diagnostic factor for screening cancers. In peripheral blood, the ANXA2 serum level has been evaluated in patients with HCC (median, 69.6 ng/ml) [25], early-stage HCC (median, 150 ng/ml) [26], gastric cancer (median, 211.0 ng/ml) [27], lung cancer [28], and oral squamous cell carcinoma (median, 27.1 ng/ml) [29] (Table 2). However, serum levels of ANXA2 in NPC patients have not yet been reported.
Additionally, circulating tumor cell (CTC) detection in peripheral blood has become a non-invasive way to predict the risk of developing metastasis in cancer patients. In 2015, Pomila Singh and her colleagues further used cancer stem cell (CSC) markers (e.g., doublecortin-like kinase 1 (DCLK1) and leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5)) with epithelial cell markers (CD44 and ANXA2) to detect circulating CSCs in patients with malignant colon adenocarcinomas. CTC detection is an assay for on-going metastasis or relapse, while circulating CSC detection is a novel diagnosis during the initiation of metastasis. Detection of circulating CSCs provides a chance to diagnose metastasis and prevent it at earlier time points [30]. Due to broad approaches of biopsies, and serum and circulating CSC detection, ANXA2 might be a short-term clinical indicator for cancer patients.
Proliferation
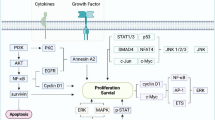
There are two types of ANXA2 in cells. The ANXA2 monomer exists in cell cytoplasm and nuclei, while the ANXA2 heterotetramer (consisting of two ANXA2 and two S100A10 molecules) exists on cell membranes. In nuclei, the ANXA2 monomer combines with 3-phosphoglycerate kinase (PGK) to become a complex. This complex performs the same function as primer-recognition proteins to stimulate DNA polymerase alpha, which contributes to the initiation of DNA replication in the early S phase in cervical cancer cells (Fig. 1a) [31]. In addition to promoting DNA replication, nuclear ANXA2 disrupts coilin causing it to abnormally localize to centromeres, thereby precipitating chromosome instability (CIN) which was demonstrated in human colorectal cancer (CRC) cells (Caco-2, HCT116, SW480, DLD-1, and RKO). Chromosome instability was reported to accelerate tumor growth and contribute to cellular resistance to chemotherapy [32,33,34].

Annexin A2 (ANXA2) in cancer progression. a The ANXA2-3-phosphoglycerate kinase (ANXA2-PGK) complex serves as a primer recognition protein to initiate DNA replication with the support from DNA polymerase alpha, which contributes to cell proliferation. b MIEN1 phosphorylates ANXA2 and supports ANXA2’s binding to actin filaments to modulate cytoskeletal change, thus resulting in cell migration. c ANXA2 initiates the endothelial-mesenchymal transition (EMT) via the Twist/Snail pathway. After initiation of the EMT, cells changed to a mesenchymal-like morphology, and cell junctions dissolved. d The ANXA2 heterotetramer complex links to the plasminogen and tissue plasminogen activator (tPA). After plasminogen is cleaved into plasmin, plasmin activates pro-matrix metalloproteases (MMPs) to become MMPs. MMPs digest the extracellular matrix and fibronectin, thus resulting acceleration of invasion. e ANXA2 increases stemness-related transcription factors (Oct4, Sox2, and Nanog) through the Akt signaling pathway, which activates cancer stem cell formation
Migration
Phosphorylation of the tyrosine 23 (Tyr23) residue on ANXA2 is a key regulator controlling cell mobility. The migration and invasion enhancer 1 (MIEN1) interacts with ANXA2 to enhance Tyr23 phosphorylation on ANXA2. Phosphorylated ANXA2 binds to actin filaments on cell membranes, and modulates cell scattering and cytoskeletal changes via actin remodeling in human breast cancer cells (SK-BR-3 and BT-474). A phosphorylation deficiency of Tyr23 and Tyr50 causes cells to lose the ability to migrate in in vitro wound healing assays [35,36,37,38]. However, a different report showed that the extracellular matrix metalloproteinase (MMP) inducer (also known as CD147) prohibits Tyr23 phosphorylation on ANXA2, and promotes cell migration via suppressing ANXA2-DOCK3-β-catenin-WAVE2 step-by-step signaling in human hepatoma cells (SMMC-7721, HuH-7, and HepG2) [39]. It seems that either the promotion of Tyr23 phosphorylation by MIEN1 or the inhibition of Tyr23 phosphorylation by CD147 eventually contributes to cell migration (Fig. 1b). The precise role of Tyr23 phosphorylation in ANXA2’s actions requires further investigation.
Endothelial-mesenchymal transition (EMT)
The EMT is a normal morphogenic process during embryonic development and tissue restructuring; however, the EMT is also the initial step in metastasis [7]. Twist and Snail are two critical transcription factors that promote the EMT in cancers. In EMT initiation, Twist and Snail decrease epithelial proteins (i.e., E-cadherin) and increase mesenchymal proteins (i.e., N-cadherin, fibronectin, β-catenin, and vimentin). Cancer cells without adherent junctions (i.e., E-cadherin) can gain an advantage to metastasize from one organ to a different indirectly connected one [40]. Phosphorylation of the Tyr23 residue on ANXA2 was reported to be an initiator of the EMT (mediated by Rho or induced by transforming growth factor (TGF)-β) [12, 37, 41]. In ANXA2-knockdown NPC cells, expression levels of Twist and Snail decreased. ANXA2-knockdown NPC cells retained the endothelial-like phenotype rather than changing to a mesenchymal-like one. This result indicated that ANXA2 is a critical factor in initiation of the EMT via the Twist/Snail signal pathway (Fig. 1c) [8].
Invasion and metastasis
ANXA2 promotes the invasion and metastasis of different cancers (ovarian cancer, HCC, gliomas, pancreatic cancer, renal cell carcinoma, lung cancer, breast cancer, and NPC) [8,9,10,11,12, 42,43,44]. The mechanism of the association of the ANXA2/S100A10 heterotetramer with extracellular matrix (ECM) digestion was described in previous studies [45,46,47]. Here, we update recent advances and more precisely summarize the mechanism. First, cytoplasmic phospholipase A2 (cPLA2) binds to the ANXA2/S100A10 heterotetramer in the cytoplasm to assist ANXA2 in translocating to cell membranes. On cell membranes, S100A10 links to the plasminogen and tissue plasminogen activator (tPA). S100A10 is required for the plasminogen-dependent oxidation of the ANXA2/S100A10 heterotetramer. Oxidation facilitates the catalytic cleavage of plasminogen to plasmin between arginine (Arg)561 and valine (Val)562 by the active tPA [48]. Plasmin cleaves the tissue inhibitor of metalloproteinases on pro-matrix metalloproteases (MMPs), and then the pro-MMPs become the active forms of MMPs. MMPs (e.g., MMP-2 and MMP-9) degrade fibronectin and the ECM, thus resulting in acceleration of invasion and metastasis (Fig. 1d) [49,50,51,52].
CSC formation
CSCs are cancer cells with stem-like properties to initiate self-renewal and differentiation. CSCs promote invasion, metastasis, suppress chemotoxicity and radiotoxicity [53,54,55]. In the previous section on aberrant expression in cancers, we described how ANXA2 can be a novel marker to detect circulating CSCs in the clinic; however, the role of ANXA2 in CSCs is not yet clearly understood. As we know, the Akt protein upregulates stemness-related transcription factors (Oct4, Sox2, and Nanog) [56,57,58] and is involved in CSC maintenance in different cancer types (gliomas, esophageal carcinoma, and non-small cell lung cancer) [59,60,61]. To further determine the mechanism between Akt and ANXA2, we established ANXA2-knockdown NPC cells. After ANXA2 silencing, the amount of active Akt decreased, thus indirectly suppressing protein levels of stemness-related transcription factors (Oct4, Sox2, and Nanog). Without sufficient stemness-related transcription factors, ANXA2-knockdown NPC cells are likely to lose their CSC properties (Fig. 1e) [8].
Therapeutic resistance
Correlations between the ANX family and therapeutic resistance
Radiotherapy and concurrent chemoradiotherapy are general strategies for NPC; however, we found resistance to both radiotoxicity and chemotoxicity in NPC cells with high ANXA2 expression. In addition to ANXA2, high expressions of other Annexin proteins were also reported to be associated with therapeutic resistance in different cancers, such as ANXA1 against chemo−/radiotherapy in the CNE2 NPC cell line [14, 15], ANXA2 against multiple drugs in NPC, gastric cancer, breast cancer, and pancreatic cancer [8, 62,63,64], ANXA3 against chemotoxicity in HCC patients (n = 34) [65], and ANXA4 against chemotherapeutic drugs in human ovarian (OVTOKO and OVISE), lung (H460) and colorectal (HCT15) cancer cells [66, 67]. Conversely, low ANXA11 expression is related to ovarian cancer with cisplatin resistance [68]. A correlation between therapeutic resistance and the Annexin protein family was found; nevertheless, the mechanism is still unclear. To improve ANAX2’s therapeutic efficacy against NPC, it is necessary to understand the mechanism of how it enables tumor cells to suppress radiotoxicity or chemotoxicity.
Resistance to chemotherapy
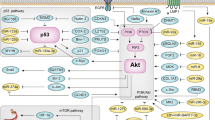
Human NPC cells with high ANXA2 expression can potentially resist different chemotherapeutic drugs (cisplatin, 5-fluorouracil, docetaxel, and vincristine). ANXA2-coated enlargeosomes widely exist in various cell types to regulate Ca2+-dependent cell exocytosis. Knockdown of ANXA2 in tumor cells might decrease the number of and limit the function of enlargeosomes. Dysfunctional enlargeosomes allow chemotherapeutic drugs to condense in the cytoplasm, thus resulting in tumor death (Fig. 2a) [8, 69,70,71]. Furthermore, intracellular ANXA2 binds the p50 subunit of nuclear factor (NF)-κB to become the ANXA2-p50 complex when pancreatic cancer cells (MIA-PaCa-2) are exposed to genotoxic agents (such as gemcitabine). This complex can be translocated to nuclei to activate the NF-κB signaling pathway. Activated NF-κB has multiple roles in cancer progression through modulating cell apoptosis and drug resistance. On the other hand, Qing-Yong Ma and his colleagues discovered that the phosphatidylinositol-3-kinase (PI3K)/Akt/NF-κB signaling pathway is activated by the interaction of ANXA2 and tenascin-C on the surface of pancreatic cancer cells (AsPC-1, PANC-1, and MIA-PaCa-2), which suppresses gemcitabine-induced cytotoxicity [72,73,74].

Annexin A2 (ANXA2) in therapeutic resistance. a ANXA2-coated enlargeosomes widely exist in various cell types to regulate Ca2+-dependent cell exocytosis. Enlargeosomes exocytose chemotherapeutic drugs to prevent their chemotoxic accumulation inside tumor cells, thus resulting in chemotherapeutic resistance. b The phosphorylated ANXA2 protein is imported into nuclei to protect against DNA damage by irradiation. ANXA2 also mediates the Akt protein to form the Akt-heat shock protein 27 (Akt-HSP27) complex, which ameliorates radiotoxicity-induced DNA damage and apoptosis. c When tumor-infiltrating dendritic cells (DCs) are attached to nasopharyngeal carcinoma (NPC) cells, the interaction between DC-SIGN and ANXA2 causes DCs to lose mature major histocompatibility complex (MHC), and release high levels of the immunosuppressive cytokine interleukin (IL)-10. IL-10 causes consecutive immunosuppressive responses including DC immaturity, inhibition of IL-12 synthetic, CD8+ T cell dysfunction, and regulatory T cell expansion
Resistance to radiotherapy
In 2012, David M. Waisman and his colleagues indicated that the ANXA2 protein was imported into nuclei to protect against DNA damage caused by irradiation in human breast and lung cancer cells. ANXA2 is mainly localized in the cytoplasm and plasma membranes, and only a small amount of it is imported into nuclei. ANXA2 contains a leucine-rich nuclear export signal (NES) in its N-terminal domain. In the NES motif, lysine (Lys)10 and Lys12 are two critical residues which prevent ANXA2 from accumulating in nuclei. However, inducers such as gamma-radiation, ultraviolet radiation, etoposide, chromium VI, hydrogen peroxide (H2O2), and reactive oxygen species (ROS) may induce ANXA2 phosphorylation, which allows it to be translocated into nuclei [75, 76]. Phosphorylation regulates ANXA2’s export from and import into nuclei. Phosphorylation of the serine (Ser)11 and Ser25 residues allows ANXA2 to be exported from nuclei; however, phosphorylation at the Tyr23 residue contrastingly allows it to be imported into nuclei (Fig. 2b) [77, 78]. Different phosphorylation sites on ANXA2 can cause totally opposite results. In 2015, we investigated the downstream signaling pathway of ANXA2 in NPC cells. ANXA2 is involved in the Akt pathway and indirectly increases the number of Akt proteins [8]. When exposed to irradiation, the Akt protein binds to heat shock protein 27 (HSP27) to become the Akt-HSP27 complex, which ameliorates radiotoxicity-induced DNA damage and apoptosis (Fig. 2b) [79].
Resistance to immunotherapy
A range of novel immunotherapies for cancers are under evaluation. Strategies for NPC were reported and are divided into two streams. First, Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes (CTLs) have become an effective adoptive cell therapy (ACT). The EBV latent membrane protein 1 (LMP1) is expressed on EBV-infected carcinoma cells. In 2014, Dennis J Moss and his colleagues transferred autologous CTLs that specifically targeted LMP1-expressing carcinoma cells into patients with recurrent NPC. After an injection, most of the pulmonary lesions disappeared, but the primary tumor did not regress [80]. To improve the disadvantage of targeting LMP1 alone, a new adenoviral vector was designed. This new adenoviral vector inserted EBV LMPs and EBV nuclear antigen-1 (EBNA1), which expanded specific CTLs against LMP- and/or EBNA1-expressing NPCs [80,81,82,83]. Second, DC-based immunotherapy is another option. DCs present tumor antigens to naïve CD8+ T cells in draining lymph nodes, and then naïve CD8+ T cells turn into tumor-specific CTLs [84,85,86,87,88]. However, NPC cells could give rise to suppressive responses after cell-cell interactions with DCs, thus resulting in immune escape [16, 89,90,91]. DCs can sense tumor-derived factors through receptors in both extracellular and intracellular milieus. Receptors include intracellular helicases, surface/intracellular toll-like receptors (TLRs), and surface C-type lectin receptors (CLRs). CLRs capture pathogen-associated molecular patterns (PAMPs) and endogenous ligands. DC-SIGN, a kind of C-type lectin, is composed of a carbohydrate recognition domain (CRD), a neck region with seven repeats, and a transmembrane region with a cytoplasmic tail [92,93,94,95,96]. DC-SIGN recognizes N-acetylglucosamine, mannose, fucose, and non-sialylated Lewis structures by CRD [95]. Mannosylated lipoarabinomannan (ManLAM) induces DC-SIGN downstream transcription factors (such as Ras, Raf-1, and NF-κB) to increase interleukin (IL)-10 promoter activity [97, 98]. After DC-SIGN is ligated by mannose- or fucose-containing oligosaccharides, it indirectly increases IL-10 production via the Th2 pathway in DCs. Normally, IL-10 suppresses prolonged and exaggerated immune responses [99, 100]. However, when DCs attach to NPC cells with high ANXA2 expression, the strong interaction between DC-SIGN and ANXA2 causes DCs to release extremely high levels of IL-10 (Fig. 2c). Once IL-10 spreads into the tumor environment, it causes consecutive immunosuppressive responses including DC immaturity (losing the major histocompatibility complex and the co-stimulatory molecules), the IL-12 synthetic inhibition, CD8+ T cell dysfunction, and regulatory T cell expansion [101,102,103,104,105,106,107].
Prospective ANXA2-targeted therapy
From 2013, different research teams began to develop ANXA2-targeted therapy. C Ricciardelli’s team used an anti-ANXA2 antibody to reduce both tumor growth and metastasis in an ovarian cancer mice model (SK-OV3) [9]. One year later, Mandip Singh and his colleagues inserted short hairpin (sh)RNA targeting ANXA2 (shANXA2) into a cationic ligand-guide (CLG, a liposomal carrier) to construct a CLG-ANXA2 compound. The CLG-ANXA2 was designed to recognize cancer cells and CSCs in a lung cancer mouse model (H1650). After CLG-ANXA2 was taken up by tumor cells, shANXA2 prohibited ANXA2 messenger (m)RNA expression and decreased its protein level. The CLG-shANXA2 group showed inhibited tumor growth (reduced 72%~ 75% relative to the control, p < 0.001) [108]. To verify the above results, we established ANXA2-knockdown NPC cell lines by shRNA. Proliferation, migration, adhesion, and CSC formation were indeed reduced in ANXA2-knockdown NPC cells. Moreover, ANXA2-knockdown NPC cells lost the ability to suppress chemotoxicity, radiotoxicity, and immune responses [8, 16]. Targeting ANXA2 raises the possibility of being able to overcome the low therapeutic efficacy of cancers with high ANXA2 expression.
In addition to ANXA2, high expression of the epidermal growth factor receptor (EGFR) was previously discovered in NPC cells [109, 110]. In 2005, combined therapy with cetuximab (as the first chimeric anti-EGFR antibody) and carboplatin was used against NPC in 60 patients. After treatment, only 11.7% of patients had a response, and 48.3% had a stable disease rate [111]. It seems that treatment with cetuximab and carboplatin needs more consideration due to its low efficacy. In 2017, Liming Huang and his colleagues reported appealing results that silencing ANXA2 would reverse the EGF-induced EMT and inhibit cell migration in epidermoid cervical carcinoma cells (Ca-Ski, HeLa, and SiHa) [112]. ANXA2 helps the tyrosine-protein kinase transforming protein, Src (v-Src), mediate actin-cytoskeletal rearrangement which enhances proliferation, migration, and viability through the EFGR pathway [113]. The above findings gave us a new selection to combine anti-ANXA2 and anti-EGFR antibodies to fight against double-positive NPC (EGFR+/ANXA2+).
Nevertheless, ANXA2-deficient (ANXA2−/−) mice showed an increased risk of thrombosis and a decreased ability of neoangiogenesis [47]. Although ANXA2-targeted therapy suppresses cancers, it may produce side effects in patients. Thus, it is necessary to consider the expression titer of ANXA2, the dose level of ANXA2-targeted antibodies (or carrier with shANXA2), and the patient’s health condition before using ANXA2-targeted therapy.
Conclusions
This review reveals the cancerous and suppressive mechanisms of ANXA2. First, we stepwise described the mechanisms of how ANXA2 promotes proliferation, migration, the EMT, metastasis, invasion, and CSC formation. On cell membranes, the ANXA2-S100A10 heterotetramer promotes activation of MMPs to increase the invasive ability. In cytoplasm, ANXA2, after being phosphorylated at Tyr23, binds to actin filaments to enhance migration. Inside cell nuclei, ANXA2 promotes both EMT- and CSC-related transcriptional factors (Snail, Twist, Oct4, Sox2, and Nanog). Second, we illustrated different therapeutic resistances by ANXA2. After phosphorylation, ANXA2 is translocated into nuclei and prevents DNA damage due to radiotoxicity. ANXA2-coated enlargeosomes exocytose chemotherapeutic drugs to decrease the level of chemotoxicity in cancer cells. Moreover, the molecular interaction of ANXA2 and DC-SIGN triggers immunosuppression, which results in tumor immune escape. The above findings gave us a deeper understanding of the molecular aspects of cancer progression, and provided a great opportunity to improve the therapeutic efficacy against NPC and other cancers with high ANXA2 expression. Although the ANXA2-targeted therapy has not been examined in clinical trials yet, it is expected to produce promising treatment outcomes.
Abbreviations
- ANXA2:
-
Annexin A2
- Arg:
-
Arginine
- CLG:
-
Cationic ligand-guided
- CLR:
-
C-type lectin receptor
- cPLA2:
-
Cytoplasmic phospholipase A2
- CRC:
-
Colorectal cancer
- CRD:
-
Carbohydrate recognition domain
- CSC:
-
Cancer stem cell
- CTC:
-
Circulating tumor cell
- DC:
-
Dendritic cell
- DC-SIGN:
-
Dendritic cell-specific EBV
Epstein-Barr virus
- DFS:
-
Disease-free survival
- DLCK1:
-
Doublecortin-like kinase 1
- EBNA1:
-
EBV nuclear antigen-1
- ECM:
-
Extracellular matrix
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Endothelial-mesenchymal transition
- H2O2 :
-
Hydrogen peroxide
- HCC:
-
Hepatocellular carcinoma
- HR:
-
Hazard ratio
- HSP27:
-
Heat shock protein 27
- ICAM:
-
Intracellular adhesion molecule
- ICAM-3:
-
Grabbing non-integrin
- IL-10:
-
Interleukin-10
- LGR5:
-
Leucine-rich repeat-containing G-protein coupled receptor 5
- LMP:
-
Latent membrane protein
- Lys:
-
Lysine
- MIEN1:
-
Migration and invasion enhancer 1
- MMP:
-
Matrix metalloproteases
- NES:
-
Nuclear export signal
- NPC:
-
Nasopharyngeal carcinoma
- PAMP:
-
Pathogen-associated molecular pattern
- PGK:
-
Phosphoglycerate kinase
- PI3K:
-
Phosphatidylinositol-3-kinase
- ROS:
-
Reactive oxygen species
- Ser:
-
Serine
- shANXA2:
-
Short hairpin RNA targeting Annexin A2
- TGF:
-
Transforming growth factor
- tPA:
-
Tissue plasminogen activator
- Tyr:
-
Tyrosine
- Val:
-
Valine
References
Sun LM, Epplein M, Li CI, Vaughan TL, Weiss NS. Trends in the incidence rates of nasopharyngeal carcinoma among Chinese Americans living in Los Angeles County and the San Francisco metropolitan area, 1992-2002. Am J Epidemiol. 2005;162(12):1174–8.
Cho WC. Nasopharyngeal carcinoma: molecular biomarker discovery and progress. Mol Cancer. 2007;6:1.
Ma BB, Chan AT. Recent perspectives in the role of chemotherapy in the management of advanced nasopharyngeal carcinoma. Cancer. 2005;103(1):22–31.
Zhang L, Chen QY, Liu H, Tang LQ, Mai HQ. Emerging treatment options for nasopharyngeal carcinoma. Drug Des Devel Ther. 2013;7:37–52.
Caponigro F, Longo F, Ionna F, Perri F. Treatment approaches to nasopharyngeal carcinoma: a review. Anti-Cancer Drugs. 2010;21(5):471–7.
Lee AW, Fee WE Jr, Ng WT, Chan LK. Nasopharyngeal carcinoma: salvage of local recurrence. Oral Oncol. 2012;48(9):768–74.
Liu X, Ma D, Jing X, Wang B, Yang W, Qiu W. Overexpression of ANXA2 predicts adverse outcomes of patients with malignant tumors: a systematic review and meta-analysis. Med Oncol. 2015;32(1):392.
Chen CY, Lin YS, Chen CL, Chao PZ, Chiou JF, Kuo CC, Lee FP, Lin YF, Sung YH, Lin YT, et al. Targeting annexin A2 reduces tumorigenesis and therapeutic resistance of nasopharyngeal carcinoma. Oncotarget. 2015;6(29):26946–59.
Lokman NA, Elder ASF, Ween MP, Pyragius CE, Hoffmann P, Oehler MK, Ricciardelli C. Annexin A2 is regulated by ovarian cancer-peritoneal cell interactions and promotes metastasis. Oncotarget. 2013;4(8):1199–211.
Zhai H, Acharya S, Gravanis I, Mehmood S, Seidman RJ, Shroyer KR, Hajjar KA, Tsirka SE. Annexin A2 promotes glioma cell invasion and tumor progression. J. Neurosci. Off. J. Soc. Neurosci. 2011;31(40):14346–60.
Zhang HJ, Yao DF, Yao M, Huang H, Wang L, Yan MJ, Yan XD, Gu X, Wu W, Lu SL. Annexin A2 silencing inhibits invasion, migration, and tumorigenic potential of hepatoma cells. World J. Gastroenterol. 2013;19(24):3792–801.
Zheng L, Foley K, Huang LQ, Leubner A, Mo GL, Olino K, Edil BH, Mizuma M, Sharma R, Le DT, et al. Tyrosine 23 phosphorylation-dependent cell-surface localization of Annexin A2 is required for invasion and metastases of pancreatic Cancer. PLoS One. 2011;6(4)
Sharma MR, Koltowski L, Ownbey RT, Tuszynski GP, Sharma MC. Angiogenesis-associated protein annexin II in breast cancer: selective expression in invasive breast cancer and contribution to tumor invasion and progression. Exp Mol Pathol. 2006;81(2):146–56.
Zeng GQ, Cheng AL, Tang J, Li GQ, Li MX, Qu JQ, Cao C, Liao L, Xiao ZQ. Annexin A1: a new biomarker for predicting nasopharyngeal carcinoma response to radiotherapy. Med Hypotheses. 2013;81(1):68–70.
Chow BHY, Chua DTT, Sham JST, Zhang MY, Chow LWC, Bi J, Ma NF, Xie D, Loo WTY, Fung JMW, et al. Increased expression of annexin I is associated with drug-resistance in nasopharyngeal carcinoma and other solid tumors. Proteom Clin Appl. 2009;3(6):654–62.
Chao PZ, Hsieh MS, Cheng CW, Hsu TJ, Lin YT, Lai CH, Liao CC, Chen WY, Leung TK, Lee FP, et al. Dendritic cells respond to nasopharygeal carcinoma cells through annexin A2-recognizing DC-SIGN. Oncotarget. 2015;6(1):159–70.
Wang CY, Lin CF. Annexin A2: its molecular regulation and cellular expression in cancer development. Dis Markers. 2014;2014:308976.
Xu XH, Pan W, Kang LH, Feng H, Song YQ. Association of annexin A2 with cancer development (review). Oncol Rep. 2015;33(5):2121–8.
Grindheim AK, Saraste J, Vedeler A. Protein phosphorylation and its role in the regulation of Annexin A2 function. Biochim Biophys Acta. 2017;1861(11 Pt A):2515–29.
Deng Y, Chen C, Hua M, Xi Q, Liu R, Yang S, Liu J, Zhong J, Tang M, Lu S, et al. Annexin A2 plays a critical role in epithelial ovarian cancer. Arch Gynecol Obstet. 2015;292(1):175–82.
Zhang Q, Zhao ZS, Ma YY, Wang HJ, Ma J, He XJ, Zhang DH. Combined expression of S100A4 and Annexin A2 predicts disease progression and overall survival in patients with urothelial carcinoma. Urol Oncol-Semin Ori. 2014;32(6):798–805.
Zhang H, Yao M, Wu W, Qiu L, Sai W, Yang J, Zheng W, Huang J, Yao D. Up-regulation of annexin A2 expression predicates advanced clinicopathological features and poor prognosis in hepatocellular carcinoma. Tumour Biol.: J Int. Soc. Oncodev. Biol. Med. 2015;36(12):9373–83.
Lokman NA, Pyragius CE, Ruszkiewicz A, Oehler MK, Ricciardelli C. Annexin A2 and S100A10 are independent predictors of serous ovarian cancer outcome. Transl Res. 2016;171:83–95. e81-82
Alonso-Alconada L, Santacana M, Garcia-Sanz P, Muinelo-Romay L, Colas E, Mirantes C, Monge M, Cueva J, Oliva E, Soslow RA, et al. Annexin-A2 as predictor biomarker of recurrent disease in endometrial cancer. Int J Cancer. 2015;136(8):1863–73.
El-Abd N, Fawzy A, Elbaz T, Hamdy S. Evaluation of annexin A2 and as potential biomarkers for hepatocellular carcinoma. Tumor Biol. 2016;37(1):211–16.
Shaker MK, Fattah HIA, Sabbour GS, Montasser IF, Abdelhakam SM, El Hadidy E, Yousry R, El Dorry AK. Annexin A2 as a biomarker for hepatocellular carcinoma in Egyptian patients. World J Hepatol. 2017;9(9):469–76.
Tas F, Tilgen Yasasever C, Karabulut S, Tastekin D, Duranyildiz D. Circulating annexin A2 as a biomarker in gastric cancer patients: correlation with clinical variables. Biomed Pharmacother. 2015;69:237–41.
Yang J, Yang F, Nie JQ, Zou XH, Tian HQ, Qin YE, Liu CQ. Evaluation of Annexin A2 as a novel diagnostic serum biomarker for lung cancer. Cancer Biomark. 2015;15(2):205–11.
Zhang W, Gao C, Zhang S, Fang G. Serum Annexin A2 level is associated with diagnosis and prognosis in patients with oral squamous cell carcinoma. J Oral Maxillofac Surg. 2017;75(5):1081–7.
Kantara C, O'Connell MR, Luthra G, Gajjar A, Sarkar S, Ullrich RL, Singh P. Methods for detecting circulating cancer stem cells (CCSCs) as a novel approach for diagnosis of colon cancer relapse/metastasis. Lab Investig. 2015;95(1):100–12.
Vishwanatha JK, Jindal HK, Davis RG. The role of primer recognition proteins in DNA replication: association with nuclear matrix in HeLa cells. J Cell Sci. 1992;101(Pt 1):25–34.
Kazami T, Nie H, Satoh M, Kuga T, Matsushita K, Kawasaki N, Tomonaga T, Nomura F. Nuclear accumulation of annexin A2 contributes to chromosomal instability by coilin-mediated centromere damage. Oncogene. 2015;34(32):4177–89.
Bakhoum SF, Compton DA. Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest. 2012;122(4):1138–43.
Wang TL, Diaz LA Jr, Romans K, Bardelli A, Saha S, Galizia G, Choti M, Donehower R, Parmigiani G, Shih Ie M, et al. Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proc Natl Acad Sci U S A. 2004;101(9):3089–94.
Hubaishy I, Jones PG, Bjorge J, Bellagamba C, Fitzpatrick S, Fujita DJ, Waisman DM. Modulation of annexin II tetramer by tyrosine phosphorylation. Biochemistry-Us. 1995;34(44):14527–34.
Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6(6):449–61.
de Graauw M, Tijdens I, Smeets MB, Hensbergen PJ, Deelder AM, van de Water B. Annexin A2 phosphorylation mediates cell scattering and branching morphogenesis via cofilin activation. Mol Cell Biol. 2008;28(3):1029–40.
Kpetemey M, Dasgupta S, Rajendiran S, Das S, Gibbs LD, Shetty P, Gryczynski Z, Vishwanatha JK: MIEN1, a novel interactor of Annexin A2, promotes tumor cell migration by enhancing AnxA2 cell surface expression. Mol Cancer. 2015;14:156
Cui HY, Wang SJ, Miao JY, Fu ZG, Feng F, Wu J, Yang XM, Chen ZN, Jiang JL. CD147 regulates cancer migration via direct interaction with Annexin A2 and DOCK3-beta-catenin-WAVE2 signaling. Oncotarget. 2016;7(5):5613–29.
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90.
Rescher U, Ludwig C, Konietzko V, Kharitonenkov A, Gerke V. Tyrosine phosphorylation of annexin A2 regulates rho-mediated actin rearrangement and cell adhesion. J Cell Sci. 2008;121(Pt 13):2177–85.
Yang SF, Hsu HL, Chao TK, Hsiao CJ, Lin YF, Cheng CW. Annexin A2 in renal cell carcinoma: expression, function, and prognostic significance. Urol Oncol. 2015;33(1):22 e11–21.
Wang YQ, Zhang F, Tian R, Ji W, Zhou Y, Sun XM, Liu Y, Wang ZY, Niu RF. Tyrosine 23 phosphorylation of Annexin A2 promotes proliferation, invasion, and Stat3 phosphorylation in the nucleus of human breast Cancer SK-BR-3 cells. Cancer Biol Med. 2012;9(4):248–53.
Wang CY, Chen CL, Tseng YL, Fang YT, Lin YS, Su WC, Chen CC, Chang KC, Wang YC, Lin CF. Annexin A2 silencing induces G2 arrest of non-small cell lung cancer cells through p53-dependent and -independent mechanisms. J Biol Chem. 2012;287(39):32512–24.
Kassam G, Choi KS, Ghuman J, Kang HM, Fitzpatrick SL, Zackson T, Zackson S, Toba M, Shinomiya A, Waisman DM. The role of annexin II tetramer in the activation of plasminogen. J Biol Chem. 1998;273(8):4790–9.
Bharadwaj A, Bydoun M, Holloway R, Waisman D. Annexin A2 heterotetramer: structure and function. Int J Mol Sci. 2013;14(3):6259–305.
Grewal T, Wason SJ, Enrich C, Rentero C. Annexins - insights from knockout mice. Biol Chem. 2016;397(10):1031–53.
Kwon M, Yoon CS, Jeong W, Rhee SG, Waisman DM. Annexin A2-S100A10 heterotetramer, a novel substrate of thioredoxin. J Biol Chem. 2005;280(25):23584–92.
Bandorowicz-Pikula J, Wos M, Pikula S. Do annexins participate in lipid messenger mediated intracellular signaling? A question revisited. Mol Membr Biol. 2012;29(7):229–42.
Wang YX, Lv H, Li ZX, Li C, Wu XY. Effect of shRNA mediated down-regulation of Annexin A2 on biological behavior of human lung adencarcinoma cells A549. Pathol Oncol Res. 2012;18(2):183–90.
Zhang W, Zhao P, Xu XL, Cai L, Song ZS, Cao DY, Tao KS, Zhou WP, Chen ZN, Dou KF. Annexin A2 promotes the migration and invasion of human hepatocellular carcinoma cells in vitro by regulating the shedding of CD147-harboring microvesicles from tumor cells. PLoS One. 2013;8(8):e67268.
Song H, Pan D, Sun W, Gu C, Zhang Y, Zhao P, Qi Z, Zhao S. SiRNA directed against annexin II receptor inhibits angiogenesis via suppressing MMP2 and MMP9 expression. Cell Physiol Biochem. 2015;35(3):875–84.
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–68.
Colak S, Medema JP. Cancer stem cells--important players in tumor therapy resistance. FEBS J. 2014;281(21):4779–91.
Cojoc M, Mabert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol. 2015;31:16–27.
Mimeault M, Batra SK. Altered gene products involved in the malignant reprogramming of cancer stem/progenitor cells and multitargeted therapies. Mol Asp Med. 2014;39:3–32.
Qin J, Ji J, Deng R, Tang J, Yang F, Feng GK, Chen WD, Wu XQ, Qian XJ, Ding K, et al. DC120, a novel AKT inhibitor, preferentially suppresses nasopharyngeal carcinoma cancer stem-like cells by downregulating Sox2. Oncotarget. 2015;6(9):6944–58.
Zhao QW, Zhou YW, Li WX, Kang B, Zhang XQ, Yang Y, Cheng J, Yin SY, Tong Y, He JQ, et al. Aktmediated phosphorylation of Oct4 is associated with the proliferation of stemlike cancer cells. Oncol Rep. 2015;33(4):1621–9.
Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–35.
Li H, Gao Q, Guo L, Lu SH. The PTEN/PI3K/Akt pathway regulates stem-like cells in primary esophageal carcinoma cells. Cancer Biol Ther. 2011;11(11):950–8.
Singh S, Trevino J, Bora-Singhal N, Coppola D, Haura E, Altiok S, Chellappan SP. EGFR/Src/Akt signaling modulates Sox2 expression and self-renewal of stem-like side-population cells in non-small cell lung cancer. Mol Cancer. 2012;11:73.
Zhang ZD, Li Y, Fan LQ, Zhao Q, Tan BB, Zhao XF. Annexin A2 is implicated in multi-drug-resistance in gastric cancer through p38MAPK and AKT pathway. Neoplasma. 2014;61(6):627–37.
Zhang F, Zhang H, Wang Z, Yu M, Tian R, Ji W, Yang Y, Niu R. P-glycoprotein associates with Anxa2 and promotes invasion in multidrug resistant breast cancer cells. Biochem Pharmacol. 2014;87(2):292–302.
Takano S, Togawa A, Yoshitomi H, Shida T, Kimura F, Shimizu H, Yoshidome H, Ohtsuka M, Kato A, Tomonaga T, et al. Annexin II overexpression predicts rapid recurrence after surgery in pancreatic cancer patients undergoing gemcitabine-adjuvant chemotherapy. Ann Surg Oncol. 2008;15(11):3157–68.
Pan QZ, Pan K, Weng DS, Zhao JJ, Zhang XF, Wang DD, Lv L, Jiang SS, Zheng HX, Xia JC. Annexin A3 promotes tumorigenesis and resistance to chemotherapy in hepatocellular carcinoma. Mol Carcinog. 2015;54(8):598–607.
Han EK, Tahir SK, Cherian SP, Collins N, Ng SC. Modulation of paclitaxel resistance by annexin IV in human cancer cell lines. Br J Cancer. 2000;83(1):83–8.
Mogami T, Yokota N, Asai-Sato M, Yamada R, Koizume S, Sakuma Y, Yoshihara M, Nakamura Y, Takano Y, Hirahara F, et al. Annexin A4 is involved in proliferation, chemo-resistance and migration and invasion in ovarian clear cell adenocarcinoma cells. PLoS One. 2013;8(11):e80359.
Song J, Shih Ie M, Salani R, Chan DW, Zhang Z, Annexin XI. Is associated with cisplatin resistance and related to tumor recurrence in ovarian cancer patients. Clin. Cancer Res. 2007;13(22 Pt 1):6842–9.
Lorusso A, Covino C, Priori G, Bachi A, Meldolesi J, Chieregatti E. Annexin2 coating the surface of enlargeosomes is needed for their regulated exocytosis. EMBO J. 2006;25(23):5443–56.
Cocucci E, Racchetti G, Podini P, Meldolesi J. Enlargeosome traffic: exocytosis triggered by various signals is followed by endocytosis, membrane shedding or both. Traffic. 2007;8(6):742–57.
Prada I, Cocucci E, Racchetti G, Meldolesi J. The Ca2+−dependent exocytosis of enlargeosomes is greatly reinforced by genistein via a non-tyrosine kinase-dependent mechanism. FEBS Lett. 2007;581(25):4932–6.
Gong XG, Lv YF, Li XQ, Xu FG, Ma QY. Gemcitabine resistance induced by interaction between alternatively spliced segment of tenascin-C and Annexin A2 in pancreatic Cancer cells. Biol Pharm Bull. 2010;33(8):1261–7.
Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-kappaB addiction and its role in cancer: 'one size does not fit all. Oncogene. 2011;30(14):1615–30.
Jung H, Kim JS, Kim WK, Oh KJ, Kim JM, Lee HJ, Han BS, Kim DS, Seo YS, Lee SC, et al. Intracellular annexin A2 regulates NF-kappaB signaling by binding to the p50 subunit: implications for gemcitabine resistance in pancreatic cancer. Cell Death Dis. 2015;6:e1606.
Madureira PA, Hill R, Lee PWK, Waisman DM. Genotoxic agents promote the nuclear accumulation of Annexin A2: role of Annexin A2 in mitigating DNA damage. PLoS One. 2012;7(11)
Grindheim AK, Hollas H, Raddum AM, Saraste J, Vedeler A. Reactive oxygen species exert opposite effects on Tyr23 phosphorylation of the nuclear and cortical pools of annexin A2. J Cell Sci. 2016;129(2):314–28.
Eberhard DA, Karns LR, VandenBerg SR, Creutz CE. Control of the nuclear-cytoplasmic partitioning of annexin II by a nuclear export signal and by p11 binding. J Cell Sci. 2001;114(Pt 17):3155–66.
Liu J, Rothermund CA, Ayala-Sanmartin J, Vishwanatha JK. Nuclear annexin II negatively regulates growth of LNCaP cells and substitution of ser 11 and 25 to glu prevents nucleo-cytoplasmic shuttling of annexin II. BMC Biochem. 2003;4:10.
Kanagasabai R, Karthikeyan K, Vedam K, Qien W, Zhu Q, Ilangovan G. Hsp27 protects adenocarcinoma cells from UV-induced apoptosis by Akt and p21-dependent pathways of survival. Mol. Cancer Res. 2010;8(10):1399–412.
Lutzky VP, Crooks P, Morrison L, Stevens N, Davis JE, Corban M, Hall D, Panizza B, Coman WB, Coman S, et al. Cytotoxic T cell adoptive immunotherapy as a treatment for nasopharyngeal carcinoma. Clin Vaccine Immunol. 2014;21(2):256–9.
Li J, Chen QY, Mo H, Zhang YL, Huang ZF, Zeng YX. Immunophenotyping at the time of diagnosis distinguishes two groups of nasopharyngeal carcinoma patients: implications for adoptive immunotherapy. Int J Biol Sci. 2011;7(5):607–17.
Smith C, Khanna R. A new approach for cellular immunotherapy of nasopharyngeal carcinoma. Oncoimmunology. 2012;1(8):1440–2.
Smith C, Tsang J, Beagley L, Chua D, Lee V, Li V, Moss DJ, Coman W, Chan KH, Nicholls J, et al. Effective treatment of metastatic forms of Epstein-Barr virus-associated nasopharyngeal carcinoma with a novel adenovirus-based adoptive immunotherapy. Cancer Res. 2012;72(5):1116–25.
Giannini A, Bianchi S, Messerini L, Gallo O, Gallina E, Asprella Libonati G, Olmi P, Zampi G. Prognostic significance of accessory cells and lymphocytes in nasopharyngeal carcinoma. Pathol Res Pract. 1991;187(4):496–502.
Zong YS, Zhang CQ, Zhang F, Ruan JB, Chen MY, Feng KT, Yu ZF. Infiltrating lymphocytes and accessory cells in nasopharyngeal carcinoma. Jpn. J. Cancer Res.: Gann. 1993;84(8):900–5.
Hartmann E, Wollenberg B, Rothenfusser S, Wagner M, Wellisch D, Mack B, Giese T, Gires O, Endres S, Hartmann G. Identification and functional analysis of tumor-infiltrating plasmacytoid dendritic cells in head and neck cancer. Cancer Res. 2003;63(19):6478–87.
Yufeng D, Guocheng Z, Dongliang X, Rong F, Yuhong C, Ruying L, Jingshi Z, Xuhong Z. Whole-tumor-antigen-pulsed dendritic cells elicit cytotoxic T-cell response against pediatric nasopharyngeal carcinoma in vitro. Med Oncol. 2009;26(1):78–85.
Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–52.
Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, O'Garra A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J Immunol. 1991;146(10):3444–51.
Steinbrink K, Wolfl M, Jonuleit H, Knop J, Enk AH. Induction of tolerance by IL-10-treated dendritic cells. J Immunol. 1997;159(10):4772–80.
Hiltbold EM, Vlad AM, Ciborowski P, Watkins SC, Finn OJ. The mechanism of unresponsiveness to circulating tumor antigen MUC1 is a block in intracellular sorting and processing by dendritic cells. J Immunol. 2000;165(7):3730–41.
Geijtenbeek TBH, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GCF, Middel J, Cornelissen ILMHA, Nottet HSLM, KewalRamani VN, Littman DR, et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100(5):587–97.
Kwon DS, Gregorio G, Bitton N, Hendrickson WA, Littman DR. DC-SIGN-mediated internalization of HIV is required for trans-enhancement of T cell infection. Immunity. 2002;16(1):135–44.
Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6(6):476–83.
Svajger U, Anderluh M, Jeras M, Obermajer N. C-type lectin DC-SIGN: an adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell Signal. 2010;22(10):1397–405.
Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. 2011;12(10):959–65.
Grutz G. New insights into the molecular mechanism of interleukin-10-mediated immunosuppression. J Leukoc Biol. 2005;77(1):3–15.
Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Geijtenbeek TBH. Carbohydrate-specific signaling through the DC-SIGN signalosome tailors immunity to Mycobacterium tuberculosis, HIV-1 and helicobacter pylori. Nat Immunol. 2009;10(10):1081–U1058.
den Dunnen J, Gringhuis SI, Geijtenbeek TBH: Innate signaling by the C-type lectin DC-SIGN dictates immune responses. Cancer Immunol Immun 2009, 58(7):1149-1157.
Hsu SC, Chen CH, Tsai SH, Kawasaki H, Hung CH, Chu YT, Chang HW, Zhou YF, Fu JR, Plunkett B, et al. Functional interaction of common allergens and a C-type lectin receptor, dendritic cell-specific ICAM3-grabbing non-integrin (DC-SIGN), on human dendritic cells. J Biol Chem. 2010;285(11):7903–10.
Chang J, Kunkel SL, Chang CH. Negative regulation of MyD88-dependent signaling by IL-10 in dendritic cells. Proc Natl Acad Sci U S A. 2009;106(43):18327–32.
Heidt S, Segundo DS, Chadha R, Wood KJ. The impact of Th17 cells on transplant rejection and the induction of tolerance. Curr. Opin. Organ Transplant. 2010;15(4):456–61.
Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170(6):2081–95.
de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174(5):1209–20.
Ding L, Linsley PS, Huang LY, Germain RN, Shevach EM. IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the up-regulation of B7 expression. J Immunol. 1993;151(3):1224–34.
Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, Muller W, Trinchieri G, Sher A. In the absence of endogenous IL-10, mice acutely infected with toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol. 1996;157(2):798–805.
Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765.
Andey T, Marepally S, Patel A, Jackson T, Sarkar S, O'Connell M, Reddy RC, Chellappan S, Singh P, Singh M. Cationic lipid guided short-hairpin RNA interference of annexin A2 attenuates tumor growth and metastasis in a mouse lung cancer stem cell model. J Control Release. 2014;184:67–78.
Chua DT, Nicholls JM, Sham JS, Au GK. Prognostic value of epidermal growth factor receptor expression in patients with advanced stage nasopharyngeal carcinoma treated with induction chemotherapy and radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59(1):11–20.
Ma BB, Poon TC, To KF, Zee B, Mo FK, Chan CM, Ho S, Teo PM, Johnson PJ, Chan AT. Prognostic significance of tumor angiogenesis, Ki 67, p53 oncoprotein, epidermal growth factor receptor and HER2 receptor protein expression in undifferentiated nasopharyngeal carcinoma--a prospective study. Head & neck. 2003;25(10):864–72.
Chan AT, Hsu MM, Goh BC, Hui EP, Liu TW, Millward MJ, Hong RL, Whang-Peng J, Ma BB, To KF, et al. Multicenter, phase II study of cetuximab in combination with carboplatin in patients with recurrent or metastatic nasopharyngeal carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005;23(15):3568–76.
Cui L, Song J, Wu L, Cheng L, Chen A, Wang Y, Huang Y, Huang L. Role of Annexin A2 in the EGF-induced epithelial-mesenchymal transition in human CaSki cells. Oncol Lett. 2017;13(1):377–83.
Shetty PK, Thamake SI, Biswas S, Johansson SL, Vishwanatha JK. Reciprocal regulation of annexin A2 and EGFR with Her-2 in Her-2 negative and herceptin-resistant breast cancer. PLoS One. 2012;7(9):e44299.
Acknowledgements
None.
Funding
This work was supported by a research project of the Ministry of Science and Technology (MOST104-2314-B-038-060).
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
CYC, YJC, and CHC were involved in the design, wrote the manuscript, and supervised the research. YSL and CHC commented on the study and provided material and/or technical support. All authors read and gave final approval of the submitted and published versions.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
None.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, CY., Lin, YS., Chen, CH. et al. Annexin A2-mediated cancer progression and therapeutic resistance in nasopharyngeal carcinoma. J Biomed Sci 25, 30 (2018). https://doi.org/10.1186/s12929-018-0430-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-018-0430-8