
Abstract
Background
Noopept (N-phenyl-acetyl-L-prolylglycine ethyl ester) was constructed as a dipeptide analog of the standard cognition enhancer, piracetam. Our previous experiments have demonstrated the cognition restoring effect of noopept in several animal models of Alzheimer disease (AD). Noopept was also shown to prevent ionic disbalance, excitotoxicity, free radicals and pro-inflammatory cytokines accumulation, and neurotrophine deficit typical for different kinds of brain damages, including AD. In this study, we investigated the neuroprotective action of noopept on cellular model of AD, Aβ25–35-induced toxicity in PC12 cells and revealed the underlying mechanisms.
Results
The neuroprotective effect of noopept (added to the medium at 10 μM concentration, 72 hours before Аβ25–35) was studied on Аβ25–35-induced injury (5 μM for 24 h) in PC12 cells. The ability of drug to protect the impairments of cell viability, calcium homeostasis, ROS level, mitochondrial function, tau phosphorylation and neurite outgrowth caused by Аβ25–35 were evaluated.
Following the exposure of PC12 cells to Аβ25–35 an increase of the level of ROS, intracellular calcium, and tau phosphorylation at Ser396 were observed; these changes were accompanied by a decrease in cell viability and an increase of apoptosis. Noopept treatment before the amyloid-beta exposure improved PC12 cells viability, reduced the number of early and late apoptotic cells, the levels of intracellular reactive oxygen species and calcium and enhanced the mitochondrial membrane potential. In addition, pretreatment of PC12 cell with noopept significantly attenuated tau hyperphosphorylation at Ser396 and ameliorated the alterations of neurite outgrowth evoked by Аβ25–35.
Conclusions
Taken together, these data provide evidence that novel cognitive enhancer noopept protects PC12 cell against deleterious actions of Aβ through inhibiting the oxidative damage and calcium overload as well as suppressing the mitochondrial apoptotic pathway. Moreover, neuroprotective properties of noopept likely include its ability to decrease tau phosphorylation and to restore the altered morphology of PC12 cells. Therefore, this nootropic dipeptide is able to positively affect not only common pathogenic pathways but also disease-specific mechanisms underlying Aβ-related pathology.
Similar content being viewed by others


Background
Alzheimer’s disease (AD) is the most common form of neurodegenerative disease, accompanied by age-related dementia, affecting 27 million individuals worldwide [1]. Mechanisms underlying the progression of late-onset AD consist of a number of interacting events including excessive accumulation of amyloid, aberrant tau-protein phosphorylation, oxidative stress, chronic inflammatory conditions, excitotoxicity, disruption of neurotrophine signaling, impairments in cytoskeleton stability and axonal transport, synaptic and neuronal loss [2]. Pharmacological treatment of AD currently involves cholinesterase inhibitors and NMDA receptor antagonists. Unfortunately, according to most investigators therapeutics of both these groups provide mainly symptomatic benefits without counteracting the progression of the disease [3].
Drug research in the last decade has attempted to develop disease-modifying drugs hopefully able to delay the onset or counteract the progression of AD. Strategies targeting at Aβ pathology include decreasing of Aβ production, preventing aggregation of Aβ into amyloid plaques, stimulating clearance of Aβ. Neither inhibitors of β-secretase or γ-secretase, nor stimulators of α-secretase have demonstrated satisfactory potency combined with low toxicity. Drugs targeting tau-protein are known to be divided into several groups: modulators of tau phosphorylation, inhibitors of tau-phosphorylating kinases (e.g. glycogen-synthase-kinase-3β, cyclin-dependent kinase-5, p70-S6-kinase) and compounds that prevent tau aggregation and misfolding [4].
AD is a complex multifactorial pathology, including multiple cycles and subcycles of self-amplifying neurodegenerative process [5],[6]. Monotherapy targeting single steps in this complicated cascade may explain disappointments in trials with agents affecting only one chain of this “circulus vituosus“. So it would be advantageous to explore the possibilities of novel multi-target therapy, aimed to affect different disease-related mechanisms, resulting in additive or synergic therapeutic responses [7].
Neuropeptides have drawn special attention as potential multitarget drugs because of their high biological activity (several orders higher than that of nonpeptide ones), availability of several recognising sites supposed to be complimentary to various targets, the ability to interact with different signal molecules, and minimal side effects. However, their usage as drugs is hindered by the poor blood–brain barrier penetration and low biological stability [8].
Design of dipeptides is one of the promising approaches taking into account high biological stability of these short molecules and presence of specific ATP-dependent transport systems for di/tripeptides in the intestine (PEPT1) and in the blood–brain barrier (PEPT2) [9]. This provides a basis for brain availability of dipeptides in case of systemic route of administration, including peroral one.
Original approach to the design of active dipeptides is being developed for many years at V.V. Zakusov Institute of Pharmacology. Searching for dipeptides with cognitive enhancing activity Gudasheva et al. based on the idea to get the structures conformationally close to piracetam as a standard cognition enhancer [10]. This drug-based peptide design led us to the series of acyl-prolyl-containing dipeptides possessing pronounced cognitive enhancing and neuroprotective activities [11]. Noopept (N-phenyl-acetyl-L-prolylglycine ethyl ester, GVS-111, Noopept®) (Figure 1) was chosen from this series because of its pronounced nootropic activity [12], high bioavailability for brain tissues in case of peroral administration [13] and specificity of its mechanism of action [14]. Noopept demonstrated wide spectrum of cognition improving effects [15] as well as pronounced neuroprotective activities both in vivo [15] and in vitro conditions [16]. Compared to piracetam noopept produces a cognition enhancing effect at much lower concentrations and demonstrates activity over a wider range of cognition disturbances and neuronal damages [17]. Noopept showed effectiveness in several animal models of AD: olfactory bulbectomy [18], administration of amyloid into Meinert nucleus [19] and intracerebroventricul administration of diabetogenic toxin streptozotocine [20]. Moreover, the experimental data on cognitive improving effect of noopept have been confirmed in clinic (Phase III and postregistration trials) demonstrating beneficial effect on cognitive functions in patients with MCI of cerebro-vascular or posttraumatic origin [21], and in particular in patients with amnestic form of MCI carrying APOE ε 4+ allele [22]. Taken together these findings prompted us to continue the investigation of noopept on the cellular AD-related model. In the present study we investigated the protective effect of noopept against Аβ25–35-mediated damage of PC12 cells, measuring the cellular viability, apoptosis, intracellular Ca2+, ROS, mitochondrial membrane potential, tau protein phosphorylation level and neurite outgrowth. Aβ25–35 fragment was used as a peptide mimicking several of the toxic effects of the full-length amyloid-β peptide and therefore widely exploiting in both in vitro and in vivo Alzheimer’s disease models [23].

Chemical structures of piracetam and noopept. The structural similarity of piracetam (A) to noopept (B). Both molecules contain pyrrolidine ring, acylated nitrogen in this ring, amide moiety and the fragment of glycine.
Methods
Cell cultures and treatments
PC12 cells were cultured routinely at 37°C in DMEM medium, supplemented with 10% fetal bovine serum (FBS), 5% horse serum, 2 mM L-glutamine, 50 μg/ml gentamicin. To induce PC12 differentiation, NGF (50 ng/ml; Sigma-Aldrich Inc., USA) was added to the DMEM containing 1% FBS, followed by a 5-day incubation. Differentiated PC12 (dPC12) cells were pretreated with noopept at concentration of 10 μM for 72 h, then cells were rinsed with the medium and exposed to amyloid-β-peptide (Аβ25–35, 5 μM; Tocris Bioscience, UK) for 24 h. Untreated cells were used as control.
Cell viability and apoptosis measurements
Cell viability was determined by conventional MTT assay. dPC12 cells were plated in 24-well plates with 500 μl DMEM medium at the density of 1 × 104 cells/well. After treatment with noopept (10 μM) for 72 h followed by Аβ25–35 (5 μM) for 24 h, cells were incubated with 200 μl MTT solution (0.5 mg/ml) at 37°C for additional 4 h. Thereafter the cells were solubilized with 200 μl dimethylsulfoxide. After mixing for 10 min absorbance was measured at 540 nm using the microplate spectrophotometer (EnSpire® Multimode Plate Reader; Perkin Elmer, USA). Cell viability was expressed as the percentage to cell viability in control. Flow cytometry analysis was used to identify the apoptotic cells. dPC12 cells (5 × 104) in 6-well plates were treated as described above. Cells were harvested, washed out with cold phosphate-buffered saline (PBS) and stained with the Annexin V/PI (Annexin V-FITC Kit, Beckman Coulter Inc., USA) according to the manufacturer’s instructions. The data were processed using the FCS Express 4 software (De novo Software, USA) and the Cytomics FC 500 flow cytometer (Beckman Coulter, USA).
Measurement of intracellular Ca2+
After incubation with noopept and Аβ25–35 dPC12 cells (1 × 104 cells/well) were washed in Ca2+-free HBSS, containing 2.5 mM probenecid (Tocris Bioscience, UK). Then cells were loaded with 4 μM of Ca2+ indicator Fluo-4 AM and 0.02% pluronic acid (Invitrogen, USA) and incubated for 20 min at 30°C. Cells were washed out twice in buffer without dye, and incubated for further 15 min. The fluorescence of samples in 0.1 ml of buffer in new 96-well plates was monitored by the microplate spectrophotometer, using 485 nm excitation filter and 520 nm emission filter.
Measurement of intracellular reactive oxygen species (ROS)
The generation of ROS was measured by the oxidative conversion of cell permeable 2,7-dichlorofluorescein diacetate (H2DCFDA; Invitrogen, USA) to fluorescent dichlorofluorescein. dPC12 cells (5 × 103 cells/well) in 96-well plates were cultured for 72 h in 10% DMEM medium with noopept at concentrations of 10 μM. H2DCFDA was then added directly to the growth medium at a final concentration of 5 μM; cells were incubated for 1 h at 37°C. Cells were rinsed twice with PBS, placed in a fresh medium and treated with Аβ25–35 (5 μM) for 24 h. After this treatment cells were washed out with PBS. The plates were then read on the microplate spectrophotometer with 485 nm excitation and 535 nm emission wavelengths.
Assessment of mitochondrial function
dPC12 cells were plated at a density of 5 × 103 cells/well in 96-well plates. After treatment with noopept (10 μM) for 72 h and Аβ25–35 (5 μM) for 24 h changes in the mitochondrial membrane potential (MMP) were determined by incubating with 10 μM of JC-1 reagent (Invitrogen, USA) for 20 min at 37°C in the darkness. Then the cells were washed with PBS three times, and the fluorescent intensity was determined by microplate reader.
Western blotting
dPC12 cells (5 × 104 cells/per well) were treated as described above and after incubation the cells were harvested and suspended in lysis buffer (10 mM Tris, 1 mM EDTA, 1% SDS, pH 7.5). Protein concentrations were determined by the Bradford assay and equivalent amounts (10–15 μg) of total cellular proteins were separated by electrophoresis on a 12% SDS - polyacrylamide gel. Proteins were transferred to PVDV membrane and probed with anti-p-tau (Ser396; 1:800 v/v; Abcam, England) antibodies. After incubation with horseradish peroxidase–conjugated secondary antibody (1:10000; BioRad, Hercules, USA), immunoblots were developed using “Pierce ECL Western Blotting Substrate” (Thermo Scientific, USA). Membranes were stripped off and reprobed with anti-β-tubulin antibody (1:2000 v/v; Cell Signaling, USA) for loading control. Immunoblots were quantified by densitometry (ImageJ, http://rsbweb.nih.gov/ij/). Data were normalized to β-tubulin and the corresponding control was taken as 100%.
Immunocytochemistry and morphometry
dPC12 cells (1 × 104 cells/well) were plated onto poly-L-lysine coated coverslips in 24-well plates. After the treatment, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 for 10 min and stained with mouse monoclonal antibody to neuron specific beta III tubulin (1:100; Abcam, England), followed by AlexaFluor 488-conjugated secondary antibody (1:1000; Invitrogen, USA). Coverslips were then incubated with Hoechst 33258 (1 μg/ml) for 5 min at room temperature. After washing out with PBS, samples were mounted in Mowiol 4–88 based media (Sigma, USA). Fluorescent images were obtained with Axio Imager (Carl Zeiss, Germany) microscope with acquisition settings at the maximal resolution (1024 × 1024 pixels) with 20x objective. Morphological analysis of cells (the number of neurites per cell and average neurite length) was carried out with Sholl analysis (Sholl analysis plug-in for ImageJ, http://rsbweb.nih.gov/ij/). Cells with at least one visible process equal to or greater than one cell body were considered as positive for neurite formation. All neurites of individual PC12 cell were traced, and the number of pixels was automatically converted to micrometers. Comparison of the number of processes between the experimental groups was carried out at a distance of 55 μm from the body of the cell. 50 randomly chosen cells were photographed and examined in each of three coverslips for each experimental condition. Results were obtained from three independent experiments.
Statistical analysis
Each of the above listed parameters was measured in 3 to 5 independent experiments with 3–5 technical replicates per separate experiments. Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by Turkey’s post-hoc test (Statistica v.6.0., StatSoft Inc., OK, USA). Data represent the mean ± SEM. A difference was considered statistically significant if the p < 0.05.
Results
Effect of noopept on cell viability and apoptosis in Aβ25–35-treated PC12 cells
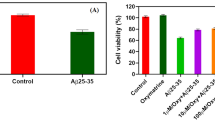
A 24-h incubation of PC12 cells with Aβ25–35 (5 μM) decreased cell viability measured by MTT-test up to 32 ± 17.35%. Exposure of PC12 cells to noopept (10 μM, 72 h) significantly (p = 0.025) reduced cell death caused by Aβ25–35, increasing the cell viability to 230 ± 60.45% (Figure 2A). Therefore exposure of PC12 cells to noopept (10 μM, 72 h) not only attenuated the cytotoxic effect of Aβ25–35, but significantly (by about twofold comparing to intact control) increased the cell viability.

Prevention of Aβ 25–35 - induced cytotoxicity by noopept. (A) The cells were pre-treated with noopept (10 μM) for 72 h before exposure to 5 μM of Аβ25–35 for 24 h. Cell viability was determined by MTT assay. Data are expressed as means ± SEM. Five independent experiments were carried out in triplicate. (B) Apoptosis was assessed by double staining of cells with Annexin-V-FITC and propidium iodide. The bar chart represents the percentage distribution of apoptotic cells. Data are expressed as means ± SEM. Three independent experiments were carried out in triplicate.
Apoptosis was quantified by double staining of cells with Annexin-V/PI (Figure 2B) to distinguish healthy PC12 cells (Annexin V-negative, PI-negative) from early apoptotic cells (Annexin V-positive, PI-negative) and late apoptotic cells (Annexin V-positive, PI-positive). Annexin V/PI staining revealed an increase in the percentage of early and late apoptotic cells from 5.1 ± 0.4 and 1.1 ± 0.4 in the control group to 13.1 ± 1.2 and 8.3 ± 0.5 respectively following incubation with Aβ25–35. Pretreatment of PC12 cells with noopept (10 μM for 72 h) prior to Aβ25–35 exposure, significantly decreased the percentage of Annexin V +/PI – (up to 6.9 ± 1.3; p = 0.0023) and Annexin V +/PI + cells (up to 4.9 ± 0.9; p = 0.0027), thus demonstrating the normalizing drug effect on early as well as on late apoptotic events.
Effect of noopept on Ca2+ level, ROS production and mitochondrial membrane potential
It is well known that Aβ25–35-caused cell death is accompanied by the rise of Ca2+, ROS accumulation and mitochondrial membrane potential disturbance in different neuronal and neuron-like cells. Exposure of differentiated PC12 cells to Aβ25–35 resulted in a 25% elevation of [Ca2+]I, while noopept statistically significantly (p = 0.027) inhibited calcium rise (Figure 3A). By using of the ROS fluorescent dye H2DCF-DA we were able to show that Aβ25–35 caused a moderate increase in ROS level, which was abolished by noopept (p = 0.0024) (Figure 3B). The noopept ability to counteract the Aβ25–35-induced cytotoxicity was also assessed by monitoring of the changes in the mitochondrial membrane potential using fluorescent dye JC-1. When PC12 cells were incubated with Aβ25–35 (5 μM for 24 h) a reduction of MMP was detected. Noopept was shown to protect the mitochondrial membrane potential against Aβ25–35 – induced mitochondrial disturbance (p = 0.0023) (Figure 3C). Taken together data obtained suggest that neuroprotective effect of noopept against beta amyloid neurotoxicity involves the limiting of oxidative stress, calcium disregulation and mitochondrial dysfunction.

Effect of noopept on Аβ 25–35 -evoked disturbances of intracellular calcium level, ROS accumulation and mitochondrial function. (A) Pre-treatment with noopept reduces the rate of intracellular calcium in PC12 cells exposed to Aβ. (B) Noopept diminishes Аβ25–35 - induced enhancement of reactive oxygen species generation. (C) Noopept exposure ameliorates the mitochondrial membrane potential of PC12 cells after Аβ25–35-caused stress. Results represent means ± SEM. The values were obtained from three independent experiments with five technical replicates (A) and from five independent experiments with four technical replicates (B and C).
Noopept decreased tau phosphorylation induced by Aβ25–35
The effect of Aβ25–35 on tau protein phosphorylation level was measured by evaluating of the changes in immunoreactivity using anti-phospho-Ser396-tau antibodies. An increased level of tau phosphorylation at Ser396 was observed in the presence of 5 μM Aβ25–35, while the pretreatment with noopept caused the decline of p-tau Ser396 level (p = 0.0024) (Figure 4). Thus, the protective effect of noopept on Aβ25–35 toxicity apparently involves the attenuation of tau protein phosphorylation.

Noopept decreases the tau phosphorylation induced by Аβ in PC12 cells. Western blot analysis and graphs showed the changes in the content of the phosphorylated tau (Ser396) in PC12 cells pre-treated with noopept following by Аβ25–35 incubation. Densitometry values were normalized using the β-tubulin as internal control and expressed as means ± SEM. Four independent experiments were carried out using three replicate wells.
Noopept ameliorates Aβ-induced impairment of PC12 cells morphology
To further characterize the neuroprotective features of noopept we investigated the effect of the drug on morphology of differentiated PC12 cells. Quantification of neuritic complexity by determination of the average number and length of β-III-tubulin-immunopositive processes and neurites number at different distances from soma showed that PC12 cell treated with Aβ25–35 exhibited unfavorable changes in their cytoarchitecture. These changes were manifested in decreased number of neurites per cell (2.3 in control cultures versus 1.6 in Aβ-exposed cells), significantly reduced neurite length (from 302 μM up to 129 μM) (Figure 5A, B) and a decrease of neurites number with increasing distance from soma resulted in simplification of cells. The pretreatment of cells with noopept tended to overcome these detrimental effects of Aβ. In particular, the drug restored the number of neurites (2.44 versus 1.64; p = 0.0022) and increased their length compared to those in Aβ-treated group (from 129 μM up to 203 μM; p = 0.011) (Figure 5A, B). Overall the amount of longer neurites increased in noopept treated cells, compared to cells exposed to Aβ25–35 alone.

Noopept protects the Аβ 25–35 - induced impairments of cells morphology. (A) Quantification of number of IIIβ-tubulin - immunopositive neurites and (B) the average neurites length of PC12 cells after noopept pre-treatment following by Аβ25–35 addition. Data expressed as means ± SEM. Data from three coverslips (50 cells per coverslip) for each experimental group in three independent experiments were evaluated.
Discussion
Present study revealed, for the first time, that the dipeptide cognition enhancing drug noopept protects differentiated PC12 cells against Aβ-mediated toxicity as evidenced by enhanced cell viability. While Aβ25–35 (5 μM) decreased cell viability, exposure of PC12 cells to noopept has not only overcome the depressing effect of amyloid on cells survival, but even increased it by about twofold compared to intact control. Our results further indicate that pre-treatment of the cells with noopept reduced the percentage of apoptotic cells observed following incubation with the Aβ25–35 peptide. Using Annexin V-FITC/PI double staining for the distinction of early- and late-apoptotic cells, we demonstrated that noopept attenuates both early and late apoptotic events induced by Aβ. Our findings of antiapoptotic effect of noopept against Aβ induced apoptosis are consistent with those obtained with this dipeptide in SH-SY5Y cells underwent to the toxic effect of another misfolded protein, α-synuclein amyloids [24].
Numerous in vivo and in vitro studies indicate that beta-amyloid triggers both common to different brain damages pathogenic pathways by inducing the rise of intracellular calcium level, reactive oxygen species production, alteration of mitochondrial function, and Aβ-specific signaling pathways resulted in increased tau phosphorylation [25]. Moreover, biochemical disturbances caused by Aβ are accompanied by substantial cytoskeleton abnormalities and consequently impaired axonal transport [26]. Particularly, prominent reductions of neurite outgrowth and neuritic elongation have been observed in different neuronal and neuron-like cells after oligomeric as well as fibrillar Aβ treatment [27],[28].
In this study, noopept was found to mitigate the intracellular calcium influx and excessive production of ROS, suggesting that the neuroprotective effects of the drug in this cellular model are probably associated with inhibition of Aβ-induced overload of calcium and antioxidant properties. Another mechanism involved in the neuroprotective action of noopept likely includes its ability to ameliorate mitochondrial dysfunction following Aβ25–35 exposure thereby interfering with mitochondrial apoptotic pathway.
These data are in accordance with our previous findings reporting neuroprotective action of noopept in various in vivo and in vitro studies. Noopept was shown to be able to normalize main secondary events by blocking the voltage dependent calcium channels [29], attenuating the neurotoxic effects of glutamate on granular cerebellar neurons [30], decreasing the glutamate release by cortical neurons [31]. Noopept significantly increased neuronal survival and prevented the accumulation of intracellular free radicals and apoptosis in experiments on cultured Down’s syndrome neurons [16]. The drug counteracted also the free radicals accumulation caused by α-synuclein on cultured neuroblastoma SH-SY5Y cells [24]. Interestingly, noopept was demonstrated to increase immunoreactivity to β-amyloid in mice with olfactory bulbectomy, considered as one of AD animal models [18].
Here we have shown for the first time that noopept can protect cells against Aβ-mediated toxicity by attenuating an increased tau phosphorylation at Ser396. Moreover, while Aβ-treated cells demonstrated decrease of neurites number and their length, noopept was shown to restore the number of neurites and significantly augment their processes length. It is known that extensively phosphorylated tau protein forms pathologic inclusions containing fibrillar aggregates were found in the brain of patients suffering from certain neurodegenerative disorders associated with dementias [32]. Tau protein is considered as one of the microtubules stabilizing proteins playing important role in facilitation of tubulin assembly into microtubules, thus contributing to the neurite outgrowth and maintenance of normal cellular morphology [33],[34]. Multiple studies provide evidence that the phosphorylation of tau at distinct serine/threonine residues by different protein kinases affects the ability of tau to promote microtubule polymerisation and stability [35]. Abnormally hyperphosphorylated tau possesses lower affinity for microtubules; it promotes the cytoskeleton rearrangements with consequent impairments of axonal transport and intracellular trafficking [36]. Neurite outgrowth of neuronal cells requires the assembly of tubulin into microtubules. The stability of microtubule network depends, at least in part, on the rate and extent of tau phosphorylation. Particularly, neurite outgrowth of neuronal and neuron-like cells was shown to correlate with the phosphorylation of tau at Ser262, Ser356, Ser396/404; these modifications reduce the ability of tau to bind to microtubules [37],[35]. A number of studies suggest that Aβ peptides under in vitro conditions can cause the increased phosphorylation of tau protein at different sites, thus provoking microtubules destabilization and cytoskeleton network degeneration [38],[26],[39]–[41]. Indeed, exposure of neuronal or neuron-like cells to the β-amyloid results in pronounced neurite retraction and reduced cell complexity [42]–[45] concomitant with a significant increase in tau phosphorylation at the Ser 396 whereas other serine/threonine sites – Ser199, Ser202, Thr205 and Ser404 show no significant alteration [46],[47]. Results from the present study suggest that abrogation of tau hyperphosphorylation at Ser396 by noopept eventually may play a role in restoration and even improvement of PC12 cell morphology. Neurite outgrowth promoting activity of noopept found in this cellular model, probably depends on drug’s ability to decrease the level of tau phosphorylation, thus affecting tau binding to microtubules. It should be mentioned that our previous experiments demonstrated noopept’ ability to increase the expression of NGF and BDNF in hippocampal and hypothalamic neurons in streptozotocin - intracerebroventricularly treated rats known to be an experimental model of sporadic AD [20]. PC12 cells express TrkA and respond to NGF by neurite outgrowth [48]. Findings of present study of noopept ability to exert antiapoptotic effect and to increase number and length of neuritis are in line with our supposition on the NGF involvement in above described effects of noopept on PC12 cells.
Recent studies provided evidence that both types of medicines currently used for AD treatment, NMDA receptor antagonists and AchE inhibitors, affect positively at least some of AD-related mechanisms. For example memantine was shown to inhibit the abnormal hyperphosphorylation of tau [49] and protected the neurons from Aβ-induced reduction of neurite outgrowth [50]. AchE inhibitor galantamine decreases the neuronal apoptosis induced by Aβ25–35, as well as membrane potential dissipation, suppressing the activity of caspase-9, caspase-12 and caspase-3 [51]. Results comparable to those obtained for noopept were observed for its conformationally related analog, piracetam. This cognitive enhancer attenuates the Aβ-caused alterations of mitochondrial membrane potential of PC12 cells and inhibited the negative effect of Aβ on neurite outgrowth [52].
Taken together findings obtained in this study suggest that noopept affects positively the core pathogenic mechanisms underlying the Aβ-mediated toxicity and provide new insights into the neuroprotective action of this drug and its possible beneficial effect in amyloid-related pathology. Further studies to confirm the neuroprotective effect of noopept against Aβ-induced neurotoxicity in AD animal model need to be conducted.
Conclusions
Cognitive enhancer noopept exerts a protective effect against Aβ25–35-induced toxicity in PC12 cells. The protective ability of noopept most likely results from moderate suppression of oxidative stress and intracellular calcium influx, stabilization of mitochondrial function and reducing of apoptosis. Another possible mechanism by which this compound protects cells from amyloid toxicity may be related to the decrease of tau phosphorylation and, eventually, neurite stabilization and outgrowth.
Abbreviations
- Aβ:
-
β-Amyloid
- AchE:
-
Acetylcholinesterase E
- AD:
-
Alzheimer disease
- APOE:
-
Apolipoprotein E
- APP:
-
Amyloid precursor protein
- BDNF:
-
Brain-derived neurotrophic factor
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- FBS:
-
Fetal bovine serum
- FITC:
-
Fluorescein isothiocyanate
- HBSS:
-
Hank’s Balanced Salt Solution
- H2DCFDA:
-
2’,7’-dichlorodihydrofluorescein diacetate
- JC-1:
-
5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimi- dazolylcarbocyanine iodide
- MCI:
-
Mild cognitive impairment
- MMP:
-
Mitochondrial membrane potential
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- NGF:
-
Nerve growth factor
- NMDA:
-
N-methyl-D-aspartate
- PBS:
-
Phosphate buffered saline
- PEPT1:
-
Peptide transporter 1
- PEPT2:
-
Peptide transporter 2
- ROS:
-
Reactive oxygen species
- TrkA:
-
Neurotrophic tyrosine kinase receptor type 1
References
Thies W, Bleiler L: Alzheimer’s Association, 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011, 7: 208-244.
Krstic D, Knuesel I: Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2013, 9 (1): 25-34.
Schneider LS, Dagerman KS, Higgins JP, McShane R: Lack of evidence for the efficacy of memantine in mild Alzheimer Disease. Arch Neurol. 2011, 68: 991-998.
Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M: Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 2010, 9 (7): 702-716.
Longo FM, Massa SM: Neuroprotective strategies in Alzheimer’s Disease. NeuroRx. 2004, 1: 117-127.
Buccafusco JJ: Emerging cognitive enhancing drugs. Expert Opin Emerg Drugs. 2009, 14: 577-589.
Frautschy SA, Cole GM: Why pleiotropic interventions are needed for Alzheimer’s Disease. Mol Neurobiol. 2010, 41: 392-409.
Kaidanovich O, Eldar-Finkelman H: Peptides targeting protein kinases: strategies implications. Physiology. 2006, 21: 411-418.
Sala-Rabanai M, Loo DDT, Hirayama BA, Turk E, Wright EMJ: Molecular interactions between dipeptides, drugs and the human intenstinal H+ oligopeptide cotransporter hPEPT 1. J Physiol. 2006, 574: 149-166.
Gudasheva TA, Voronina TA, Ostrovskaya RU, Rozantsev GG, Vasilevich NI, Trofimov SS, Kravchenko EV, Skoldinov AP, Seredenin SB: Synthesis and antimnestetic activity of a series of N-acylprolyl-containing dipeptides. Eur J Med Chem. 1996, 31: 151-157.
Seredenin SB, Voronina TA, Gudasheva TA, Ostrovskaya RU, Rozantsev GG, Skoldinov AP, Trofimov SS, Halikas J, Garibova TL: Biologically active N-acylprolyldipeptides having antiamnestic, antihypoxic effects.US patent 1995. No. 5.439.930.,
Ostrovskaya RU, Gudasheva TA, Trofimov SS, Kravchenko EV, Firova FV, Molodavkin GM, Voronina TA, Seredenin SB: GVS-111, an Acylprolyl-Containing Dipeptide With Nootropic Properties. Biological Basis of Individual Sensitivity to Psychotropic Drugs. Edited by: Seredenin SB, Longo V, Gaviraghi G. 1994, Graffhan Press Ltd, UK, 79-91.
Boiko SS, Ostrovskaya RU, Zherdev VP, Korotkov SA, Gudasheva TA, Voronina TA, Seredenin SB: Pharmacokinetics of new nootropic acylprolyldipeptide and its penetration across the blood–brain barrier after oral administration. Bull Exp Biol Med. 2000, 129: 359-361.
Gudasheva TA, Bojko SS, Voronina TA, Akparov VK, Trofimov SS, Rozantsev GG, Skoldinov AP, Zherdev VP, Seredenin SB: The major metabolite of dipeptide piracetam analogue GVS-111 in rat brain and its similarity to endogenous neuropeptide cyclo-L-prolylglycine. Eur J Drug Metab Pharmacokinet. 1997, 22: 245-252.
Ostrovskaya RU, Romanova GA, Barskov IV, Shanina EV, Gudasheva TA, Victorov IV, Voronina TA, Seredenin SB: Memory restoring and neuroprotective effects of the proline containing dipeptide, GVS-111, in a photochemical stroke model. Behav Pharmacol. 1999, 10: 549-553.
Pealsman A, Hoyo-Vadillo C, Seredenin SB, Gudasheva TA, Ostrovskaya RU, Busciglio J: GVS-111 prevents oxidative damage and apoptosis in normal and Down’s syndrome human cortical neurons. Int J Dev Neurosci. 2003, 21: 117-124.
Ostrovskaia RU, Gudasheva TA, Voronina TA, Seredenin SB: The original novel nootropic and neuroprotective agent Noopept. Eksp Klin Farmakol. 2002, 65 (5): 66-72.
Ostrovskaya RU, Gruden MA, Bobkova NA, Sewell RDE, Gudasheva TA, Samokhin AN, Seredenin SB, Noppe W, Sherstnev VV, Morozova-Roche LA: The nootropic and neuroprotective proline-containing dipeptide Noopept restores spatial memory and increases immunoreactivity to amyloid in an Alzheimer’s Disease model. J Psychopharmacol. 2007, 21: 611-619.
Ostrovskaya RU, Belnik AP, Storozheva ZI: Noopept efficiency in experimental Alzheimer Disease (cognitive deficiency caused by beta-amyloid 25–35 injection into Meynert basal nuclei of rats). Bull Exp Biol Med. 2008, 146: 77-80.
Ostrovskaya RU, Tsaplina AP, Vakhitova YV, Salimgareeva MK, Yamidanov RS: Effect of the novel cognition enhancing and neuroprotective dipeptide Noopept on the streptozotocin-induced model of sporadic Alzheimer Disease in rats. Eksp Klin Farmakol. 2010, 73: 2-6.
Neznamov GG, Teleshova ES: Comparative studies of Noopept and piracetam in the treatment of patients with mild cognitive disorders in organic brain diseases of vascular and traumatic origin. Neurosci Behav Physiol. 2009, 39 (3): 311-321.
Gavrilova SI, Kolihalov IV, Fedorova JB, Kaljn JB, Selezneva ND: Expirience of Noopept clinical usage in the treatment of mild cognitive impairment syndrome. Modern Therapie of Psychiatric Disorders. 2008, 1: 27-32.
Iversen LL, Mortishire-Smith RJ, Pollack SJ, Shearman MS: The toxicity in vitro of beta-amyloid protein. Biochem J. 1995, 311 (Pt 1): 1-16.
Jia X, Gharibyan AL, Öhman A, Liu Y, Olofsson A, Morozova-Roche LA: Neuroprotective and nootropic drug Noopept rescues α-synuclein amyloid cytotoxicity. J Mol Biol. 2011, 414: 699-712.
Canevari L, Abramov AY, Duchen MR: Toxicity of amyloid beta peptide: tales of calcium, mitochondria, and oxidative stress. Neurochem Res. 2004, 29 (3): 637-650.
Henriques AG, Vieira SI, Da Cruz e Silva EF, Da Cruz e Silva OAB: Aβ promotes Alzheimer’s disease-like cytoskeleton abnormalities with consequences to APP processing in neurons. J Neurochem. 2010, 113: 761-771.
Spires TL, Hyman BT: Neuronal structure is altered by amyloid plaques. Rev Neurosci. 2004, 15 (4): 267-278.
Postuma RB, He W, Nunan J, Beyreuther K, Masters CL, Barrow CJ, Small DH: Substrate-bound beta-amyloid peptides inhibit cell adhesion and neurite outgrowth in primary neuronal cultures. J Neurochem. 2000, 74 (3): 1122-1130.
Solntseva EI, Bukanova JV, Ostrovskaya RU, Gudasheva TA, Voronina TA, Skrebitsky VG: The effects of piracetam and its novel peptide analogue GVS-111 on neuronal voltage-gated calcium and potassium channels. Gen Pharmacol. 1997, 29: 85-89.
Andreeva NA, Stel’mashuk EV, Isaev NK, Ostrovskaya RU, Gudasheva TA, Viktorov IV: Neuroprotective effects of nootropic dipeptide GVS-111 on the model of glucose-oxygen deprivation, glutamate toxicity and oxidative stress in vitro. Bull Exp Biol Med. 2000, 130: 418-421.
Us KS, Klodt PM, Kudrin VS, Sapronova AY, Ostrovskaya RU, Ugryumov MV, Rayevsky KS: The effect of the synthetic neuroprotective dipeptide Noopept on glutamate slices. J Neurochem. 2007, 1: 138-142.
Iqbal K, Liu F, Gong CX, Grundke-Iqbal I: Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010, 7 (8): 656-664.
Esmaeli-Azad B, McCarty JH, Feinstein SC: Sense and antisense transfection analysis of tau function: tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J Cell Sci. 1994, 107 (Pt 4): 869-879.
Takei Y, Teng J, Harada A, Hirokawa N: Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000, 150 (5): 989-1000.
Wang JZ, Xia YY, Grundke-Iqbal I, Iqbal K: Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J Alzheimers Dis. 2013, 33 (Suppl 1): S123-S139.
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K: Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994, 91 (12): 5562-5566.
Biernat J, Mandelkow EM: The development of cell processes induced by tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol Biol Cell. 1999, 10 (3): 727-740.
Busciglio J, Lorenzo A, Yeh J, Yankner BA: β-Amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995, 14: 879-888.
Avila J, Pérez M, Lim F, Gómez-Ramos A, Hernández F, Lucas JJ: Tau in neurodegenerative diseases: tau phosphorylation and assembly. Neurotox Res. 2004, 6 (6): 477-482.
Gendron TF, Petrucelli L: The role of tau in neurodegeneration. Mol Neurodegener. 2009, 4: 13-
Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S: Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002, 115 (1): 201-211.
Drechsel DN, Hyman AA, Cobb MH, Kirschner MW: Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992, 3 (10): 1141-1154.
Salinero O, Moreno-Flores MT, Ceballos ML, Wandosell F: beta-Amyloid peptide induced cytoskeletal reorganization in cultured astrocytes. Neurosci Res. 1997, 47: 216-223.
Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A: Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002, 99 (9): 6364-6369.
Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ: Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011, 108 (14): 5819-5824.
Takashima A, Honda T, Yasutake K, Michel G, Murayama O, Murayama M, Ishiguro K, Yamaguchi H: Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25–35) enhances phosphorylation of tau in hippocampal neurons. Neurosci Res. 1998, 31 (4): 317-323.
Johansson S, Jämsä A, Vasänge M, Winblad B, Luthman J, Cowburn RF: Increased tau phosphorylation at the Ser396 epitope after amyloid beta-exposure in organotypic cultures. Neuroreport. 2006, 17 (9): 907-911.
Shimoke K, Sasaya H, Ikeuchi T: Analysis of the role of nerve growth factor in promoting cell survival during endoplasmic reticulum stress in PC12 cells. Methods Enzymol. 2011, 490: 53-70.
Li L, Sengupta A, Haque N, Grundke-Igbal I, Igbal K: Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett. 2004, 566: 261-269.
Hu M, Schurdak ME, Puttfarcken PS, El Kouhen R, Gopalakrishnan M, Li J: High content screen microscopy analysis of A beta 1-42-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of alpha 7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 2007, 1151: 227-235.
Liu X, Xu K, Yan M, Wang Y, Zheng X: Protective effects of galantamine against Ab-induced PC12 cell apoptosis by preventing mitochondrial dysfunction and endoplasmic reticulum stress. Neurochem Int. 2010, 57: 588-599.
Kurz C, Ungerer I, Lipka U, Kirr S, Schütt T, Eckert A, Leuner K, Müller WE: The metabolic enhancer piracetam ameliorates the impairment of mitochondrial function and neurite outgrowth induced by beta-amyloid peptide. Br J Pharmacol. 2010, 160 (2): 246-257.
Acknowledgements
This work was partially supported by the Grant for the state support of leading scientific schools of the Russian Federation (№ 5923.2014.4 to VAV). We are grateful Prof. Grivennikov I.A. (Institute of Molecular Genetics, Russian Academy of Sciences, Moscow) for provision of rat pheochromocytoma cell line.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
SBS, RUO and TAG conceived the experiments. YVV and VAV designed the experiments. USK, MKS, LFZ performed the experiments and analyzed the data. RUO and YVV interpret the data and wrote the paper. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Ostrovskaya, R.U., Vakhitova, Y.V., Kuzmina, U.S. et al. Neuroprotective effect of novel cognitive enhancer noopept on AD-related cellular model involves the attenuation of apoptosis and tau hyperphosphorylation. J Biomed Sci 21, 74 (2014). https://doi.org/10.1186/s12929-014-0074-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-014-0074-2