
Abstract
Background
Pathogenic variants in the ATCAY gene are associated with a rare autosomal recessive disorder called Cayman cerebellar ataxia. Affected individuals display psychomotor retardation, cerebellar dysfunction, nystagmus, intention tremor, ataxic gait and dysarthric in some cases.
Case presentation
Whole exome sequencing was performed for a 21-month-old girl suffering from developmental delay specifically in motor and language aspects, hypotonia, nystagmus, pes planus and strabismus. The detected variant in the patient was confirmed by family segregation analysis by Sanger sequencing in both of her parents. Previously three homozygous variants in the ATCAY gene (missense, splice site and frameshift deletion) have been reported in patients with Cayman cerebellar ataxia. Here we report the fourth homozygous variant and the second homozygous frameshift deletion in this gene to be associated with autosomal recessive Cayman cerebellar ataxia.
Conclusion
The identification of this novel homozygous frameshift deletion in the ATCAY gene expands our understanding of the genetic landscape underlying Cayman cerebellar ataxia. Furthermore, the occurrence of this variant in Iran, in addition to Pakistan, signifies the importance of considering genotypic and phenotypic factors beyond ethnicity when studying this disorder. These findings contribute to the ongoing efforts to unravel the molecular basis of Cayman cerebellar ataxia and improve diagnostic approaches and potential therapeutic interventions.
Similar content being viewed by others


Background
The Cayman cerebellar ataxia is an autosomal recessive neurologic disorder represented by birth-onset hypotonia, variable psychomotor retardation, and cerebellar dysfunction, including nystagmus, intention tremor, dysarthria, ataxic gait, and truncal ataxia in the patients [1, 2].
The disorder was initially identified in an isolated region on Grand Cayman Island and believed to be restricted to this area. First, Brown et al., in 1986 and then Nystuen et al., in 1996 reported 26 and 19 individuals, respectively with Cayman cerebellar ataxia on Grand Cayman Island. All patients had psychomotor retardation, prominent cerebellar dysfunction manifest by nystagmus, intention tremor, ataxic gait and dysarthric was present in some affected individuals. Life span was normal. Brain computed tomography (CT) scan, performed in 4 patients, showed marked cerebellar hypoplasia, with the vermis more involved than the hemispheres [1, 3]. In the previous case reports, the mutated gene and the variants related to Cayman ataxia cerebellar disorder were not detected. For the first time, Bomar et al., investigated the ATCAY gene locus 19p13.3 as the associated gene with the disorder. They also detect two homozygous variants in the ATCAY gene including a missense p.(Ser301Arg) and a splice site variants (c.965 + 3G > T) in the affected individuals. The variants segregation analysis and the carrier status of 40 family members were confirmed [2]. Manzoor et al., in 2018 reported a multigenerational highly consanguineous Pakistani family (RDHR-04) in which 6 people had severe gait ataxia with onset in infancy. All also had pes planus. Additional more variable features included strabismus, possible ocular apraxia, bibrachial dystonia, bradykinesia, and distal muscle atrophy primarily in the feet. Brain imaging in 1 patient showed marked cerebellar atrophy. There was suspicion of cognitive impairment. It was the first observation of the disorder in a place other than Grand Cayman Island and also the first frameshift deletion in ATCAY gene p.(Asp201AlafsTer20) [4]. In this article, we report the fourth homozygous variant and the second homozygous frameshift deletion in this gene p.(Lys295AspfsTer52) to be associated with autosomal recessive Cayman cerebellar ataxia.
The ATCAY gene encodes a neuron-restricted protein called caytaxin. Caytaxin has a CRAL-TRIO motif named after cellular retinal and the TRIO guanine exchange factor. This motif is common to proteins that bind to lipophilic molecules. Variants in such proteins cause a vitamin-E responsive ataxia. Three dimensional studies of the caytaxin showed its ligand is more polar than vitamin E [2]. Caytaxin contains 371 amino acids and is also called BNIP-H, a brain-specific member of the BNIP-2 family. BNIP-H interacts directly with kidney-type glutaminase (KGA) and both have broadly overlapping expression in the hippocampus and cerebellum. KGA converts glutamine to glutamate as an important source of neurotransmitter [5]. So, BNIP-H binds to KGA and regulates the synthesis of glutamate at synapses during neurotransmission. Variants in the ATCAY and loss of its function lead to glutamate excitotoxicity and deregulation of glutamatergic activation, causing Cayman [6].
Case presentation
Clinical and neuroimaging findings
The patient is a 21-month-old girl who was presented to Myelin Disorders Clinic at Tehran University of Medical Sciences due to developmental delay; particularly in motor, and language aspects. She was the sixth child of consanguineous parents and was born at 40 weeks of gestational age through an uneventful vaginal delivery with birth weight and head circumference of 3000 g (Z-score -0.5 SD) and 33 cm (Z-score -0.7 SD), respectively.
Her mother had a history of two abortions with unknown etiology. One of her sisters had behavioral problems and one of her brothers had stuttering speech. The patient didn’t achieve her normal developmental milestones completely until now. At this age, she has acceptable social interactions, obeys one-step commands and need to be supported significantly in order to sit, and has the ability to make sounds. Her maximum speech ability does not exceed sounds’ expression. One of the chief complaints reported by her mother was her feeding and swallowing problems.
During the last clinical examination, she had a white subconjunctival lesion named lipodermoid on the lateral aspect of the left eye (Fig. 1A, white arrow), diffuse melanosis on the back (Fig. 1B), atrophy of buttock muscles in addition to strabismus, pendular nystagmus, truncal hypotonia, pes planus, and hyporeflexia with + 1 deep tendon reflexes (DTRs). Gross Motor Function Classification System (GMFCS) score was 3/5. Her weight was 7 kg (less than 3rd centile, Z-score -3.8 SD) and HC was 43.5 cm (Z-score -2.34 SD).

A a white subconjunctival lesion (lipodermoid), B diffuse melanosis on the back, C-E Brain MRI at the age of 16 months, axial and sagittal T2-Weighted Imaging sequence, illustrates delayed myelination; high in T2 WI is more marked in subcortical white matter in temporal and frontal lobes (C, D; black arrow), and hyperintensity of globus pallidus (C, white arrows). Cerebellar hemisphere (D, white circle) and cerebellar vermis (E, black circle) atrophy are additional findings
Basic metabolic tests including plasma lactate level, serum ammonia, Tandem Mass Spectroscopy (MS/MS), urine organic acid analysis and creatine kinase were all normal.
At 16 months of age, brain magnetic resonance imaging (MRI) revealed delayed myelination, along with the presence of hyperintensity in the globus pallidus, which had not been reported in previously documented cases. Additionally, there was observed atrophy of the cerebellar hemisphere and vermis (Fig. 1 C, D, E).
Genetics findings
Whole exome sequencing and bioinformatics analysis was preformed as described previously [7]. Called variants were filtered considering minor allele frequency (MAF) > 0.01 based on the Gnome Aggregation Database (gnomAD v3) Consortium, and Iranome population databases [8, 9]. Benign annotated variants according to American College of Medical Genetics (ACMG) recommendations and clinical databases including Human Genome Mutation Database (HGMD) Professional and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) were excluded from the analysis [10, 11]. The remaining variants were once again filtered based on the clinical information of the patient. Human Phenotype Ontology (HPO) database was used to extract associated genes with the patient’s phenotypes. The following HPO terms [12] were applied in the analysis: Generalized hypotonia (HP:0001290), Dysphasia (HP:0002357), Cerebellar atrophy (HP:0001272), Neurodevelopmental delay (HP:0012758), Abnormal corpus callosum morphology (HP:0001273), Brain imaging abnormality (HP:0410263), Fever (HP:0001945), Scanning speech (HP:0002168), Abnormality of the kidney (HP:0000077). The remaining variants were analyzed considering ACMG recommendations and the disease associated with each gene based on the Online Mendelian Inheritance in Men (OMIM) database. The candidate variant was checked in Integrative Genomics Viewer (IGV) software in order to see the depth of coverage and blat score of the reads showing the variant [13]. We ended up with a novel homozygous frameshift 2 nucleotides deletion in the ATCAY gene [NM_033064.5:c.883_884del, p.(Lys295AspfsTer52)] (Table 1).
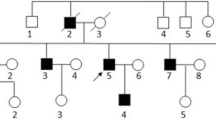
Genomic DNA of the proband’s parents were extracted. The genomic region covering the variant in the ATCAY gene was PCR-amplified using Forward primer: CATGGCTCTGTCTGGACCTA, and Reverse primer: GGCCCCTTAAACCTCTTTTG designed by online primer3Plus tool (https://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi). The PCR products were sequenced using a 3500 Applied Biosystems capillary electrophoresis machine. The sequencing chromatographs were analyzed using the CodonCode aligner software. The identified variant was confirmed by the segregation analysis in her parents (Fig. 2). It was classified as pathogenic based on the ACMG recommendations and considered as the cause of the disease in the proband.

Sanger sequencing chromatographs shows heterozygous frameshift deletion in the parents
Discussion and conclusion
In 1986, Brown et al. were the first to report on Cayman, documenting 24 individuals with psychomotor retardation and cerebellar dysfunction on Grand Cayman Island. Following their work, Nystuen et al. reported 19 patients exhibiting similar phenotypes affected by Cayman cerebellar ataxia in 1996. Later in 2003, Bomar et al. investigated the ATCAY gene locus 19p13.3 as the associated gene locus with Cayman cerebellar ataxia, identifying two homozygous pathogenic variants: a missense variant [NM_033064.5:c.903C > G, p.(Ser301Arg)] and a splice site variant [NM_033064.5:c.965 + 3G > T] in the patients. Subsequently, in 2018, Manzoor et al. described six affected individuals from a highly consanguineous Pakistani family, displaying severe gait ataxia and cerebellar dysfunction. They identified a homozygous pathogenic frameshift variant with a 7-nucleotide deletion in the ATCAY gene [NM_033064.5:c.602_608del, p.(Asp201AlafsTer20)]. Here, we present a novel homozygous pathogenic frameshift variant involving a 2-nucleotide deletion in the ATCAY gene [NM_033064.5:c.883_884del, p.(Lys295AspfsTer52)] in a patient from Iran. This represents the second homozygous frameshift variant in the ATCAY gene and the second location, after Pakistan, outside of Grand Cayman Island (Table 2).
Besides common phenotypes observed in our patient and previously reported cases, she exhibited additional neurological features that warrant attention. These include the presence of a subconjunctival lipodermoid, diffuse melanosis on the back, atrophy of buttock muscles, failure to thrive, and a novel finding in the brain MRI indicating involvement of the globus pallidus, which has not been reported previously and may potentially represent a coincidental occurrence. Obtaining further reports on phenotypes from patients with similar gene mutations will provide valuable insights for a more accurate characterization of this disease and aid in making informed decisions. Mongolian spots or dermal melanosis are a type of birthmark that is commonly seen in infants and young children, especially in the Caucasian population, that is typically found on the lower back, buttocks, and sometimes other areas of the body, but persistent diffuse dermal melanosis is a diagnostic clue for some inborn errors of metabolism and few rare disorders. Although this finding may be an incidental sign in our patient we suggest that it may be an associated finding with Cayman cerebellar ataxia that was not reported till now. [14, 15]. This finding highlights the importance of further investigation and documentation of unique features associated with Cayman ataxia cerebellar. Understanding these additional neurological manifestations, such as the involvement of globus pallidus and ectopic melanocytes, can contribute to a more comprehensive understanding of the disorder and potentially aid in future diagnostic and therapeutic approaches.
Availability of data and materials
All data and materials applied in this study are stored in databases under the supervision of DeNA laboratory (http://dna-lab.ir/) and Pardis Gene Technology company (https://pardisgene.com/).
Abbreviations
- CT:
-
Computed Tomography
- CRAL-TRIO:
-
Cellular Retinal and the TRIO guanine exchange factor
- BNIP-H:
-
Brain-specific member of the BNIP-2 family
- KGA:
-
Kidney-type Glutaminase
- DTRs:
-
Deep Tendon Reflexes
- GMFCS:
-
Gross Motor Function Classification System
- MS/MS:
-
Tandem Mass Spectroscopy
- MRI:
-
Magnetic Resonance Imaging
- MAF:
-
Minor Allele Frequency
- gnomAD v3:
-
Gnome Aggregation Database
- ACMG:
-
American College of Medical Genetics
- HGMD:
-
Human Genome Mutation Database
- HPO:
-
Human Phenotype Ontology
- OMIM:
-
Online Mendelian Inheritance in Men
- IGV:
-
Integrative Genomics Viewer
References
Nystuen A, et al. A cerebellar ataxia locus identified by DNA pooling to search for linkage disequilibrium in an isolated population from the Cayman Islands. Hum Molec Genet. 1996;5(4):525–31. https://doi.org/10.1093/hmg/5.4.525. (PMID: 8845847).
Bomar JM, et al. Mutations in a novel gene encoding a CRAL-TRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nature Genet. 2003;35(3):264–9. https://doi.org/10.1038/ng1255. (PMID: 14556008).
Brown LM, et al. A nonprogressive cerebellar ataxia on Grand Cayman Island. (Abstract) Neurology. 1984;34 (1):273.
Manzoor H, et al. Novel homozygous variants in ATCAY, MCOLN1, and SACS in complex neurological disorders. Parkinsonism Relat Disord. 2018;51:91–5. https://doi.org/10.1016/j.parkreldis.2018.02.005. (PMID: 29449188).
Buschdorf JP, et al. Brain-specific BNIP-2-homology protein Caytaxin relocalises glutaminase to neurite terminals and reduces glutamate levels. J Cell Sci. 2006;119(6):3337–50. https://doi.org/10.1242/jcs.03061. (PMID: 16899818).
Buschdorf1 JP, et al. Brain-specific BNIP-2-homology protein Caytaxin relocalises glutaminase to neurite terminals and reduces glutamate levels. J Cell Scie. https://doi.org/10.1242/jcs.03061.
Mohammadi P, et al. Whole-exome sequencing identified first homozygous frameshift variant in the COLEC10 gene in an Iranian patient causing 3MC syndrome type 3. Molecul Genetics Genom Medic. 2021;9(11):e1834. https://doi.org/10.1002/mgg3.1834. (PMID: 34636477).
Fattahi Z, et al. A catalog of genomic variations in the Iranian population. Hum Mutat. 2019;40(11):1968–84. https://doi.org/10.1002/humu.23880.
Wang Q, et al. Landscape of multi-nucleotide variants in 125,748 human exomes and 15,708 genomes. Nature Commun. 2020;11(1):2539. https://doi.org/10.1038/s41467-019-12438-5.
Richards CS, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genetics in Medicine. 2008;10(4):294–300. https://doi.org/10.1097/GIM.0b013e31816b5cae. (PMID: 18414213).
Maffucci P, et al. Blacklisting variants common in private cohorts but not in public databases optimizes human exome analysis. Proceed Nation Acad Scie United States America. 2019;116(3):950–9. https://doi.org/10.1073/pnas.1808403116. (PMID: 30591557).
Köhler S, et al. The human phenotype ontology in 2021. Nucleic Acids Res. 2021;49(D1):D1207–17. https://doi.org/10.1093/nar/gkaa1043. (PMID: 33264411).
Robinson JT, et al. Integrative genomics viewer. Nature Biotechnology. 2011;29(1):24–61. https://doi.org/10.1038/nbt.1754. (PMID: 21221095).
Ashrafi RM, et al. Extensive mongolian spots: a clinical sign merits special attention. Pediatric Neurol. 2006;34(2):143–5. https://doi.org/10.1016/j.pediatrneurol.2005.07.010. (PMID: 16458829).
Ashrafi RM, et al. Diffuse dermal melanocytosis in two patients with Sandhoff disease and mucopolysaccharidosis VI. Int J Dermatol. 2014;53(6):736–8. https://doi.org/10.1111/ijd.12303. (PMID: 24134161).
Acknowledgements
We thank all the participants in this research. We are specifically grateful for all the supports from DeNA laboratory (http://dna-lab.ir/) and Pardis Gene Technology company (https://pardisgene.com/) in this study. We are also thankful to the staffs of Pediatric Neurology Department and Myelin Disorders Clinic of Children’s Medical Center, Tehran University of Medical Science, Tehran, Iran and Medical Genetics Department of Medical Sciences Faculty, Tarbiat Modares University, Tehran, Iran.
Funding
Funding of this project was provided by NIMAD (grant number: 971846), DeNA laboratory (http://dna-lab.ir/) and Pardis Gene Technology company (https://pardisgene.com/). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
MG and MH conceived and designed the experiments. ESS, MRA, and HGH conducted the experiments. ESS, MG, and MH analyzed and interpreted the data. MG and ESS contributed materials and analysis tools. ESS, MG, and MH wrote the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the ethics committee of the National Institute for Medical Research Development (NIMAD), Tehran, Iran (Ethics ID: IR.NIMAD.REC.1397.508). The written informed consent was received from both guardians in order to participate in this study.
Consent for publication
Both guardians provided a signed written consent to publish all personal and medical details included in this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Siavashani, E.S., Ashrafi, M.R., Ghabeli, H. et al. Novel homozygous frameshift variant in the ATCAY gene in an Iranian patient with Cayman cerebellar ataxia; expanding the neuroimaging and clinical features: a case report. BMC Med Genomics 16, 226 (2023). https://doi.org/10.1186/s12920-023-01643-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01643-3