
Abstract
Background
SALL4, a member of the SALL genes family, encodes a zinc-finger transcriptional factor that either activates or represses gene transcription depending on cell type during embryonic development. SALL4 mutations cause extremely variable conditions including Duane-radial ray (DRR), Okihiro, Holt-oram, Acro-renal ocular and IVIC syndromes, all with autosomal dominant inheritance pattern. However, all these syndromes with different terminologies are actually the same entity termed SALL4 related disorders.
Case presentation
Herein, we examine an Iranian patient suspected to DRR syndrome which has not been previously described in the population. Whole-exome sequencing (WES) was performed to examine pathogenic genes in the proband. Subsequently, Sanger sequencing was used to confirm the mutation found. To elucidate the effects of the identified mutation, clinical data of patient was collected. Morever, the possible impact of the mutation found on the corresponding protein was evaluated using bioinformatics tools. WES identifed a novel de novo heterozygous nonsense mutation in exon 2 of SALL4 gene (c.712 C > T:p.Q238X). Subsequently, segregation and phenotype-genotype correlation analysis as well as in-silico approaches confirmed the autosomal dominance inheritance and disease-causing nature of the identified mutation. In addition, studied patient had features not described previously, including kyphoscoliosis, dimple presacral sinus, barrel chest and artric disc (C6–C7). These manifestations could be additional characteristics of the growing phenotypic spectrum of SALL4 related disorders.
Conclusion
Our findings could extend the pathogenic mutations and phenotypic spectrum of SALL4 related disorders. Such reports can also aid to conduct genetic counseling, prenatal diagnosis and clinical management for individuals at high risk of SALL4 related disorders.
Similar content being viewed by others

Background
The SALL genes, encoding zinc-finger transcription factors, are homologous to the Drosophila spalt gene and also present in mice. Four of which have been identified in humans, so far [1, 2]. The researchers have found that SALL4 gene expression is higher in the embryonic stage, while its expression decreases in adulthood and is limited to the testis and ovaries [3]. On the other hand, Yong et al. [4] through loss of function studies on SALL4 realized the vital role of this gene in cell survival and tumorigenesis. The SALL4 gene, with four exons (3159 bp) and 18.14 kb length, is located on 20q13.13–q13.2 region, and encodes a homonymous transcription factor SALL4 with 1053 amino acids [5, 6]. This protein contains eight zinc finger motifs that three highly conserved C2H2 double zinc finger domains of the protein are evenly distributed, a single C2H2 motif is attached to the second domain and at N-terminal region contains an isolated C2H2 motif [7].
SALL4-related disorders show autosomal dominant inheritance pattern, so that haploinsufficiency of SALL4 gene leads to Duane-radial ray (DRR), IVIC, Acro-renal ocular (ARO), and rarely, Holt-oram syndromes with approximately 40–50% of cases being caused by a de novo mutations [8, 9]. ARO syndrome is characterized by eye defects mainly as colobomas, radial ray malformations, and renal abnormalities [10]. DRR syndrome, also known as Okihiro syndrome, has highly variable clinical manifestations and shows incomplete penetration [11]. Additionally, it is worth mentioning that 70% of radial ray defects are associated with other abnormalities (syndromic) and 30% are isolated [12]. Duane malformation and radial ray disorders such as the limbs (especially thumb), hearing loss and anomalies of renal and anorectal, identified in this syndrome [5]. In patients with SALL4 mutation, radial ray abnormalities account for 91.3%. In addition, duane retraction syndrome (DRS) is a congenital anomaly that leads to impaired eye movement due to abnormal growth of the cranial nerve VI [13]. About 1–5% of all cases of strabismus involve duane retraction syndrome [11]. IVIC syndrome was first identified in 1980 through allelic heterogeneity in the SALL4 gene of six generations of a Venezuelan family with autosomal dominant pleiotropic traits. In general, the clinical manifestations reported in patients with IVIC syndrome are similar to those of DRRS affaceted patients, but phenotypes such as mild thrombocytopenia, leukocytosis (before age 50), and sensori-neural or conduction deafness, or both have not been observed in Okihiro syndrome. Therefore, it is difficult to distinguish between these two syndromes [10, 14]. However, all these syndromes with various names and terminologies are actually the same entity called SALL4 related disorders.
In this study, we describe a novel de novo heterozygous nonsense mutation in SALL4 gene segregating with duane radial ray syndrome in an Iranian family. Furthermore, the present study further expands the clinical symptoms associated with DRRS by describing the novel abnormal phenotypes manifested in the affected individual. A review of the literature is also provided. Beyond any doubt, such studies show that rapid progress in the field of high throughout sequencing allows more accurate and less expensive diagnosis of rare inherited disorders [15,16,17].
Case presentation
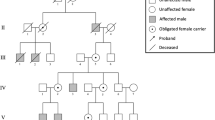
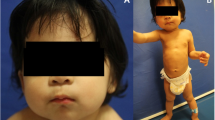
At the present study we explored the molecular mechanism of pathogenesis in a patient suspected to duane radial ray syndrome. The nonconsanguineous pedigree with an affected girl was recruited (Fig. 1a). The studied family is of Iranian origin located in Semnan province (central Iran). As shown in Fig. 1, proband was a 4-year-old girl with clinical symptoms of DRRS who results from a non-consanguineous marriage. Clinical and physical examinations of the patient showed craniofacial deformity, mild microcephaly, cleft palate, thumb aplasia of left hand, thumb hopoplasia of right hand (Fig. 1c), mild syndactyly in right hand, bilateral club foot and short leg, kyphoscoliosis, barrel chest, deep presacral sinus, anorectal abnormality, anal stenosis, constipation, and fecal incontinence.
Echocardiography diagnosed the patient with atrial septal defect. Additionally, the patient underwent audiometric and ophthalmic evaluation. This assessment showed moderate and severe hearing impairment in the right and left ear (Fig. 1b), respectively, as well as type III duan anomaly as deep-set eyes with partial strabismus of right eye. Cleft palate was surgically repaired. She did not show any impairments in the ultrasound evaluation of the kidney, liver, gallbladder and bladder. Transfontanelle ultrasound showed no evidence of hydrocephalus, as well. In addition, Fig. 1c shows the phenotypes of craniofacial deformity, mild microcephaly, mild syndactyly and thumb abnormalities in patient III-1. On the other hands, the patient underwent MRI of the brain, which showed normal structure in the supra, infratentorial, and midline areas. The cortical and white matter signals of the brain, pituitary gland, parasellar regions, base of skull, orbits and 7-8th nerve complexes were also unchanged. MRI of the cervical spine and thoracolumbar spine showed a partial atretic disc at the C7-C6 leve and the presence of lipoma at the end of the filum, however, other regions were normal. According to the experiment performed at 4 year of age of patient, the concentrations of IGF1, TSH and T4 were in the normal range (101 ng/ml, 3.4 µIU/ml and 10.5 µg/dl respectively), but the T3 resin uptake was slightly higher than the normal range (37.4%).

Family pedigree and results of audiologic evaluation of the proband (III-1). a Pedigree and segregation analysis of c.712 C > T: p.Q238X mutation in the SALL4 gene. Sanger sequencing displayed that patient (III-1) is heterozygous and her healthy parents (II-4 and II-5) are normal for the identified mutation. b Audiograms of the affected proband which were obtained using pure tone audiometry with air conduction from frequencies 250 to 8000 Hz. c Patient's dysmorphic phenotypes. Left thumb aplasia, hypoplasia of right thumb, and mild syndactyly are evident
After obtaining informed consent, karyotype test was performed for the patient. Since no chromosomal abnormalities were detected, genomic DNA was extracted from blood sample of patient and her healthy parents using QIAamp DNA Blood Mini Kit (Germany) according to the manufacturer's instructions. To investigate the genetic cause of the wide range of abnormalities reported in the affected patient, whole-exome sequencing was used to enrich all exons of the protein-coding genes and a few other important genomic regions. The WES was performed for about 100 million reads, using the Illumina Hiseq2000 sequencer platform and Agilent SureSelect Human All Exon V7 kit (Agilent, Santa Clara, USA). The output raw data were converted from bcl to fastq files by Bcl2Fastq software (Bcl2Fastq 2.18.0.12; Illumina, Inc.). Then, Illumina sequencing adapters and all lowquality reads (< 80 bp) were filtered using fastp software (https://github.com/OpenGene/fastp). The clean reads (fastqc files) were mapped to the UCSC hg19 human reference genome using BWA software (v0.7.12r1044; http://biobwa.sourceforge.net/). Subsequently, the duplicated reads were removed using Picard software (v2.2.3; http://broadinstitute.github.io/picard/) and mapped reads were used for the detection of variants/mutations. For this purpose, data filtering was first performed based on frequency and then according to intronic, upstream, downstream, 3ʹ-UTR, 5ʹ-UTR, intergenic and other non-coding variants. At final step, synonymous mutations were also fltered. In general, test platform examined more than 95% of the targeted regions with sensitivity of > 99%. The results of WES were analyzed by bioinformatics tools BWA aligner [18], GATK [19], and Annovar [20] and public databases ClinVar, gnomAD, Kaviar, and GME. In addition, ACMG (American College of Medical Genetics) guidelines and local population database (BayanGene) with more than 4000 unrelated individuals were utilized. As control, 250 healthy individuals with the same ethnicity as the studied patient were also screened for the mutation found. Online bioinformatics tools MutationTaster, SIFT, and CADD_phred software were used to predict the likely pathogenic effects of the mutation. To investigate the effect of identified mutation on the SALL4 protein in terms of possible structural and functional changes compared to wild-type protein, SMART tool was used. In order to confirm the new mutation found, PCR and Sanger sequencing was performed. Primers was designed using the Primer3 program (https://primer3.ut.ee) as: F-5ʹ GGCTTCCAGCTTTCTGGCTG 3ʹ and R-5ʹ ACCAAGGTGGCGGTGAATCAG 3ʹ (PCR product: 280 bp). Subsequently, Chromas software was applied to analyze the results of Sanger sequencing.
According to WES technique results, we identified a novel heterozygous stop-gain mutation in exon 2 of SALL4 (NM_001318031); chr20:51791771: c.712 C > T: p.Q238X in the proband (patient III-1). The review of public databases and the local population database (bayangen) did not identify any previous reports of the identified mutation in the SALL4 gene. In addition, none of the 250 healthy individuals (controls) with the same ethnicity as the studied patient showed the identified mutation, emphasizing the novelity of the mutation found. The identified mutation was predicted to be disease-causing by in silico approaches (Table 1). Furthermore, genotype-phenotype correlation analysis matched the observed phenotypes in patient with the detected novel mutation in the SALL4 gene. As shown in Fig. 1a, the Sanger sequencing confirmed the existence of the mutation in the proband, and its absent in her healthy parents showing de novo origin of the mutation found. After audiological evaluation, moderate conductive hearing impairment was reported in the right ear and severe mixed hearing impairment in the left ear of the proband. In this assessment, the pure tone of the patient was measured at 250–8000 Hz (Fig. 1b). These data suggest that this new mutation could be the cause of DRR and IVIC syndromes with a wide range of abnormalities in the studied patient. As depicted in Fig. 2, the SALL4 protein has eight zinc finger motifs: three C2H2 double zinc finger domains which are highly conserved, the second of which has a single C2H2 zinc finger attached at its C-terminal end, and also an N-terminal C2HC zinc finger motif [6].

Schematic representation of the SALL4gene/protein structure. a Previous SALL4 mutations were reported and the new mutation found (c.712 C > T;p.Q238X) is shown with a different color. b SALL4 consists of three C2H2 double zinc finger domains (orang) and two seperated C2H2 motifs (blue)
Discussion and conclusions
In this study, we identified a new SALL4 mutation (NM_001318031: exon 2: c.712 C > T: p.Q238X) in an Iranian affected patient suspected to DRRS. As far as we know, this is the first genetic study with respect to the SALL4 gene mutations in Iranian populations. The identified mutation leads to the substitution of glutamine 238 with a stop codon (Q238X) in the SALL4 protein. Hence, this substitution alters the amino acid sequence and leads to a premature stop codon at position 238 with the complete loss of 819 out of 1053 amino acids in the wild type protein sequence. The deleted sequence contains 7 out of 8 C2H2 zinc finger domains including three highly conserved C2H2 double zinc finger domains and a single C2H2 zinc finger domain (Fig. 3) resulting to disruption of whole protein structure and function. In fact, this mutation is predicted to be responsible for the disease pathogenesis by either a truncated SALL4 protein affecting 7 functional C2H2 zinc finger domains or haploinsufficiency due to nonsense-mediated mRNA decay.

Schematic illustration of gene and protein changes due to nonsense mutation c:712 C > T:p.Q238X in SALL4gene. a The normal sequence of the gene and the corresponding amino acid sequence have displayed on the left. The nucleotide conversion of C to T is shown in different colors (yellow). The altered sequence and the corresponding truncated protein are depicted on the right. b 7 out of 8 C2H2 zinc finger domains of SALL4 protein are deleted due to novel de novo nonsense mutation c:712 C > T (p.Q238X)
As previously pointed out, the SALL4 gene belongs to a group of evolutionarily conserved genes called the spalt transcription factor family, which plays a critical role in regulating embryonic growth in many organisms. In addition, SALL4 transcription factor is known as a homeotic factor which is essential for the early growth of the posterior head and anterior tail regions in Drosophila [21]. As mentioned earlier, in human, mutations in the SALL4 gene can cause DRR, IVIC, ARO, and HO syndromes that are associated with various malformations of several organ systems [10]. Based on observed phenotypes in the studied patient which previously listed in the patient section, these characteristics are highly matched with DRRS. Although, renal abnormalities have also been usual in affected patients [1, 11, 22], but this feature was not present in the patient we studied. Interestingly, new phenotypic features including kyphoscoliosis, dimple presacral sinus, barrel chest and artric disc (C6–C7) were also observed in our patient. However, it seems that all SALL4 gene related disorders are allelic diseases with highly overlap phenotypes. Therefore, from now on, it is better to use the term SALL4-related disorders instead of referring to a specific disorder caused by the SALL4-gene mutation. In the case of our patient, since her parents did not show the mutation found, the origin of the mutation was de novo.
So far, 43 pathogenic and likely pathogenic variants have been reported in patients from Venezuela [23], Brazil [22], Germany [24], China [6, 25], Italy [26], Chile [3] and Turkey [12]. Recently, a frameshift mutation in exon 4 has been reported that leading to increasing in the length of the SALL4 protein from 1053 to 1076 amino acids [25]. However, like more than 73% of the mutations found in the SALL4, which occur mainly in the exon 2 of the gene, the mutation found in this study (as the forty-fourth identified mutation) is also located in exon 2. It confirms that the exon 2 is hotspot of mutation throughout the SALL4 gene. However, it is not yet clear why different mutations in this pleiotropic gene lead to extremely variable characteristics in different affected patients. Except the type of SALL4 mutation, it seems that involvment of SALL4 transcription factor in epigentics phenomena by recruiting the nucleosome remodeling and histone deacetylase complex (NuRD) [27], its different interactions with distinct genes and proteins [2, 28] and thus the genetic background of the SALL4 dependent protein network/pathways in patients are at least part of the answer to this question. Therefore, considering the precise pathogenesis mechanism and upstream/downstream target genes of SALL4 is of great importance in future studies to elucidate the exact cause of highly variable clinical symptoms of SALL4 related patients.
In conclusion, we have identified a novel pathogenic heterozygous SALL4 mutation in an Iranian family. The patient showed wide range of SALL4 related disorders. In addition, new reported manifestations including kyphoscoliosis, dimple presacral sinus, barrel chest and artric disc (C6–C7) were expanded the phenotypic range of SALL4 mutations. However, future reports would be essential in order to confirm these novel manifestations of the mutation found. Regarding to critical functions of SALL4 transcription factor in a wide variety of biological processes, future studies on either genetics or epigenetics aspects of upstream and downstream rugulations of SALL4 might shed light the exact cause of these extremely variable characteristics in affected patients. Altogether, the results of this research provide a better understanding of SALL4 mutations on phenotypic outcome and strengthens the clinical importance of this gene in affected patients of SALL4 related disorders.
Availability of data and materials
The identified variant in this research is accessible on the ClinVar repository under accession number "SCV002550878" for the SALL4 gene mutation. Also, in addition to the pathogenic mutation found in SALL4 gene, all other filtered variants/mutations found are available in the Additional file 1.
Abbreviations
- SALL4 :
-
Spalt like transcription factor 4
- DRRS:
-
Duane-radial ray syndrome
- ARO:
-
Acro-renal ocular
- HOS:
-
Holt-oram syndrome
- AROS:
-
Acro-renal ocular syndrome
- IVIC:
-
Instituto Venezolano de Investigaciones Cientificas
- WES:
-
Whole-exome sequencing
- DRS:
-
Duane retraction syndrome
- ACMG:
-
American College of Medical Genetics
References
Borozdin W, Boehm D, Leipoldt M, Wilhelm C, Reardon W, Clayton-Smith J, et al. SALL4 deletions are a common cause of Okihiro and acro-renal-ocular syndromes and confirm haploinsufficiency as the pathogenic mechanism. J Med Genet. 2004;41(9):1–9.
Sakaki-Yumoto M, Kobayashi C, Sato A, Fujimura S, Matsumoto Y, Takasato M, et al. The murine homolog of SALL4, a causative gene in Okihiro syndrome, is essential for embryonic stem cell proliferation, and cooperates with Sall1 in anorectal, heart, brain and kidney development. Development. 2006;133(15):3005–13.
Huserman J, Diaz C. Duane-radial ray syndrome a SALL4-related disorder. Report of a case in Chile. Glob J Rare Dis. 2020;5(1):022–4.
Yong KJ, Gao C, Lim JSJ, Yan B, Yang H, Dimitrov T, et al. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med. 2013;368(24):2266–76.
Cox R, Bouzekri N, Martin S, Southam L, Hugill A, Golamaully M, et al. Okihiro syndrome is caused by SALL4 mutations. Hum Mol Genet. 2002;11(23):2979–87.
Yang MM, Ho M, Lau HHW, Tam POS, Young AL, Pang CP, et al. Diversified clinical presentations associated with a novel Sal-like 4 gene mutation in a chinese pedigree with Duane retraction syndrome. Mol Vis. 2013;19:986–94.
Kohlhase J, Chitayat D, Kotzot D, Ceylaner S, Froster UG, Fuchs S, et al. SALL4 mutations in Okihiro syndrome (Duane-radial ray syndrome), acro-renal-ocular syndrome, and related disorders. Hum Mutat. 2005;26(3):176–83.
Kohlhase J. SALL4-Related Disorders. GeneReviews. University of Washington. 2015. https://www.ncbi.nlm.nih.gov/books/NBK1373/. Accessed 28 July 2021.
Matyskiela ME, Couto S, Zheng X, Lu G, Hui J, Stamp K, et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat Chem Biol. 2018;14(10):981–7.
Becker K, Beales PL, Calver DM, Matthijs G, Mohammed SN. Okihiro syndrome and acro-renal-ocular syndrome: clinical overlap, expansion of the phenotype, and absence of PAX2 mutations in two new families. J Med Genet. 2002;39:68–71.
Chacón-Camacho OF, Cabral-Macías J, Ayala-Ramírez R, Arteaga-Vázquez J, Svyryd Y, Helmes K, et al. Clinical and genetic findings in Mexican patients with Duane anomaly and radial ray malformations/okihiro syndrome. Rev Inves Clin. 2016;68:269–74.
Avci S, Toksoy G, Bagirova G, Altunoglu U, Karaman B, Basaran S, et al. Clinical classification of radial ray defects and research into etiopathogenesis. J Istanb Facul Med. 2018;81(4):127–38.
Borozdin W, Graham JM, Böhm D, Bamshad MJ, Spranger S, Burke L, et al. Multigene deletions on chromosome 20q13.13-q13.2 including SALL4 result in an expanded phenotype of Okihiro syndrome plus developmental delay. Hum Mutat. 2007;28(8):830.
Arias S, Penchaszadeh VB, Pinto-Cisternas J, Larrauri S. The IVIC syndrome: a new autosomal dominant complex pleiotropic syndrome with radial ray hypoplasia, hearing impairment, external ophthalmoplegia, and thrombocytopenia. Am J Med Genet. 1980;6(1):25–59.
Fahimi H, Behroozi S, Noavar S, Parvini F. A novel recessive PDZD7 bi-allelic mutation in an iranian family with non-syndromic hearing loss. BMC Med Genom. 2021;14(1):4–11.
Noavar S, Behroozi S, Tatarcheh T, Parvini F, Foroutan M, Fahimi H. A novel homozygous frame-shift mutation in the SLC29A3 gene: a new case report and review of literature. BMC Med Genet. 2019;20(1):4–10.
Karimi AH, Karimi MR, Farnia P, Parvini F, Foroutan M. A homozygous truncating mutation in NALCN causing IHPRF1: detailed clinical manifestations and a review of literature. Appl Clin Genet. 2020;13:151–7.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26(5):589–95.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):1–7.
Wang Q, Li D, Cai B, Chen Q, Li C, Wu Y, et al. Whole-exome sequencing reveals SALL4 variants in premature ovarian insufficiency: an update on genotype–phenotype correlations. Hum Genet. 2019;138(1):83–92.
Alves LU, Perez ABA, Alonso LG, Otto PA, Mingroni-Netto RC. Novel frameshift variant in gene SALL4 causing Okihiro syndrome. Eur J Med Genet. 2016;59(2):80–5.
Paradisi I, Arias S. IVIC syndrome is caused by a c. 2607delA mutation in the SALL4 locus. Med Genet. 2007;332:326–32.
Kohlhase J, Schubert L, Liebers M, Rauch A, Becker K, Mohammed SN, et al. Mutations at the the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. J Med Genet. 2003;40:473–8.
Ma X, Huang R, Li G, Zhang T, Ma J. A de novo mutation of SALL4 in a chinese family with Okihiro syndrome. Mol Med Rep. 2022;25(4):1–7.
Miertus J, Borozdin W, Frecer V, Tonini G, Bertok S, Amoroso A, et al. A SALL4 zinc finger missense mutation predicted to result in increased DNA binding affinity is associated with cranial midline defects and mild features of Okihiro syndrome. Hum Genet. 2006;119(1–2):154–61.
Kong NR, Bassal MA, Tan HK, Kurland JV, Yong KJ, Young JJ, et al. Zinc finger protein SALL4 functions through an AT-rich motif to regulate gene expression. Cell Rep. 2021;34(1):108574.
Lu J, Jeong H, Kong N, Yang Y, Carroll J, Luo HR, et al. Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS One. 2009;4(5):1–13.
Acknowledgements
We are so grateful to patient and her respected family who kindly consented to join the study. We thank Dr. Bahar Ashjaei (from Children's Hospital Medical Center, Tehran University of Medical Science, Tehran, Iran), Dr. Bahar Allahverdi (from Childrens Growth and Development Research Center, Tehran University of Medical Science, Tehran, Iran), Dr. Seyede Mona Salehi (Radiologist, Semnan, Iran) and Mr. Mohammad-Reza Erfanian (Audiologist, Semnan, Iran) for technical collaborations, as well. The authors also thank Semnan University and Kharazmi University for their facilities and cooperation.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
MAH performed experimental assays, literature review and drafted the manuscript. FP and AA, organized this research, performed data analysis, reviewed clinical and laboratory data, and finalized this manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Consent for publication
Written informed consent for publication was obtained from parents and father of the patient, as her legal guardian.
Ethics approval and consent to participate
This research had been approved by the ethics committee of the pharmaceutical sciences branch of Islamic Azad University, Tehran, Iran (ethics approval code no. IR.IAU.PS.REC.1396.91). Written informed consent was obtained from parents and father of the patient, as her legal guardian, to participate in this study. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
All variants detected in the studied patient.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ajam-Hosseini, M., Parvini, F. & Angaji, A. A novel de novo nonsense mutation in SALL4 causing duane radial ray syndrome: a case report and expanding the phenotypic spectrum. BMC Med Genomics 16, 33 (2023). https://doi.org/10.1186/s12920-023-01467-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01467-1