
Abstract
Introduction
Dilated cardiomyopathy (DCM) is characterized by the dilation and impaired contraction of 1 or both ventricles and can be caused by a variety of disorders. Up to 50% of idiopathic DCM cases have heritable familial diseases, and the clinical screening of family members is recommended. Identifying a genetic cause that can explain the DCM risk in the family can help with better screening planning and clinical decision-making. Whole-exome sequencing (WES) has aided significantly in the detection of causative genes in many genetically heterogeneous diseases. In the present study, we applied WES to identify the causative genetic variant in a family with heritable DCM.
Methods
WES was applied to identify genetic variants on a 26-year-old man as the proband of a family with DCM. Subsequently, Sanger sequencing was performed to confirm the variant in the patient and all the available affected and unaffected family members. The pathogenicity of the variant was evaluated through co-segregation analysis in the family and employment of in silico predictive software.
Results
WES demonstrated the missense pathogenic heterozygous nucleotide variant, c.1907G > A, (p.Arg636His, rs267607004, NM_0011343), in exon 9 of the RBM20 gene in the proband. The variant was co-segregated in all the affected family members in a heterozygous form and the unaffected family members. The in silico analysis confirmed the variant as pathogenic.
Conclusion
Pathogenic RBM20 nucleotide variants are associated with arrhythmogenic DCM. We believe that our report is the first to show an RBM20 variant in Iranian descent associated with DCM.
Similar content being viewed by others


Introduction
Dilated cardiomyopathy (DCM), a myocardial disorder, is characterized by enlarged cardiac ventricles and impaired systolic function, leading to heart failure and premature death [1, 2]. DCM affects 1 in 250–500 people in the general population and is one of the most common forms of inherited cardiomyopathies [3, 4]
DCM phenotypes are determined by genetic and nongenetic causes [5]. Nongenetic forms of DCM may occur with hypertension, valvular heart disease, viral/inflammatory myocarditis, extravagant alcohol consumption, illicit substance use, toxins, and metabolic diseases [6,7,8,9]. Genetic variations play a prominent role in the pathogenesis of DCM. Currently, laboratories in the United States and Europe offer different panels ranging from 30 to more than 150 DCM-related genes; nonetheless, most of the genes are only anecdotally associated with the disease or have a putative link on the basis of biological connections with known genes [10, 11]. Moreover, pathogenic gene variants can be determined in 25 to 40% and 10 to 25% of familial and sporadic DCM cases, respectively [6, 12, 13]. In this regard, familial DCM primarily demonstrates an autosomal dominant inheritance pattern, in which a single pathogenic mutation can cause the disease [14, 15]. Autosomal recessive and X-linked inheritances have been shown as well [13].
Whole-exome sequencing (WES) is a viable and powerful technique that beyond traditional candidate gene and locus-mapping methods provides an efficient approach to find the genetic cause of heterogeneous diseases [16, 17] like DCM [18].
In this study, WES was performed on an Iranian family with an autosomal dominant inheritance pattern of DCM.
Methods
Subjects
Clinical data were acquired via a thorough cardiac assessment of each individual in the Outpatient Heart Failure Clinic of Rajaie Cardiovascular Medical and Research Center. The diagnosis of DCM was made according to the diagnostic criteria of the European Society of Cardiology (ESC) Working Group on Myocardial and Pericardial Diseases [9]. Cardiac magnetic resonance images and echocardiography and electrocardiography data, as well as blood samples, were obtained.
The study was approved by the Ethics Committee of Rajaie Cardiovascular Medical and Research Center (IR.RHC.REC.1399.078) and was conducted in accordance with the Helsinki Declaration. The proband and 9 of his family members (affected and unaffected) were enrolled in the study.
DNA extraction and WES
Genomic DNA was isolated from 200 µL of peripheral blood samples from the proband and all the available family members in the pedigree by using a DNA extraction kit (DNPTM Kit, Iran).
WES was done with the SOLIDv4 platform (SureSelect X kit, Macrogen South Korea) following the manufacturer’s instructions. Sequences obtained from WES were aligned to the GRCh37/hg19 human reference genome, and the WES-identified variants were filtered. Briefly, filtering was performed as follow: variants were filtered due to minor allele frequency (MAF) of 1000 Genomes, ExAc, gnomAD, esp6500, Greater Middle East, and Iranome databases, respectively. Then bioinformatics software platforms including Combined Annotation Dependent Depletion (CADD Phred > 15) (https://cadd.gs.washington.edu/), PolyPhen-2 (score = 0–0.15: Benign; score = 0.15–0.85: Possibly damaging; score = 0.85–1:Probably damaging) (http://genetics.bwh.harvard.edu/pph2/), SIFT (score ≤ 0.05: Deleterious; score > 0.05: Tolerable) (https://sift.bii.a-star.edu.sg/), PROVEAN (score ≤ -2.5: Deleterious; score > -2.5: Natural.) (http://provean.jcvi.org/index.php), and MutationTaster (http://www.mutationtaster.org/), were employed to predict the variant’s pathogenicity. The identified variant was classified (graded) in accordance with the American College of Medical Genetics and Genomics (ACMG) guidelines.5
Polymerase chain reaction (PCR), primer design, and sanger sequencing
The confirmation of the putative pathogenic variant detected by WES was carried out via Sanger sequencing with a 3500 Genetic Analyzer (Applied Biosystems, USA) in the proband and all the available family members. Briefly, specific primers were designed to amplify a 940-bp fragment encompassing the candidate pathogenic variant in RBM20 with the following sequences: forward primer: 5'-TGTGTGGTTCTGTAGAGTTGGG-3' and reverse primer: 5'-CCTAGCGCATAGTAAATAGCCAG -3'.
The cycling conditions for amplifying the region were as follows: initial denaturation at 94 °C for 5 min, followed by 30 cycles of 94 °C for 30 s, 63 °C for 30 s, and 72 °C for 30 s, with a final extension at 72 °C for 10 min. Then, the forward primer was utilized to sequence the part of interest.
Bioinformatics analysis
Gene Runner (Gene Runner 6.5.50) was used to design the primers. The sequencing results were analyzed by using BioEdit (BioEdit 7.2.1). The identified nucleotide variant was studied through the UCSC Genome Browser (https://genome.ucsc.edu) and ClinVar (www.ncbi.nlm.nih.gov/clinvar) and Iranome (http://www.iranome.ir/)databases.
Results
Clinical information
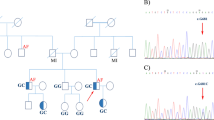
The family of interest comprised a 2-generation pedigree with DCM (Fig. 1). A positive family history of DCM, myocardial infarction, and sudden cardiac death was reported from the maternal side of the family.

The pedigree of the index family with hereditary dilated cardiomyopathy is illustrated herein. The green-filled square and circle indicate affected males and females, respectively. The gray squares and circles with diagonal lines indicate the deceased males and females, respectively. A thick red arrow in the pedigree specifies the proband. For the pathogenic nucleotide variation in RBM20, c.G1907A, the wild type allele is shown by G, and the potentially pathogenic variant is indicated by A. The genotypes pinpoint the co-segregation of c.G1907A (p.R636H) in the heterozygous form (GA) in all the affected members (III-2, IV-4, and IV-7), whereas the 2 unaffected family members (III-10 and IV-2) were wild type homozygous states (GG)
The proband in the family (IV-7) was a 26-year-old man who presented initially with retrosternal chest pain, a slightly elevated serum troponin level, and mild left ventricular systolic dysfunction. He denied any palpitation, lightheadedness, syncope, shortness of breath, or weakness. His past medical history was also unremarkable. Of note, the proband’s mother (III-4) and maternal uncle (III-5) had passed away (sudden death), and his maternal aunt was a known case of nonischemic DCM.
One of the proband’s cousins, a 33-year-old woman (IV-4), was also under medical care because of frequent premature ventricular ectopy and mildly reduced left ventricular systolic function. Electrocardiography showed normal sinus rhythm with normal QRS morphology, normal QT intervals, and no pathologic ST-segment or T-wave changes.
Transthoracic echocardiography revealed a normal left ventricular size, mildly reduced systolic function (ejection fraction: 45–50%), and no significant valve problems. Additionally, diastolic function and pulmonary artery pressure were normal. The patient also had reduced longitudinal strain, especially in the basal myocardial segments, with a global longitudinal strain value of − 6.1%.
Cardiac magnetic resonance imaging confirmed the mildly reduced systolic function (ejection fraction: 44%) and showed a left ventricular end-diastolic volume index of 121 mL/m2. The right ventricle was mildly enlarged, with a right ventricular end-diastolic volume index of 113 mL/m2 and a right ventricular ejection fraction of 40%. Tissue characterization revealed no remarkable myocardial edema or fibrosis.
A 24-h Holter monitoring test was negative for any sustained supra- or ventricular arrhythmia.
With the impression of familial DCM, the patient was started on a goal-directed medical treatment, including the administration of bisoprolol, spironolactone, and sacubitril/valsartan. Empagliflozin was added to the medications subsequently. He was also counseled concerning the implantation of a cardioverter-defibrillator considering his high-risk family history (sudden death in the mother and the uncle); however, he refused to undergo the procedure. He was compliant with his medications for 6 months and remained stable clinically with no symptoms of chest pain, shortness of breath, palpitation, or syncope. Still, he refused to continue with the goal-directed medical treatment. (He found the treatment futile and avoided medical follow-up for fear of the ongoing COVID-19 pandemic.) Eleven months after the diagnosis, he sought medical care because of coffee-ground emesis, which was evaluated via esophagogastroduodenoscopy. A course of pantoprazole was prescribed. Two months later and in a physical altercation, the patient suddenly collapsed and passed away.
The other enrolled family members (III-10, IV-b2, IV-5, IV-6, IV-8, and IV-9) were apparently normal. Screening echocardiography was performed on all these individuals and showed a borderline ejection fraction (50%–55%) and a mildly reduced global longitudinal strain value (− 19.7%) in the 22-year-old sister of the patient (IV-8).
Genetic finding
An unbiased next-generation DNA sequencing, encompassing the entire coding exons (WES), was carried out to scrutinize the causative genetic variation in the family. WES was accomplished with a mean target coverage rate of 100 × . The results were consecutively analyzed with Bowtie [19], FreeBayes [20], and SnpSift. Regarding to databases filtering and pathogenicity evaluation, three variants were chosen for further analysis.
The WES outcome revealed a missense pathogenic/likely pathogenic variant, c.1907G > A (rs267607004, NM_0011343), in the RBM20 gene in the index patient (Fig. 2) along with two other nucleotide variations (Additional file 1: Table S1). The c.1907G > A transition was harbored in a very highly conserved part of exon 9 of the RBM20 gene located on Chr10: 112,572,062 (GRCh37/hg19), resulting in an Arg636-to-His substitution in the arginine-serine (RS) domain (Fig. 2). This variant is reported neither in the 1000 Genomes Project nor in Iranome databases.

The image depicts the identification of a RBM20 nucleotide change in a family with dilated cardiomyopathy. A The image shows the confirmation of the variant by Sanger sequencing. B and C The images demonstrate the genomic organization of the human RBM20 gene and present a schematic representation of the RBM20 protein with the predicted functional domains, and amino acid alignment of the RBM20 arginine-serine–rich domain among vertebrate
The in silico analysis of this variant through CADD (25.2), PolyPhen-2 (D), SIFT (D), PROVEAN (D), and MutationTaster (D) predicted that it would be damaging. This variant also met the ACMG criteria (PM1, PM2, PM5, PP3, and PP5) and was itemized as a pathogenic variant.
Sanger sequencing of this variant was performed in the proband and all 8 available family members in order to confirm the presence and the pattern of inheritance. The Sanger sequencing upshots demonstrated that the index patient (IV-7) and 1 of his aunts (III-2, who had an implantable cardioverter-defibrillator) carried the pathogenic variant in a heterozygous status (GA), while the other unaffected aunt (III-10) did not carry this pathogenic variant (GG) (Fig. 1).
The proband’s brother (IV-9) and older sister (IV-6) were a wild type for the variant. In addition, his younger sister (IV-8) also carried the variant in a heterozygous form and presented with a borderline left ventricular ejection fraction (50%–55%) and a mildly reduced global longitudinal strain value (− 19.7%) in her echocardiographic examination.
The proband’s aunt (III-2) had 3 children. Two of them (1 boy [IV-5] and 1 girl [IV-4]) were genetically heterozygous for the RBM20 variant (GA), while the other daughter (IV-2) was a wild type for the variant (Fig. 1). The proband’s male first cousin (IV-5), 30 years of age, was a carrier of the pathogenic variant, but he exhibited no DCM symptoms. In contrast, the proband’s female first cousin (IV-4), 33 years old, was a carrier of the pathogenic variant and needed an implantable cardioverter-defibrillator due to ventricular arrhythmias.
Discussion
DCM can be caused by mutations in genes that mostly encode the cytoskeletal, sarcomeric, or contractile proteins of myocytes [6, 21, 22]. These genes are almost directly involved in the generation and/or transmission of contractile force via protein–protein interactions. In addition, pathogenic mutations in unsuspected DCM genes like LMNA and EYA4 lead to DCM as well [23, 24]. Nucleotide transitions in Lamin A/C, encoded by LMNA with unknown mechanisms, cause DCM and conduction system disorders [23,24,25]. Further mutations in EYA4, a transcriptional coactivator, could cause syndromic DCM with sensorineural hearing loss [24]. Accumulating evidence indicates that alternative splicing perturbations contribute to cardiac diseases. Some splicing factors, including RBM20, RBM24, Rbfox, and SF3B1, play critical roles in developing adult cardiac tissue [26,27,28,29]. In 2009, RBM20 was introduced as a familial DCM gene [30]. More recently, RBM20 nucleotide variation have been found in familial and sporadic DCM cases [31]. Due to Iranome database 57 exonic nucleotide variants of RBM20 gene (Additional file 2: Table S2) were reported in healthy Iranian population. Additionally, 106 and 2 nucleotide variations were announced in introns and 3’-UTR of RBM20 gene.
In the current investigation, with the aid of WES, we succeeded in identifying a pathogenic variant, c.1907G > A (rs26760704), in the RBM20 gene in the members of an Iranian family with DCM. To the best of our knowledge, this is the first report of a pathogenic nucleotide variant in the RBM20 gene in Iranian patients with DCM.
The RBM20 gene, composed of 14 exons, encodes the RNA-binding motif protein of 1227 amino acid residues in several conserved domains, including -a arginine/serine (RS)-rich region just following the prototype RNA recognistion motif (RRM) domain (Fig. 2) [26, 32, 33]. The RBM20 gene has an extremely high expression in both atria and ventricles [34].
RBM20 is a key heart splice regulator that controls the process of some important transcripts. It exists in considerable amounts in striated muscles, with the upper level of expression in cardiac tissue. [35] Previous studies have demonstrated the mislocalization of RBM20 due to the aberrant phosphorylation of the RSRSP domain. [33, 36] Therefore, patients with heterozygous forms of mutations in the RS domain have lower concentrations of RBM20 in the nucleus, leading to functional deficiencies of RBM20 (Fig. 3). [36] The mis-splicing of some targets of RBM20 in consequence of mutations in the RBM20 gene results in progressive DCM with conduction diseases, including atrial and ventricular arrhythmias. Although the exact mechanism of RBM20 remains to be investigated, numerous pathogenic nucleotide transition variants, in conjunction with a mutational hot spot in an RS-rich domain, have been reported to be the causes of DCM. [31]

The image shows the mislocalization of RBM20 due to a pathologic nucleotide variation in the RS domain. In the wild-type form, all RBM20 proteins are transferred from the cytoplasm to the nucleus after phosphorylation. In contrast, in the mutant form, phosphorylation is abrupt, and most RBM20 proteins aggregate in the cytoplasm
Pathogenic nucleotide variations in the RS domain result in the disruption of binding with other splicing factors. Hence, these mutations have the potential to impair the normal processing of the splicing of transcripts such as Titin, culminating in highly penetrant DCM. [37, 38]
A hot-spot region in RBM20 includes an arginine-serine-arginine-serine-proline (RSRSP) stretch in 634 to 638 amino acids in the RS-rich region domain. Most RBM20 mutations harbored in the RS-rich region are heterozygous missense loss- or reduction-of-function mutations. [30, 31, 39, 40] Earlier investigations demonstrated R634Q, R634W, S635A, R636S, R636H, R636C, S637G, and P638L in familial and sporadic DCM patients [26, 30, 33, 39, 41,42,43]. The R636H mutation has been reported in familial and sporadic forms of DCM in several studies carried out in different countries (Table 1) [30, 39,40,41, 43,44,45,46,47,48,49,50]. Subsequent research on patients carrying the RBM20 mutations confirmed that DCM symptoms manifested themselves at an early age, followed by a high degree of morbidity and mortality even in young children due to the deterioration of cardiac systolic function toward end-stage heart failure [30, 51, 52].
In line with the aforementioned studies, we found an R636H mutation in a hot-spot domain (the RS-region domain) running in a dominant pattern in a family with familial DCM. The proband (the index patient), as well as his sister, aunt, and cousins (the affected aunt’s children), carried the mutant allele in a heterozygous form. Except for a cousin with a c.1907G > A variation, almost all the carriers showed the DCM phenotype. The other family members with the wild type of nucleotide variation were clinically normal, as expected. Unfortunately, the index patient, together with his mother and one of his uncles, passed away due to sudden cardiac death.
Although, previously reported confirmed that patients carrying the RBM20 mutations in RS domain showed the DCM phenotype at an early age with high penetrance along morbidity and mortality. Phenotype heterogeneity in the family specially the severe phenotype of the proband might be due to other heterozygous variants found in TTN (P.G25367S) and RYR1 (p.4265_4268del) which are not co-segregated in other available family members along with the existed pathogenic R636H variant in RBM20. Further experimental studies are needed to clarify the association and the impact of these variants on DCM.
Recently, more than 100 genes have been reported in association with DCM [55, 56]. There is, however, a dearth of information on the genetics of DCM in Iranian patients. Previous investigations have demonstrated associations between DCM and δSG [57], LMNA [58], FLNC [59], desmoplakin (DSP) [60], MYBPC3, troponin T type 2 (TNNT2), myosin heavy polypeptide 7 (MYH7) [61], and PLN [62]. The finding of a mutation in the RBM20 gene in the present study, along with other studies, in Iranian patients highlights the genetic heterogeneity and wide spectrum of mutations in Iranians and underscores the need for further comprehensive studies.
Availability of data and materials
The accession number of the reported variant in paper is available with the following accession number; rs267607004, NM_0011343.The data sets presented in the present study can be accessed in online repositories. The identified nucleotide transitions were investigated through the UCSC Genome Browser (https://genome.ucsc.edu) and ClinVar (www.ncbi.nlm.nih.gov/clinvar) databases. In silico predictive software tools entailing Combined Annotation Dependent Depletion (CADD Phred > 15) (https://cadd.gs.washington.edu/), SIFT (https://sift.bii.a-star.edu.sg), PROVEAN (provean.jcvi.org), PolyPhen-2 (genetics.bwh.harvard.edu/pph2), and MutationTaster (www.mutationtaster.org) were employed to study the pathogenesis of the detected nucleotide variation. The accession number and all the repositories used for the study are both mentioned in the article and declarations part as well.
References
Dadson K, Hauck L, Billia F. Molecular mechanisms in cardiomyopathy. Clin Sci. 2017;131(13):1375–92.
Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B, McKenna WJ. Classification of the cardiomyopathies: a position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. 2008;29(2):270–6.
McKenna WJ, Maron BJ, Thiene G. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res. 2017;121(7):722–30.
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, De Ferranti S, Després J-P, Fullerton HJ, Howard VJ. Executive summary: heart disease and stroke statistics—2015 update: a report from the American heart association. Circulation. 2015;131(4):434–41.
Bozkurt B, Colvin M, Cook J, Cooper L, Deswal A, Fonarow G, Francis G, Lenihan D, Lewis E, McNamara D. American heart association committee on heart failure and transplantation of the council on clinical cardiology; council on cardiovascular disease in the young; council on cardiovascular and stroke nursing; council on epidemiology and prevention; and council on quality of care and outcomes research. Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American heart association. Circulation. 2016;134(23):e579-646.
Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10(9):531–47.
Rapezzi C, Arbustini E, Caforio AL, Charron P, Gimeno-Blanes J, Heliö T, Linhart A, Mogensen J, Pinto Y, Ristic A. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2013;34(19):1448–58.
Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu M, Heliö T, Heymans S, Jahns R. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European society of cardiology working group on myocardial and pericardial diseases. Eur Heart J. 2013;34(33):2636–48.
Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M, Duboc D, Gimeno J, De Groote P, Imazio M. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37(23):1850–8.
Dal Ferro M, Severini GM, Gigli M, Mestroni L, Sinagra G. Genetics of dilated cardiomyopathy: Current knowledge and future perspectives. In: Sinagra G, Merlo M, Pinamonti B, editors. Dilated Cardiomyopathy: From genetics to clinical management. Cham: Springer International Publishing; 2019. p. 45–69.
McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res. 2017;121(7):731–48.
Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45(7):969–81.
Sweet ME, Taylor MR, Mestroni L. Diagnosis, prevalence, and screening of familial dilated cardiomyopathy. Expert Opin Orphan Drugs. 2015;3(8):869–76.
Harakalova M, Kummeling G, Sammani A, Linschoten M, Baas AF, van der Smagt J, Doevendans PA, van Tintelen JP, Dooijes D, Mokry M. A systematic analysis of genetic dilated cardiomyopathy reveals numerous ubiquitously expressed and muscle-specific genes. Eur J Heart Fail. 2015;17(5):484–93.
Pantou MP, Gourzi P, Gkouziouta A, Tsiapras D, Zygouri C, Constantoulakis P, Adamopoulos S, Degiannis D. Phenotypic heterogeneity within members of a family carrying the same RBM20 mutation R634W. Cardiology. 2018;141(3):150–5.
Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, Vertino-Bell A, Smaoui N, Neidich J, Monaghan KG. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704.
Malakootian M, Jalilian M, Kalayinia S, Hosseini Moghadam M, Heidarali M, Haghjoo M. Whole-exome sequencing reveals a rare missense variant in DTNA in an Iranian pedigree with early-onset atrial fibrillation. BMC Cardiovasc Disord. 2022;22(1):1–9.
Mahdavi M, Mohsen-Pour N, Maleki M, Hesami M, Naderi N, Houshmand G, Jazi HRR, Shahzadi H, Kalayinia S: Whole-exome sequencing identified compound heterozygous variants in the TTN gene causing Salih myopathy with dilated cardiomyopathy in an Iranian family. Cardiology in the Young 2021:1–6.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.
Garrison E, Marth G: Haplotype-based variant detection from short-read sequencing. 2012 http://arxiv.org/abs/1207.3907
Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Müller S, Kayvanpour E, Vogel B. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36(18):1123–35.
Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J, Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1(1):21–6.
Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ Jr, Spudich S, De Girolami U. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341(23):1715–24.
Schönberger J, Wang L, Shin JT, Do Kim S, Depreux FF, Zhu H, Zon L, Pizard A, Kim JB, MacRae CA. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat Genet. 2005;37(4):418–22.
Zahr HC, Jaalouk DE. Exploring the crosstalk between LMNA and splicing machinery gene mutations in dilated cardiomyopathy. Front Genet. 2018;9:231.
Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18(5):766–73.
Mirtschink P, Krishnan J, Grimm F, Sarre A, Hörl M, Kayikci M, Fankhauser N, Christinat Y, Cortijo C, Feehan O. HIF-driven SF3B1 induces KHK-C to enforce fructolysis and heart disease. Nature. 2015;522(7557):444–9.
Yang J, Hung L-H, Licht T, Kostin S, Looso M, Khrameeva E, Bindereif A, Schneider A, Braun T. RBM24 is a major regulator of muscle-specific alternative splicing. Dev Cell. 2014;31(1):87–99.
Gao C, Ren S, Lee J-H, Qiu J, Chapski DJ, Rau CD, Zhou Y, Abdellatif M, Nakano A, Vondriska TM. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J Clin Investig. 2016;126(1):195–206.
Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol. 2009;54(10):930–41.
Watanabe T, Kimura A, Kuroyanagi H. Alternative Splicing Regulator RBM20 and Cardiomyopathy. Front Mol Biosci. 2018;5:105.
Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S. Ensembl 2019. Nucleic Acids Res. 2019;47(D1):D745–51.
Murayama R, Kimura-Asami M, Togo-Ohno M, Yamasaki-Kato Y, Naruse TK, Yamamoto T, Hayashi T, Ai T, Spoonamore KG, Kovacs RJ. Phosphorylation of the RSRSP stretch is critical for splicing regulation by RNA-Binding Motif Protein 20 (RBM20) through nuclear localization. Sci Rep. 2018;8(1):1–14.
Filippello A, Lorenzi P, Bergamo E, Romanelli MG. Identification of nuclear retention domains in the RBM20 protein. FEBS Lett. 2013;587(18):2989–95.
Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, Adami E, Rintisch C, Dauksaite V, Radke MH. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Investig. 2014;124(8):3419–30.
Gaertner A, Klauke B, Felski E, Kassner A, Brodehl A, Gerdes D, Stanasiuk C, Ebbinghaus H, Schulz U, Dubowy KO. Cardiomyopathy-associated mutations in the RS domain affect nuclear localization of RBM20. Hum Mutat. 2020;41(11):1931–43.
Franaszczyk M, Chmielewski P, Truszkowska G, Stawinski P, Michalak E, Rydzanicz M, Sobieszczanska-Malek M, Pollak A, Szczygieł J, Kosinska J. Titin truncating variants in dilated cardiomyopathy–prevalence and genotype-phenotype correlations. PLoS ONE. 2017;12(1): e0169007.
Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619–28.
Li D, Morales A, Gonzalez-Quintana J, Norton N, Siegfried JD, Hofmeyer M, Hershberger RE. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci. 2010;3(3):90–7.
Refaat MM, Lubitz SA, Makino S, Islam Z, Frangiskakis JM, Mehdi H, Gutmann R, Zhang ML, Bloom HL, MacRae CA. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm. 2012;9(3):390–6.
Chami N, Tadros R, Lemarbre F, Lo KS, Beaudoin M, Robb L, Labuda D, Tardif J-C, Racine N, Talajic M. Nonsense mutations in BAG3 are associated with early-onset dilated cardiomyopathy in French Canadians. Can J Cardiol. 2014;30(12):1655–61.
Millat G, Bouvagnet P, Chevalier P, Sebbag L, Dulac A, Dauphin C, Jouk P-S, Delrue M-A, Thambo J-B, Le Metayer P. Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur J Med Genet. 2011;54(6):e570–5.
Wells QS, Becker JR, Su YR, Mosley JD, Weeke P, D’Aoust L, Ausborn NL, Ramirez AH, Pfotenhauer JP, Naftilan AJ. Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6(4):317–26.
Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenperä P, Koillinen H, Kaartinen M, Nieminen MS, Myllykangas S. Genetics and genotype–phenotype correlations in finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36(34):2327–37.
García-Molina E, Sabater-Molina M, López-Cuenca D, Olmo MC, Pérez I, Esparza CM, Blanes JRG. A study of the pathogenicity of variants in familial heart disease. The value of cosegregation. Am J Transl Res. 2019;11(3):1724.
Hey TM, Rasmussen TB, Madsen T, Aagaard MM, Harbo M, Mølgaard H, Møller JE, Eiskjær H, Mogensen J. Pathogenic RBM20-variants are associated with a severe disease expression in male patients with dilated cardiomyopathy. Circ Heart Fail. 2019;12(3):e005700.
Hey TM, Rasmussen TB, Madsen T, Aagaard MM, Harbo M, Mølgaard H, Nielsen SK, Haas J, Meder B, Møller JE. Clinical and genetic investigations of 109 index patients with dilated cardiomyopathy and 445 of their relatives. Circ Heart Fail. 2020;13(10):e006701.
Nguyen TV, Vu MTT, Do TNP, Tran THN, Do TH, Nguyen TMH, Huynh BNT, Le LA, Pham NTN, Nguyen TDA. Genetic determinants and genotype-phenotype correlations in Vietnamese patients with dilated cardiomyopathy. Circ J. 2021. https://doi.org/10.1253/circj.CJ-21-0077.
Streckfuss-Bömeke K, Tiburcy M, Fomin A, Luo X, Li W, Fischer C, Özcelik C, Perrot A, Sossalla S, Haas J. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol. 2017;113:9–21.
Sun Q, Guo J, Hao C, Guo R, Hu X, Chen Y, Yang W, Li W, Feng Y. Whole-exome sequencing reveals two de novo variants in the RBM20 gene in two Chinese patients with left ventricular non-compaction cardiomyopathy. Pediatric Investig. 2020;4(1):11–6.
Beraldi R, Li X, Martinez Fernandez A, Reyes S, Secreto F, Terzic A, Olson TM, Nelson TJ. Rbm20-deficient cardiogenesis reveals early disruption of RNA processing and sarcomere remodeling establishing a developmental etiology for dilated cardiomyopathy. Hum Mol Genet. 2014;23(14):3779–91.
Parikh VN, Caleshu C, Reuter C, Lazzeroni LC, Ingles J, Garcia J, McCaleb K, Adesiyun T, Sedaghat-Hamedani F, Kumar S. Regional variation in RBM20 causes a highly penetrant arrhythmogenic cardiomyopathy. Circ Heart Fail. 2019;12(3):e005371.
Rampersaud E, Siegfried JD, Norton N, Li D, Martin E, Hershberger RE. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog Pediatr Cardiol. 2011;31(1):39–47.
Liatakis I, Prappa E, Gouziouta A, Pantou MP, Gourzi P, Vlachos K, Mililis P, Kariki O, Degiannis D, Efremidis M. RBM20 mutation and ventricular arrhythmias in a young patient with dilated cardiomyopathy: a case report. Am J Cardiovasc Dis. 2021;11(3):398.
Chen SN, Mestroni L, Taylor MR. Genetics of dilated cardiomyopathy. Curr Opin Cardiol. 2021;36(3):288–94.
Favalli V, Serio A, Grasso M, Arbustini E. Genetic causes of dilated cardiomyopathy. Heart. 2016;102(24):2004–14.
Asadi M, Foo R, Salehi AR, Salehi R, Samienasab MR. Mutation in δ-Sg gene in familial dilated cardiomyopathy. Adv Biomed Res. 2017;6:32.
Yousefi HA, Asghari Moghaddam N, Jafari Fesharaki M. Association between the rs538089 of the LMNA gene and dilated cardiomyopathy in Iranian patients. Iran Heart J. 2020;21(4):103–10.
Nozari A, Aghaei-Moghadam E, Zeinaloo A, Mollazadeh R, Majnoon M-T, Alavi A, Firouzabadi SG, Mohammadzadeh A, Banihashemi S, Nikzaban M. A novel splicing variant in FLNC gene responsible for a highly penetrant familial dilated cardiomyopathy in an extended Iranian family. Gene. 2018;659:160–7.
Eshaghkhani Y, Mohamadifar A, Asadollahi M, Taghizadeh M, Karamzade A, Saberi M, Nourmohammadi P, Golchehre Z, Amin A, Keramatipour M. Whole-exome sequencing identified a novel variant (C. 405_422+ 39del) in DSP gene in an Iranian pedigree with familial dilated cardiomyopathy. Rep Biochem Mol Biol. 2021;10(2):280.
Petropoulou E, Soltani M, Firoozabadi AD, Namayandeh SM, Crockford J, Maroofian R, Jamshidi Y. Digenic inheritance of mutations in the cardiac troponin (TNNT2) and cardiac beta myosin heavy chain (MYH7) as the cause of severe dilated cardiomyopathy. Eur J Med Genet. 2017;60(9):485–8.
Mollanoori H, Naderi N, Amin A, Hassani B, Shahraki H, Teimourian S. A novel human T17N-phospholamban variation in idiopathic dilated cardiomyopathy. Gene Rep. 2018;12:122–7.
Acknowledgements
We wish to thank the patient and his family for their kind participation in the study. Many thanks are also due to Mrs Masomeh Jalilian for her technical assistance.
Funding
This work was supported by a research grant to Dr Malakootian and Dr Amin from the Research Deputyship of Rajaie Cardiovascular Medical and Research Center (9966).
Author information
Authors and Affiliations
Contributions
MHM: experiment design, data production, data interpretation, writing the first draft of manuscript, and manuscript final edit. MB: lab work, data production, and manuscript edit. SK: WES analysis and manuscript edit. MF: clinical evaluation of patients and manuscript edit. MJM(Majid Maleki): clinical evaluation and manuscript edit. PS: clinical evaluation and manuscript edit. AA: clinical evaluation of patients, experiment design, data interpretation, manuscript final edit. All the authors contributed to the article and approved the submitted version. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study protocol was approved by the Ethics Committee of Rajaie Cardiovascular Medical and Research Center (IR.RHC.REC.1399.078). The study was conducted in accordance with the Helsinki Declaration. All the individuals who joined the study signed written informed consent.
Consent for publication
Not applicable. The study doesn’t include information which may reveal identity of the person.
Competing interest
The authors hereby declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The other variants identified in an index patient.
Additional file 2.
All reported exonic nucleotide variations in RBM20 gene in Iranome database.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Malakootian, M., Bagheri Moghaddam, M., Kalayinia, S. et al. Dilated cardiomyopathy caused by a pathogenic nucleotide variant in RBM20 in an Iranian family. BMC Med Genomics 15, 106 (2022). https://doi.org/10.1186/s12920-022-01262-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01262-4