
Abstract
Background
Despite deeper understanding of the genetic landscape of acute myeloid leukemia (AML), the improvement of survival is still a great challenge. STK10 is overexpressed in several cancers with functions varying according to cancer types. But the functions of STK10 in AML has never been reported.
Methods
We analyzed the expression, prognosis and potential functions of STK10 utilizing public web servers. Metascape and the String database were used for functional and protein–protein interaction analyses.
Results
We found STK10 was enriched in blood & immune cells and overexpressed in AML. High STK10 expression was associated with poor overall survival, which was also identified in the subgroups of patients ≤ 60 years old and patients with non-high-risk cytogenetics. We demonstrated genes associated with STK10 were enriched in blood, spleen and bone marrow, influencing the immune function and biological process of AML. ITGB2 and ITGAM might directly interact with STK10 and were associated with poor prognosis. Besides, STK10 was associated with the infiltration of immune cells and immune checkpoints, like HLA-E, CD274 and GAL-9.
Conclusions
The present study was the original description of STK10 in AML and set the stage for developing STK10 as a new prognostic marker or therapeutic target for AML.
Similar content being viewed by others

Background
Acute myeloid leukemia (AML), characterized by infiltration of the clonal, abnormally or poorly differentiated and highly proliferative cells in bone marrow (BM) or blood, is the second commonly known leukemia in adults. Despite progress in the genetic landscape and the followed transformation into promising therapies, the improvement of prognosis is still a great challenge for clinicians owing to the enormous molecular diversity [1]. Therefore, there is still an urgent need to identify new prognostic factors or therapeutic targets for AML.
The serine/threonine kinase 10 (STK10), also called lymphocyte-oriented kinase (LOK) and PRO2729, is located on nucleoplasm and plasma membrane [2]. The production of STK10 is mainly synthesized in lymphocytes and shows homology to the STE20 family members, notably involved in the mitogen-activated protein kinase (MAPK) cascades [3]. It has been demonstrated that the STK10 might act as a tumor suppressor in the aggressive lymphoma [4] but implicated an pro-tumor action in ewing sarcoma [5], indicating that STK10 might play diverse functions according to different tumor types.
A vital function of STK10 is to phosphorylate the polo-like kinase 1 (PLK1) [3], which has been found to be involved in the regulation of the cell cycle, especially in the G2/M transition and during mitosis [6,7,8,9]. PLK1 is overexpressed in several solid tumors and leads to poor prognosis, as well as in AML [7, 9, 10]. STK10 also functions as an ezrin-radixin-moesin (ERM) kinase, activating the ERM family of proteins which are involved in the development and metastasis of various types of cancers, like prostate cancer, breast cancer and rhabdomyosarcoma, etc. [3, 11,12,13]. The knockout or caspase cleavages of STK10 result in dephosphorylation of ERM, further inhibiting cell migration [3, 11]. According to a recent study, ezrin (EZR), a member of ERM, has been identified as a marker of poor prognosis in AML. EZR inhibitors successfully inhibited the viability and autonomous clonal growth in AML cells [14]. These observations may suggest an indication of the regulating function of STK10 in AML. However, the potential functions of STK10 in AML have not been explored so far.
In the present study, we uncovered the characteristic of STK10 expression in AML, as well as the prognostic values of STK10. Then we investigated the functions of STK10-associated genes and the interaction between STK10 and the STK10-related proteins. Finally, the relationship between STK10 expression and immune cell infiltration in AML was also explored. Our work has made some contributions to our understanding of the biological functions of STK10 in AML.
Methods
Human Protein Atlas (HPA) analysis
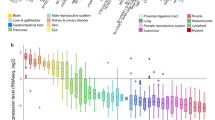
HPA (https://www.proteinatlas.org) is open access for users. It provides a huge atlas by various omics technologies, including antibody-based imaging, mass spectrometry-based proteomics, transcriptomics and systems biology [2]. We acquired STK10 expression in different cell types by searching the term “STK10” in the field of “Cell Type category (RNA)”. The result provides a summary of single-cell RNA, denoted by normalized expression (NX) of transcripts per million (TPM), from all normal single cell types.
Gene Expression Profiling Interactive Analysis (GEPIA) analysis
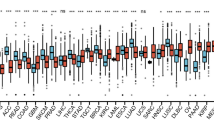
GEPIA (https://www.gepia.cancer-pku.cn) is developed based on the RNA-seq data of 9,736 tumors and 8,587 normal samples from the Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression project (GTEx) [15]. The general module on the homepage of the website provides the summary information and gene expression profile. The bar plot was adopted to present STK10 expression across all tumor samples and paired normal tissues, in which the ordinate offers TPM values of samples.
GEO, TCGA and GETx data download and process
To compare the expression of STK10 between tumor and normal tissues, we acquired the TPM format of RNA-seq data in TCGA and GTEx database that uniformly processed by Toil’s method [16] from UCSC Xena (https://xenabrowser.net/datapages/). Then we download the latest RNA-seq and clinical information from the TCGA database (https://portal.gdc.cancer.gov/). The data from the TCGA database was transformed from HTSeq-fragments per kilobase per million (FPKM) to TPM for the following analysis.
Besides, we acquired expression data of GSE9476 [17] from the Gene Expression Omnibus (GEO) repository (https://www.ncbi.nlm.nih.gov/gds/) to verify the results we concluded. GSE9476 provides expression profiling by array, collected from leukemic blasts of 26 AML patients and CD34+ selected cells of 8 BM of health donors. The log2 expression value obtained from gcRMA analyses was processed in our analysis.
Metascape analysis
Metascape (http://metascape.org) is a practical tool to infer enriched biological pathways. By submitting overlapping genes in the multiple gene list module, functional enrichments in Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were analyzed automatically [18]. Metascape database also provides enrichment analysis with Pattern Gene Database (PaGenBase) [19], providing further views in cells, tissues or organs the genes enriched in, and Transcriptional Regulatory Relationship Unraveled by Sentence-based Text mining (TRRUST) [20], offering key regulatory factors of the gene list.
String analysis and process
The String database version 11.0b (https://string-db.org/) could provide known and predicted protein–protein interactions (PPI) covering 24,584,628 proteins from 5090 organisms [21]. We employed the multiple proteins module to identify existing proteins by submitting the genes associated with STK10, and further constructed the PPI network. Through the Cytoscape software (v3.8.0) [22], we disposed of the string interaction file, produced by the String database automatically, for visualization of the network.
Immune infiltration
ssGSEA algorithm was used to assessment the level of immune infiltration [23]. The method uses a gene set which provides specific makers of immune cells to calculate the enrichment score of immune cells. After getting the level of immune infiltration in each sample, the Spearman’s test was used for exploring the correlation of STK10 with immune infiltration.
Statistical analysis
Analyses of samples were processed using R (version 3.6.3). We classified the data into two groups according to the median value of STK10 expression. Categorical variables, like sex, age, cytogenetic risk and French-American-British (FAB) classifications, were tested by Pearson’s chi-square and Fisher’s exact tests. Continuous variables mainly included White blood cell (WBC) count, BM blasts, peripheral blood (PB blasts) and gene expression. If the continuous variable does not obey normal distribution, examined by the Shapiro–Wilk test, the Mann–Whitney U test was used for the comparison, otherwise the Student-t test was adopted. Gene expression between STK10high and STK10low groups with the absolute value of log2 fold change (log2FC) > 1 and P.adj < 0.5 was considered as differentially expressed. The Spearman’s test was implemented to probe the correlation between STK10 and related genes, as well as immune cell infiltration level. Overall survival (OS) was compared using the Log-rank testing. P value < 0.05 was considered as significant (ns: P > 0.05, *: P < 0.05, **: P ≤ 0.01, ***: P ≤ 0.001).
Univariate and multivariate survival analyses was carried out using Cox regression models to test the function of STK10 in predicting patients’ survival. The multivariate survival analysis would be performed based on factors with statistical significance (P < 0.05) in the univariate analysis.
R packages adopted in this research included ggpolt2 package (v_3.3.3) and survminer package (v_0.4.9) for visualization, pROC package (v_1.17.0.1) for drawing receiver operating characteristic (ROC) curve, survival package (v_3.2-10) for analyzing OS, DEseq2 package (v_1.26.0) for figuring out the differential genes with STK10, and stat package (v_3.6.3) for correlation analysis with STK10. The correlation between STK10 and immune infiltration was analyzed by the GSVA package (v_1.34.0).
Results
STK10 overexpression in AML
To elucidate the cell type specificity of STK10 expression, we first analyzed STK10 expression based on the RNA-seq data from the HPA database. We listed the top 40 cell types based on nTPM (normalized transcripts per million) value of STK10 and numbered the top five with the value of nTPM. The levels of STK10 mRNA expression were higher in blood &immune cells, like NK cells, Dendritic cells, kupffer cells, T-cells and macrophages (Fig. 1a). Subsequently, we tested the expression of STK10 in different types of cancers by comparing the RNA-seq data of TCGA and GTEx projects in the GEPIA database. The results revealed that the level of STK10 expression was up-regulated significantly in AML (Fig. 1b). Figure 1c uncovered the difference of STK10 expression between 173 AML and 70 normal BM samples individually (P < 0.001). The ROC curve was also adopted to examine the capacity of STK10 to distinguish AML from normal samples. As presented in Fig. 1d, the area under the curve (AUC) was 1.00 (P < 0.001), suggesting the expression of STK10 could be an ideal marker to distinguish AML from normal tissues. Then we verified the expression of STK10 in GSE9476, another dataset from GEO. And the differential expression of STK10 was also observed (Fig. 1e, P < 0.001).

STK10 expression in different cell types and tumors. a STK10 expression in different cell types, analyzed by HPA. b STK10 expression across various tumors and normal tissues, analyzed by GEPIA. c The expression of STK10 between normal tissues and AML based on data from TCGA and GETx. d ROC curve based on STK10 expression in distinguishing normal tissues and AML. e STK10 expression on AML and health donors in GSE9476
Relationships between STK10 and clinicopathological characteristics of patients with AML
We stratified 151 patients into two groups based on the median value of the STK10 expression (median value, 77.64). Characteristics between the two groups were summarized in Table 1. 75 patients were classified into the STK10low group, and 76 patients were in the STK10high group. Significant differences were observed in age grouped by 60 years old (P = 0.025) and cytogenetic risk stratification (P < 0.001) between STK10low and STK10high groups. No significant differences existed in gender, WBC count, BM blast, PB blasts and FAB classification.
We next tested the expression of STK10 in different groups of age and cytogenetic risk stratification. In the patients > 60 years old, the expression of STK10 was higher when compared to the patients ≤ 60 years old (P = 0.034, Fig. 2a). STK10 was overexpressed in intermediate- (P < 0.001) and poor- (P < 0.001) risk groups compared with the favorable group (Fig. 2b). And there was no difference between intermediate- and poor- risk groups. Based on the ROC curve, we found that low expression of STK10 could serve as a diagnostic factor in predicting the favorable cytogenetic risk with medium accuracy (AUC = 0.832, CI 0.757–0.908, Fig. 2c. We further evaluated the STK10 expression in specific cytogenetics (Fig. 2d). STK10 expression was the lowest in the inv(16) group, showing no difference with that in the t(15;17) and t(8;21) groups, compared with the intermediate- or poor- risk groups. In addition, the expression of STK10 indicated no statistical significance between the intermediate- and poor- risk groups.

Correlation between STK10 expression and clinical characteristics, based on data from TCGA. a STK10 expression in AML with different ages. b STK10 expression in AML with different cytogenetics risk stratifications. c ROC curve based on STK10 expression in predicting the favorable cytogenetic risk. d STK10 expression in AML with specific cytogenetics
High STK10 expression is associated with a poor prognosis of AML
The association between STK10 expression and OS of patients with AML was evaluated by Kaplan–Meier (K-M) analysis, which indicated that low expression of STK10 was correlated with favorable OS in AML (HR 1.85, CI 1.21–2.84, P = 0.003, Fig. 3a). We further explored the relationship between STK10 expression and OS of the specific subgroups. As shown in Fig. 3b, c, among the patients ≤ 60 years old, the patients with STK10low expression had higher OS when compared with those with STK10high expression (P = 0.049). However, the OS benefit of STK10low expression could not be seen among the patients > 60 years old (P = 0.214). Similarly, the significant difference in OS between the STK10low and STK10high expression could only be found in the favorable & intermediate risk group (P = 0.009, Fig. 3d, e).

Prognostic values of STK10 in AML patients, based on TCGA datasets. a Survival curves of OS between the STK10low and STK10high groups. Prognostic values of STK10 had been shown in the subgroup of the patients ≤ 60 years (b), the patients > 60 years (c), the patients with favorable&intermediate cytogenetic risks (d) and the patients with poor cytogenetic risks (e)
We performed univariate and multivariate survival analyses based on patients’ clinical characteristics and the expression of STK10 by cox regression analysis. As presented in Additional file 1, the P values of age, cytogenetic risk and STK10 in the univariate analysis show statistical significances. In further multivariate analysis, the HR (95% CI) between STK10low and STK10high groups is 1.562 (1.001–2.438) with statistical significance (P = 0.050). This result indicates the expression of STK10 could predict patients’ survival as an independent prognostic factor.
STK10 associated gene analysis between STK10 high and STK10 low groups in AML
In order to further evaluate the functional role of STK10 in AML, we first analyzed the expression of differential genes between STK10low and STK10high groups based on the TCGA database. A total of 1999 genes had a significant difference between STK10high and STK10low groups (P.adj < 0.05, |log2FC|> 1, see Additional file 2), including 704 up-regulated genes (red triangle) and 1295 down-regulated genes (blue triangle) as shown in the volcano plot (Fig. 4a).

Genes associated with STK10 expression. a Volcano plot of different gene expression profiles between STK10high and STK10low groups. b Top ten co-expression genes positively and negatively associated with STK10 based on Spearman test’s value. c Overlapping genes that were significantly up-regulated and positively correlated with STK10. d Overlapping genes that were significantly down-regulated and negatively correlated with STK10
Then the correlation of genes in conjunction with STK10 expression was identified by the Spearman’s correlation test. A total of 5804 gens were found correlated with STK10 expression (P < 0.05, and |cor|≥ 0.3, Additional file 3). We listed the top ten of 5228 positively correlative genes and the top ten of negatively 576 correlative genes in Fig. 4b. Among the positively correlative genes, 319 genes were significantly up-regulated (Fig. 4c). And 178 genes were significantly down-regulated in the negatively correlative genes (Fig. 4d). The overlapping genes were processed into biological function analysis and PPI analysis subsequently.
Biological function of STK10-associated overlapping genes
We then investigated the biological function of the 497 overlapping genes in the Metascape database. As listed in Fig. 5a, the summary of enrichment analysis in PaGenBase revealed the overlapping genes were mainly enriched in blood, spleen and BM, indicating their potential function in the hematological system. Through the GO/KEGG pathway annotations, we found that these genes were associated with several biological processes of leukemia, like regulation of cytokine production, phagocytosis, myeloid leukocyte activation, leukocyte migration and tumor necrosis factor superfamily cytokine production (Fig. 5b). Besides, three clusters belonged to the immune system, including the immune effector process, regulation of defense response, and immunoregulatory interactions between a Lymphoid and a non-Lymphoid cell. Besides, relevant regulatory genes of the overlapping genes provided by TRRUST were presented in Fig. 5c, some of which have been identified as crucial factors in leukemogenesis, such as SPI1, CEBPA, STAT1, TP53, RARA and WT1 [24,25,26,27,28,29].

Functional analysis of the overlapping genes in AML. a Enrichment of the overlapping genes in tissues and cells based on PaGenBase. b Analysis of GO and KEGG pathways associated with STK10. c The relevant regulatory genes of the overlapping genes based on the TRRUST
PPI identifies the STK10-related proteins
The PPI analysis was conducted by inputting the 497 overlapping genes into the String database. As a result, 172 nodes and 631 edges were obtained (Additional file 4). The nodes colored yellow were the proteins that directly interacted with STK10. We showed the proteins at length in Fig. 6a, including 8 nodes and 23 edges. Among them, ITGB2 (also called LFA-1) and ITGAM were overexpressed in AML (Fig. 6b, e) and positively correlated with STK10 (P < 0.001, Fig. 6c, f). According to the K-M plot, ITGB2 and ITGAM were also related to poor OS in AML (P = 0.003 and P = 0.027 respectively, Fig. 6d, g). The expression of ITGB2 and ITGAM in the dataset GSE9476 was also higher in tumor samples compared with donors (Additional file 5a, c). Both ITGB2 and ITGAM was positively correlated with STK10 expression in GSE9476 (Additional file 5b, d).

PPI network of the overlapping genes. a Genes from (Additional file 4) interacted with STK10 directly. b, e The expression of ITGB2 and ITGAM between normal tissues and AML. c, f The co-expression of ITGB2 and ITGAM with STK10. d, g The prognostic value of ITGB2 and ITGAM in AML
In Fig. 6a, seven genes were found to be associated with STK10 directly in the protein–protein interactions analysis, including CEACAM3, ITGAM, TNFRSF1B, ADAM8, ITGB2, SH3BP5 and MS4A3. Mann–Whitney U test was adopted to compare the expression of these genes between normal tissues and tumor samples from AML. And five genes (CEACAM3, ITGAM, ADAM8, ITGB2 and SH3BP5) were selected to enter the univariate and multivariate survival analyses with STK10 to prove that whether high STK10 expression is an independent prognostic indicator in AML.As shown in Additional file 6, high level expression of ITGAM, ITGB2 and STK10 own statistical significances (P < 0.05) in the univariate analysis. In the further multivariate analysis, the HR (95% CI) between the high expression level and low expression level of STK10 is 1.610 (1.015–2.554) with statistical significance (P = 0.043), indicating STK10 could act as a prognostic indicator for patients with AML at the genetic level.
STK10 expression and immune cell infiltration in AML
The results above suggested that STK10 associated genes were involved in immunologic function (Fig. 5b). Thus, we further explored the relationship between STK10 and immune cell infiltration in AML. As shown in Fig. 7a, STK10 was significantly positively correlated with several immune cells, like regulatory T cells (TReg), CD8 T cells, T follicular helper (TFH), Th17 cells, NK CD56dim cells and cytotoxic cells (|cor|≥ 0.3, P < 0.05). Grouped by the median value of STK10 expression, the infiltration levels of all these immune cells were significantly higher in the STK10high group (Fig. 7b, d, f, h, j, l). The correlation of STK10 expression with immune cell infiltration was also evaluated (Fig. 7c, e, g, i, k, m), revealing the positive association with STK10.

The relationship between STK10 and the immune cell infiltration. a Forrest plot showing the connection between STK10 and the immune cell infiltration levels. b, d, f, h, j, l The levels of various immune cells (|cor|≥ 0.3, p < 0.05) in STK10low and STK10high groups. c, e, g, i, k, m The correlation of STK10 expression with the specific immune cell infiltration levels. Cytotoxic cells: including CD8 T cells, Tgd, and NK cells
We know that NK cells and cytotoxic T lymphocytes (CTLs) exert the antitumor effect through the combination of the receptors on the surface of the effector cells and the corresponding ligands on the surface of the target cells [30,31,32]. Therefore, we explored the association between STK10 expression and these ligands on the surface of AML cells, such as HLA-A, HLA-B, HLA-C, HLA-E, PD-L1(CD274), PD-L2 and GAL-9(produced by LGALS9), etc. Among them, HLA-E, CD274 and LGALS9 were overexpressed on tumor cells (Fig. 8a, d, g) and predicted poor OS in AML (Fig. 8b, e, h). Moreover, Spearman’s correlation test revealed the positive correlation between these ligands on tumor cells with STK10 (Fig. 8c, f, i). We also explored the expression of HLA-E and LGALS9 in GSE9476. The results were consistent with our findings above that HLA-E was overexpressed on tumor cells and positively correlated with STK10 expression (Additional file 5e, f). Although the expression of LGALS9 on tumor samples was higher as well, the spearman’s correlation test showed there was no statistical correlation between STK10 and LGALS9 (Additional file 5g, h).

The roles of specific immune checkpoints in AML. a, d, g The expression of HLA-E, CD274 and LGALS9 between normal tissue and AML. b, e, h The prognostic value of HLA-E, CD274 and LGALS9 in AML. c, f, i The co-expression of HLA-E, CD274, LGALS9 with STK10
Discussion
AML is a highly heterogeneous disease with different outcomes even among patients with the same known genetic background, indicating more accurate prognostic and therapeutic targets need to be developed. The possible mechanisms of the poor prognosis include the existence of leukemia stem cells, the interaction with BM microenvironment, immune evasion or clonal evolution leading to primary or acquired chemoresistance. The overexpression and potential pathogenetic significance of STK10 in multiple cancers have been observed [5, 11, 33]. However, the relationship between STK10 and AML has not been examined to date and needs to be further explored.
The STK10 transcript was presented in several tissues and expressed highly in hematopoietic cells [34]. It had been found that STK10 mutations were associated with hematological diseases, like peripheral T-cell lymphoma (PTCL) and Burkitt lymphoma (BL) [4, 33]. Our results confirmed STK10 possessed the cell specificity in blood & immune cells and overexpressed especially in AML, maybe serving as a promising marker in predicting the malignant transformation to AML.
Several studies related to the STK10 in cancers have defined the association of STK10 with diverse outcomes depending on the type of cancer [2]. In the current study, we demonstrated that high STK10 expression carried an increased risk of poor OS in AML. The same findings were also identified in the subgroups of the patients ≤ 60 years old or with the non-high-risk cytogenetics. Furthermore, the low expression of STK10 might become an indication of distinguishing the patients with favorable cytogenetics, including inv(16), t(8;21) and t(15;17), from the unfavorable cytogenetic risks.
A slice of studies reported STK10 was mainly expressed in spleen, thymus, BM, placenta and peripheral blood leukocytes, indicating the potential pathogenetic significance in hematopoietic tissues [3, 34]. Consistent with the above findings, our functional analysis manifested that STK10 associated genes showed functional specificity mainly in blood, spleen and BM. We also identified these genes were associated with several biological processes of leukemia, like regulation of cytokine production, phagocytosis, myeloid leukocyte activation, leukocyte migration and tumor necrosis factor superfamily cytokine production, all of which were related to the origin, pathophysiology and histopathology of AML [35]. Besides, we reported two potential genes, which directly interacted with STK10, influenced the survival of AML patients. ITGB2 and ITGAM have tissue specificity on BM [2] and take part in a portion of vital biological processes. For example, ITGB2 is involved in adhesion, migration of leukocytes [36] and immune response including NK cell-induced cytotoxicity [37,38,39]. From a bioinformatics analysis, ITGAM and ITGB2 were also up-regulated in myeloma and involved in cytokine-cytokine receptor interaction, innate immune response and inflammatory response that were similar to our results [40]. Moreover, the integrin ITGAM/ITGB2, associated with immunity and inflammation [41, 42], was gradually increased along with hematopoietic stem cells (HSC) differentiation in mice [41], suggesting the indirect role of STK10 in immune and defense. However, this view still needs to be verified by further studies.
It is universally acknowledged that immune dysregulation is a general feature in most of cancers including AML. Infiltrating immune cells participate in the regulation of quite a few biological processes, like chemotherapy, immunotherapy, immune escape and disease progression [43, 44]. Our work implicated that both CD56dim NK cells and CD8 + T cells are positively associated with STK10. The mature CD56dim/NKG2A-/KIR+/CD57 + NK cells developing from CD56bright/NKG2A + NK cells can exert greater cytotoxicity to HLA-E + target cells [30, 45, 46]. Previous studies reported that NKG2A, as an inhibitory receptor, was overexpressed in AML and associated with failure to achieve remission [46]. Consistent with these results above, we found HLA-E, the ligand for NKG2A on cancer cells, was also overexpressed indicating poor OS in AML. Moreover, our work unraveled that STK10 was positively correlated with the expression of HLA-E, potentially accounting for the crosstalk of HLA-E and STK10. Besides, NKG2A was also expressed on CD8 + T cells and identified as a new immune checkpoint [47, 48].
The rationale for widespread exploration of programmed cell death 1 protein (PD-1) inhibitors in AML is supported by the increased expression of programmed cell death ligand 1(PD-L1, CD274) and its prognostic value [49]. By blocking either the co-inhibitory receptor PD-1 or its ligand PD-L1, PD-1 inhibitors activate T cell-induced antitumor activity [32, 49]. T-cell immunoglobin mucin-3 (TIM-3) which could be activated by GAL-9 acted as an inhibitory receptor expressed on T cells [32]. Therefore, we studied ligands expression of these immune checkpoints. In our analysis, the expressions of GAL-9 and CD274 were higher in patients with AML, indicating poor prognosis as well. Moreover, a positive correlation between these immune checkpoints with STK10 expression was also observed. All the findings above suggested an immunoregulatory effect of STK10.
In summary, we clarified that STK10 was overexpressed in tumor cells and correlated with an unfavorable prognosis in AML. Functional studies revealed the close association of STK10 with several biological processes in AML. Besides, we identified two potential genes that may tightly interact with STK10 in the pathogenesis of AML, which required further investigation. Our findings also suggested that STK10 might influence immune cell infiltration and was associated with immune escape by interacting with some immune checkpoints. Taken together, our work led to the first expansion of our understanding of the significance of STK10 as a new prognostic factor or therapeutic target for AML.
Availability of data and materials
The datasets used and/or analysed during the current study are available from NCBI Gene Expression Omnibus (GEO: GSE9476) https://www.ncbi.nlm.nih.gov/geo/ and the cancer genome database (TCGA: LAML) https://portal.gdc.cancer.gov.
Abbreviations
- AML:
-
Acute myeloid leukemia
- BM:
-
Bone marrow
- STK10 :
-
Serine/threonine kinase 10
- LOK :
-
Lymphocyte-oriented kinase
- MAPK:
-
Mitogen-activated protein kinase
- PLK1 :
-
polo-like kinase 1
- ERM:
-
Ezrin-radixin-moesin
- EZR :
-
Ezrin
- HPA:
-
Human protein atlas
- TPM:
-
Transcripts per million
- GEPIA:
-
Gene expression profiling interactive analysis
- TCGA:
-
The cancer genome atlas
- GTEx:
-
The genotype-tissue expression project
- FPKM:
-
Per kilobase per million
- GEO:
-
Gene expression omnibus
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PPI:
-
Protein–protein interactions
- FAB:
-
French–American–British
- WBC:
-
White blood cell
- PB:
-
Peripheral blood
- Log2FC:
-
Log2 fold change
- OS:
-
Overall survival
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the curve
- K-M:
-
Kaplan–Meier
- TReg:
-
Regulatory T cells
- TFH:
-
T follicular helper
- CTLs:
-
Cytotoxic T lymphocytes
- PTCL:
-
Peripheral T-cell lymphoma
- BL:
-
Burkitt lymphoma
- HSC:
-
Hematopoietic stem cells
- PD-1:
-
Programmed cell death 1 protein
- PD-L1:
-
Programmed cell death ligand 1
- TIM-3:
-
T-cell immunoglobin mucin-3
References
Vakiti A, Mewawalla P. Acute myeloid leukemia. Treasure Island: StatPearls Publishing; 2021.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419.
Leroy C, Belkina NV, Long T, Deruy E, Dissous C, Shaw S, Tulasne D. Caspase cleavages of the lymphocyte-oriented kinase prevent ezrin, radixin, and moesin phosphorylation during apoptosis. J Biol Chem. 2016;291(19):10148–61.
Fukumura K, Yamashita Y, Kawazu M, Sai E, Fujiwara S, Nakamura N, Takeuchi K, Ando M, Miyazono K, Ueno T, et al. STK10 missense mutations associated with anti-apoptotic function. Oncol Rep. 2013;30:1542–8.
Arora S, Gonzales IM, Hagelstrom RT, Beaudry C, Choudhary A, Sima C, Tibes R, Mousses S, Azorsa DO. RNAi phenotype profiling of kinases identifies potential therapeutic targets in Ewing’s sarcoma. Mol Cancer. 2010;9(1):218.
Degenhardt Y, Lampkin T. Targeting Polo-like kinase in cancer therapy. Clin Cancer Res. 2010;16(2):384–9.
Cortes J, Podoltsev N, Kantarjian H, Borthakur G, Zeidan AM, Stahl M, Taube T, Fagan N, Rajeswari S, Uy GL. Phase 1 dose escalation trial of volasertib in combination with decitabine in patients with acute myeloid leukemia. Int J Hematol. 2021;113(1):92–9.
Brandwein JM. Targeting polo-like kinase 1 in acute myeloid leukemia. Ther Adv Hematol. 2015;6(2):80–7.
Liu Z, Sun Q, Wang X. PLK1, a potential target for cancer therapy. Transl Oncol. 2017;10(1):22–32.
Raab CA, Raab M, Becker S, Strebhardt K. Non-mitotic functions of polo-like kinases in cancer cells. Biochim Biophys Acta Rev Cancer. 2021;1875(1):188467.
Zhang L, Lu SY, Guo R, Ma JX, Tang LY, Wang JJ, Shen CL, Lu LM, Liu J, Wang ZG, et al. STK10 knockout inhibits cell migration and promotes cell proliferation via modulating the activity of ERM and p38 MAPK in prostate cancer cells. Exp Ther Med. 2021;22(2):851.
Wang L, Qi Y, Xiong Y, Peng Z, Ma Q, Zhang Y, Song J, Zheng J. Ezrin–radixin–moesin binding phosphoprotein 50 (EBP50) suppresses the metastasis of breast cancer and HeLa cells by inhibiting matrix metalloproteinase-2 activity. Anticancer Res. 2017;37(8):4353–60.
Proudfit A, Bhunia N, Pore D, Parker Y, Lindner D, Gupta N. Pharmacologic inhibition of Ezrin–Radixin–Moesin phosphorylation is a novel therapeutic strategy in rhabdomyosarcoma. Sarcoma. 2020;2020:9010496.
Lipreri da Silva JC, Coelho-Silva JL, Lima K, Vicari HP, Lazarini M, Costa-Lotufo LV, Traina F, Machado-Neto JA. Comprehensive analysis of cytoskeleton regulatory genes identifies ezrin as a prognostic marker and molecular target in acute myeloid leukemia. Cell Oncol (Dordr). 2021, Preprint.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–102.
Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, Pfeil J, Narkizian J, Deran AD, Musselman-Brown A, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol. 2017;35(4):314–6.
Stirewalt DL, Meshinchi S, Kopecky KJ, Fan W, Pogosova-Agadjanyan EL, Engel JH, Cronk MR, Dorcy KS, McQuary AR, Hockenbery D, et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosom Cancer. 2008;47(1):8–20.
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523.
Pan JB, Hu SC, Shi D, Cai MC, Li YB, Zou Q, Ji ZL. PaGenBase: a pattern gene database for the global and dynamic understanding of gene function. PLoS ONE. 2013;8(12):e80747.
Han H, Cho JW, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46(D1):D380–6.
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–12.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–95.
Liquori A, Ibañez M, Sargas C, Sanz MÁ, Barragán E, Cervera J. Acute Promyelocytic leukemia: a constellation of molecular events around a single PML-RARA fusion gene. Cancers (Basel). 2020;12(3):624.
Konstandin NP, Pastore F, Herold T, Dufour A, Rothenberg-Thurley M, Hinrichsen T, Ksienzyk B, Tschuri S, Schneider S, Hoster E, et al. Genetic heterogeneity of cytogenetically normal AML with mutations of CEBPA. Blood Adv. 2018;2(20):2724–31.
Shao X, Liu Y, Li Y, Xian M, Zhou Q, Yang B, Ying M, He Q. The HER2 inhibitor TAK165 sensitizes human acute myeloid leukemia cells to retinoic acid-induced myeloid differentiation by activating MEK/ERK mediated RARα/STAT1 axis. Sci Rep. 2016;6:24589.
Vagapova ER, Lebedev TD, Tikhonova AD, Goikhman BV, Ivanenko KA, Spirin PV, Prassolov VS. High expression level of SP1, CSF1R, and PAK1 correlates with sensitivity of leukemia cells to the antibiotic mithramycin. Mol Biol. 2020;54(3):522–8.
Welch JS. Patterns of mutations in TP53 mutated AML. Best Pract Res Clin Haematol. 2018;31(4):379–83.
Rossi G, Minervini MM, Carella AM, Melillo L, Cascavilla N. Wilms’ tumor gene (WT1) expression and minimal residual disease in acute myeloid leukemia. In: van den Heuvel-Eibrink MM, editor. Wilms tumor. Brisbane: Codon Publications Copyright; 2016.
Carlsten M, Jaras M. Natural killer cells in myeloid malignancies: immune surveillance, NK cell dysfunction, and pharmacological opportunities to bolster the endogenous NK cells. Front Immunol. 2019;10:2357.
Sanchez-Correa B, Campos C, Pera A, Bergua JM, Arcos MJ, Bañas H, Casado JG, Morgado S, Duran E, Solana R, et al. Natural killer cell immunosenescence in acute myeloid leukaemia patients: new targets for immunotherapeutic strategies? Cancer Immunol Immunother. 2016;65(4):453–63.
Boddu P, Kantarjian H, Garcia-Manero G, Allison J, Sharma P, Daver N. The emerging role of immune checkpoint based approaches in AML and MDS. Leuk Lymphoma. 2018;59(4):790–802.
Piccaluga PP, Navari M, De Falco G, Ambrosio MR, Lazzi S, Fuligni F, Bellan C, Rossi M, Sapienza MR, Laginestra MA, et al. Virus-encoded microRNA contributes to the molecular profile of EBV-positive Burkitt lymphomas. Oncotarget. 2016;7:224–40.
Walter SA, Cutler RE Jr, Martinez R, Gishizky M, Hill RJ. Stk10, a new member of the polo-like kinase kinase family highly expressed in hematopoietic tissue. J Biol Chem. 2003;278(20):18221–8.
Lyengar V, Shimanovsky A. Leukemia. Treasure Island: StatPearls Publishing; 2021.
Ostermann G, Weber KS, Zernecke A, Schröder A, Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. 2002;3(2):151–8.
Barber DF, Faure M, Long EO. LFA-1 contributes an early signal for NK cell cytotoxicity. J Immunol (Baltimore, Md: 1950). 2004;173(6):3653–9.
Losse J, Zipfel PF, Józsi M. Factor H and factor H-related protein 1 bind to human neutrophils via complement receptor 3, mediate attachment to Candida albicans, and enhance neutrophil antimicrobial activity. J Immunol (Baltimore, Md: 1950). 2010;184(2):912–21.
Wang J, Hao JP, Uddin MN, Wu Y, Chen R, Li DF, Xiong DQ, Ding N, Yang JH, Ding XS. Identification and validation of inferior prognostic genes associated with immune signatures and chemotherapy outcome in acute myeloid leukemia. Aging (Albany NY). 2021;13(12):16445–70.
Yang Q, Li K, Li X, Liu J. Identification of key genes and pathways in myeloma side population cells by bioinformatics analysis. Int J Med Sci. 2020;17(14):2063–76.
Herb M, Gluschko A, Schramm M. LC3-associated phagocytosis initiated by integrin ITGAM-ITGB2/Mac-1 enhances immunity to Listeria monocytogenes. Autophagy. 2018;14(8):1462–4.
Blight BJ, Gill AS, Sumsion JS, Pollard CE, Ashby S, Oakley GM, Alt JA, Pulsipher A. Cell adhesion molecules are upregulated and may drive inflammation in chronic rhinosinusitis with nasal polyposis. J Asthma Allergy. 2021;14:585–93.
Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia. 2009;23(12):2233–41.
Mendez LM, Posey RR, Pandolfi PP. The interplay between the genetic and immune landscapes of AML: mechanisms and implications for risk stratification and therapy. Front Oncol. 2019;9:1162.
Bigley AB, Simpson RJ. NK cells and exercise: implications for cancer immunotherapy and survivorship. Discov Med. 2015;19(107):433–45.
Moretta L, Montaldo E, Vacca P, Del Zotto G, Moretta F, Merli P, Locatelli F, Mingari MC. Human natural killer cells: origin, receptors, function, and clinical applications. Int Arch Allergy Immunol. 2014;164(4):253–64.
André P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, Bléry M, Bonnafous C, Gauthier L, Morel A, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175(7):1731–43.
Borst L, van der Burg SH, van Hall T. The NKG2A–HLA-E axis as a novel checkpoint in the tumor microenvironment. Clin Cancer Res. 2020;26:5549–56.
Haroun F, Solola SA, Nassereddine S, Tabbara I. PD-1 signaling and inhibition in AML and MDS. Ann Hematol. 2017;96(9):1441–8.
Acknowledgements
The authors would like to thank the UCSC, TCGA and GEO database for providing valuable gene expression matrix and clinical survival data of AML.
Funding
This study was supported by grants from the National Natural Science Foundation of China (82100218), the Project of young talents promotion of Xi’an Association for science and technology and the Project of subject promotion of Xijing hospital (XJZT19MJ27).
Author information
Authors and Affiliations
Contributions
HT and XC contributed to study design and supervision; XS and WH contributed to data collecting; LB and SJ analyzed the data and wrote the paper; HT revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Univariate and Multivariate analyses based on clinical characteristics and the expression of STK10.
Additional file 2.
The differential expressed RNA between STK10high and STK10low groups identified by R and limma.
Additional file 3.
The correlation of STK10 with other genes analyzed by the Spearman’s test.
Additional file 4.
Network of the 172 proteins from the overlapping genes, available in String database version 11.0b.
Additional file 5.
Verification of ITGB2, ITGAM, HLA-E and LGALS9 in GSE9476.
Additional file 6.
Univariate and Multivariate analyses based on the expression of genes associated with STK10 directly.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bi, L., Jia, S., Hu, W. et al. Systematic analysis of prognostic significance, functional enrichment and immune implication of STK10 in acute myeloid leukemia. BMC Med Genomics 15, 101 (2022). https://doi.org/10.1186/s12920-022-01251-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01251-7