
Abstract
Background
Recently, an increasing number of studies have reported that sperm-associated antigen (SPAG) proteins play crucial roles in solid tumorigenesis, and may serve as potentially helpful biomarkers for cancer diagnosis and prognosis. However, very few studies systematically investigated the expression of SPAG family members and their clinical significance in acute myeloid leukemia (AML).
Methods
The expression of SPAGs and their prognostic significance in AML were determined by a systematic analysis on data gathered from public databases, and the results were validated in clinical samples.
Results
Using public data, we identified only increased SPAG1 expression negatively associated with survival in AML by Cox regression (P < 0.001) and Kaplan–Meier analysis (P < 0.001). The prognostic value of SPAG1 expression was further confirmed in other independent cohorts. Clinically, higher SPAG1 expression was significantly correlated with white blood cell counts (P = 0.014) and French–American–British (FAB) subtypes (P = 0.024). Moreover, higher SPAG1 expression was more common in + 8 patients (P = 0.034), rarely found with t(8;21) (P = 0.014), and correlated with FLT3 (P < 0.001) and DNMT3A mutations (P = 0.001). Despite these associations, multivariate analysis confirmed the independent prognostic value of SPAG1 expression in AML (P < 0.001). Notably, AML patients with higher SPAG1 expression may benefit from hematopoietic stem cell transplantation (HSCT), whereas patients with lower SPAG1 expression appeared less likely to benefit. Finally, we further validated that SPAG1 expression was significantly increased in newly diagnosed AML patients compared with normal controls (P < 0.001) and with AML patients who achieved complete remission (P < 0.001). Additionally, SPAG1 expression could act as a potentially helpful biomarker for the diagnosis and prognosis of AML (P < 0.001 and = 0.034, respectively).
Conclusions
Our findings demonstrated that SPAG1 overexpression may serve as an independent prognostic biomarker and may guide the choice between HSCT and chemotherapy in patients with AML.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) represents a cohort of clonal hematopoietic malignances that originate from myeloid precursors and is a highly heterogeneous disease in terms of molecular, cytogenetic and clinical features [1]. Genetic and molecular abnormalities are closely associated with the leukemogenesis and prognosis of AML [2]. Although mutations in several genes, such as FLT3, CEBPA, NPM1, TP53, RUNX1 and ASXL1, have been well established to occur in AML, the current understanding of the molecular mechanisms involved in the development and progression of AML is still limited [3]. Precise risk stratification and prognosis assessment are of great significance in the selection of treatment for AML patients [4]. Therefore, the identification of a series of molecular alterations that can predict the clinical outcomes of AML patients may contribute to the development of AML–specific targeted therapies.
Evidence has proven that cancer–testis (CT) antigens may function in stemness due to their expression during germ cell and embryonic development, which promotes an important oncogenicity effect in cancer cells [5]. To date, a cluster of proteins named sperm-associated antigens (SPAG), of which 15 members (SPAG1, SPAG2/UAP1, SPAG3/SPAG8, SPAG4, SPAG5, SPAG6, SPAG7, SPAG9, SPAG10/MFGE8, SPAG11B, SPAG12/NHP2L1, SPAG13/SSFA2, SPAG15/SPAM1, SPAG16 and SPAG17) are CT antigens, has been identified [6]. Over the years, ample research has reported the vital role of SPAG proteins, which may function as promising new biomarkers for diagnosis and prognosis in solid tumorigenesis, yet there is a great lack of systematic investigation of SPAG family member expression and clinical evaluation of these proteins in acute myeloid leukemia (AML) [6].
To date, our research is the first to report that SPAG1 mRNA expression, among SPAG family members, is negatively associated with survival in AML. Moreover, the prognostic value of SPAG1 overexpression in AML was further confirmed by our data. High expression of SPAG1 mRNA was intrinsically connected to specific genetic (both cellular and molecular levels) abnormalities in AML. Despite these associations, SPAG1 overexpression could also function independently as a prognostic biomarker in AML, and it may serve as a reference for consolidation therapy selection between chemotherapy and hematopoietic stem cell transplantation (HSCT).
Materials and methods
Public datasets
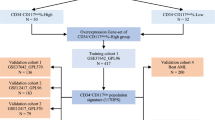
The identification cohort comprised 173 AML patients with RNA-Seq V2 data for SPAG family members (SPAG1, SPAG2/UAP1, SPAG3/SPAG8, SPAG4, SPAG5, SPAG6, SPAG7, SPAG9, SPAG10/MFGE8, SPAG11B, SPAG12/NHP2L1, SPAG13/SSFA2, SPAG15/SPAM1, SPAG16 and SPAG17) from The Cancer Genome Atlas (TCGA) [7]. The treatment regimens for these patients included induction therapy and consolidation therapy. All patients received standard chemotherapy as induction therapy. Following induction chemotherapy, a total of 100 patients underwent chemotherapy only, whereas 73 patients received HSCT as consolidation treatment. In addition, the expression of SPAG1 in AML compared with controls was analyzed in GEPIA (http://gepia.cancer-pku.cn/).
Three independent cohorts from the Gene Expression Omnibus (GEO) database (GSE12417, GSE6891 and GSE37642) were used to validate the prognostic value of SPAG1 expression in AML. Moreover, the association of the SPAG1 expression level with the prognosis of SPAG1 expression level on prognosis of 78 and 162 cytogenetically normal AML (CN-AML) patients was analyzed in the GSE12417 dataset with the public platform GenomicScape (http://genomicscape.com/microarray/survival.php) [8, 9]. The GSE6891 dataset consisted of 461 AML patients, whereas the GSE37642 dataset comprised 562 AML patients. Kaplan–Meier analysis was performed to explore the prognostic value of SPAG1 expression in two groups with median level of SPAG1 expression as the cutoff.
Patients
The validation cohort included 131 AML patients, with 86 enrolled at diagnosis and 45 at complete remission (CR), treated at our hospital. Patients with antecedent hematological diseases or therapy-related AML were eliminated. The clinical characteristics of the cases are presented in Additional file 1: Table S1. Fifteen healthy bone marrow donors served as the controls. The age of the newly diagnosed AML patients (median 52, range 18–81) was similar to that of the AML patients at CR (controls) (median 45, range 28–66). The diagnosis and classification of AML patients followed the 2016 revised World Health Organization (WHO) and French–American–British (FAB) criteria [3, 10]. The treatment regimens of these AML cases were as reported [11,12,13]. The study protocol was approved by the Institutional Ethics Committee of the Affiliated People’s Hospital of Jiangsu University, and all the volunteers provided written informed consent.
Sample preparation, RNA isolation and reverse transcription
Clinical bone marrow (BM) specimens were sampled from the validation cohort of AML cases and controls who were treated in our hospital. We separated BM mononuclear cells (BMMNCs) and then extracted total RNA by using Lymphocyte Separation Medium (Solarbio, Beijing, China) and TRIzol reagent (Invitrogen, Carlsbad, CA), respectively. cDNA was synthesized via RNA reverse transcription as described previously [11,12,13].
Real-time quantitative PCR (RT–qPCR)
Quantized data of SPAG1 and ABL1 (housekeeping gene) transcripts were unfolded by RT–qPCR via AceQ qPCR SYBR Green Master Mix (Vazyme Biotech Co., Piscataway, NJ). The primer sequences were 5′-TCTTCTGCGTCGTGCTAC-3′ (forward) and 5′-TTATCTCCACCGCCATCT-3′ (reverse) for SPAG1 as well as 5′-TCCTCCAGCTGTTATCTGGAAGA-3′ (forward) and 5′-TCCAACGAGCGGCTTCAC-3′ (reverse) for ABL1. The relative SPAG1 transcript level was calculated based on the 2−∆∆Ct method [11,12,13].
Bioinformatics analysis
All procedures referring to bioinformatics analysis were conducted as our previous reports [14, 15]. To obtain the differentially expressed genes/miRNAs (DEGs), RNA-sequencing (mRNA and microRNA) data analysis was performed according to the raw read counts with the R/Bioconductor package “edgeR” based on the filter condition: |log2 fold change (FC)|> 1.5, false discovery rate (FDR) < 0.05 and P < 0.05. All analyses were controlled for FDR by the Benjamini–Hochberg procedure. Gene Set Enrichment Analysis (GSEA) software was used for analysis, and the enrichment pathway was set to be significant based on the nominal (NOM) P < 0.05 and FDR Q < 0.05.
Statistical analysis
Comparisons of continuous and categorical variables were performed using the Mann–Whitney’s U/Kruskal–Wallis test followed by Dunn’s post–hoc test and Pearson’s χ2/Fisher’s exact test, respectively. Both the Kaplan–Meier method (log-rank test) and Cox regression were used to analyze the intrinsic connection between SPAG1 expression and survival time, including leukemia-free survival (LFS), event-free survival (EFS) and overall survival (OS). The receiver operating characteristic (ROC) curve and area under the ROC curve (AUC) were used to determine the discriminating ability of SPAG1 expression for AML and controls. Two-sided P values < 0.05 in all statistical analyses were considered statistically significant.
Results
Identification of SPAG1 among SPAG family members linked to AML prognosis in public datasets
To explore the prognostic significance of the SPAG family members (SPAG1/2/3/4/5/6/7/9/10/13/16/17) in AML, we first determined the impact of each SPAG member on survival time (both OS and LFS) by Cox regression univariate analysis among AML in TCGA datasets. When analyzing the prognostic value of the SPAG family members in AML patients, each one was evaluated according to the difference between two groups of patients, divided by the median level of SPAG expression as the cutoff. As presented in Table 1 and Additional file 1: Table S2, only SPAG1 expression had a significant connection with OS and LFS in both AML (both P < 0.001) and non-M3 AML (both P < 0.001) as well as CN-AML (P = 0.005 and 0.006, respectively). Furthermore, Kaplan–Meier analysis also revealed that AML patients with higher SPAG1 expression showed significantly shorter OS and LFS than those with lower SPAG1 expression among AML (both P < 0.001), non-M3 AML (both P < 0.001), and CN-AML (both P = 0.004) patients (Fig. 1a and b). In addition, the expression of SPAG1 was upregulated in AML patients, as analyzed by GEPIA (Fig. 1c).

The impact of SPAG1 expression on survival of AML patients. a The effect of SPAG1 expression on overall survival in whole-cohort AML, non-M3 AML, and CN-AML from TCGA dataset. b The effect of SPAG1 expression on disease/leukemia free survival in in whole-cohort AML, non-M3 AML, and CN-AML from TCGA dataset. c SPAG1 expression in AML from TCGA dataset. d The effect of SPAG1 expression on overall survival in 78 and 162 CN-AML from the GEO dataset (GSE12417) analyzed by the online web tool Genomicscape (http://genomicscape.com/microarray/survival.php). e The effect of SPAG1 expression on event-free survival and overall survival in AML from GEO datasets (GSE6891 and GSE37642)
Next, the prognostic value of SPAG1 expression in AML was further validated in GEO datasets including GSE12417, GSE6891 and GSE37642. For GSE12417, the online platform GenomicScape (http://genomicscape.com/microarray/survival.php) also confirmed the prognostic correlation of SPAG1 expression with OS in patients with CN-AML among two independent cohorts (P = 0.0035 and 0.05, respectively, Fig. 1d). For GSE6891 and GSE37642, Kaplan–Meier analysis showed that AML patients with higher SPAG1 expression had strikingly shorter EFS and/or OS times than those with lower SPAG1 expression (P = 0.025, 0.0025 and 0.045, respectively, Fig. 1e).
Clinical implications of SPAG1 expression in AML in the TCGA dataset
SPAG1 was the only remaining SPAG member linked to AML prognosis, which prompted us to analyze the associations of SPAG1 expression with the clinical/biological characteristics of AML patients. The differences between the high and low SPAG1 groups in terms of sex, age, white blood cell (WBC) counts, peripheral blood (PB)/BM blasts, FAB classifications, cytogenetics, and gene mutations are shown in Table 2. Notably, cases with higher SPAG1 expression had markedly higher WBC counts than did those with lower SPAG1 expression (P = 0.014, Table 2). Furthermore, there were marked differences between the two groups regarding the occurrence rate of each FAB classification and cytogenetics (P = 0.024, Table 2). Cases with higher SPAG1 expression were commonly classified as FAB-M4/M5 (P = 0.058 and 0.050, respectively, Table 2). Regarding cytogenetics, patients with higher SPAG1 expression more commonly exhibited + 8 (P = 0.034) and rarely t(8;21) (P = 0.014, Table 2). We further showed SPAG1 expression among groups with + 8, t(8;21) or neither (Fig. 2a). In addition, we revealed the associations of SPAG1 expression with several of the most frequent gene mutations in AML (Table 2). Higher SPAG1 expression was markedly or nearly correlated with FLT3, DNMT3A, and WT1 mutations (P < 0.001, = 0.001 and = 0.057, respectively, Table 2). Moreover, we further compared SPAG1 expression between patients carrying or not carrying these gene mutations and observed statistical significance in subgroups divided by FLT3 and DNMT3A status (P < 0.001 and = 0.015, respectively, Fig. 2b and 2c), whereas a trend was observed in subgroups divided by WT1 status (P = 0.051, Fig. 2d).

The associations of SPAG1 expression with genetic abnormalities in AML. a SPAG1 expression in AML patients with and without chromosome 8 abnormalities from TCGA datasets. b SPAG1 expression in AML patients with and without FLT3 mutations from TCGA datasets. c SPAG1 expression in AML patients with and without DNMT3A mutations from TCGA datasets. d SPAG1 expression in AML patients with and without WT1 mutations from TCGA datasets
Further confirmation of the prognostic value of SPAG1 expression in AML in the TCGA dataset
Since a significant relationship was observed between SPAG1 expression and some common prognostic factors such as WBC, cytogenetics and gene mutations, we performed multivariate analysis by Cox regression to confirm the effect of SPAG1 expression on survival rate and demonstrated that SPAG1 expression acted as a positive independent risk factor affecting OS and LFS in whole-cohort AML (both P < 0.001), non-M3 AML (P = 0.003 and 0.005, respectively), or CN-AML patients (P = 0.001 and 0.007, respectively) (Table 3 and Additional file 1: Table S3).
Mutations in FLT3, DNMT3A and WT1 are widely accepted factors that influence AML prognosis [2, 3]. According to this study, since SPAG1 expression was significantly or nearly significantly correlated with FLT3, DNMT3A and WT1 mutations, we further investigated the prognostic value of SPAG1 expression in AML independent of these gene mutations. As Fig. 3 shows, both AML and CN-AML patients with higher SPAG1 expression also exhibited markedly shorter OS and LFS times than those with poor SPAG1 expression, regardless of the mutation status of FLT3 (Fig. 3a), DNMT3A (Fig. 3b), WT1 (Fig. 3c) or all the three genes (Fig. 3d).

The impact of SPAG1 expression on survival of AML patients with specific subtypes. a Kaplan–Meier survival curves of overall survival and disease/leukemia free survival in whole-cohort AML and CN-AML without FLT3 mutation from TCGA datasets. b Kaplan–Meier survival curves of overall survival and disease/leukemia free survival in whole-cohort AML and CN-AML without DNMT3A mutation from TCGA datasets. c Kaplan–Meier survival curves of overall survival and disease/leukemia free survival in whole-cohort AML and CN-AML without WT1 mutation from TCGA datasets. d Kaplan–Meier survival curves of overall survival and disease/leukemia free survival in whole-cohort AML and CN-AML without FLT3/DNMT3A/WT1 mutation from TCGA datasets
SPAG1 expression is a prognostic indicator for AML after HSCT in the TCGA dataset and may have a guiding effect on treatment choice between chemotherapy and HSCT
HSCT is an important consolidation treatment regimen in against disease recurrence in AML. To explore whether HSCT could nullify the negative prognostic effect of higher SPAG1 expression in AML, we analyzed the effect of HSCT intervention on prognosis in both the lower and higher SPAG1 expression groups. In the SPAG1 overexpression group, HSCT for AML patients undergoing induction therapy markedly improved OS and LFS, which was not observed for those just receiving chemotherapy (both P < 0.001, Fig. 4). However, there were no obvious differences regarding OS and LFS between the HSCT and chemotherapy sets in the group of AML patients with lower SPAG1 expression (P = 0.131 and 157, respectively, Fig. 4). To sum up the results, AML patients with SPAG1 hyperexpression may profit from HSCT, which suggests that SPAG1 expression may be used to guide therapeutic selection between HSCT and chemotherapy in AML patients undergoing induction therapy.

The effect of HSCT on survival of AML patients among different SPAG1 expression groups. Kaplan–Meier survival curves of overall survival and disease/leukemia free survival among whole-cohort AML in both lower and higher SPAG1 expression group from TCGA datasets
Molecular signatures associated with SPAG1 expression in AML in the TCGA dataset
To explore the biological network in AML caused by abnormal SPAG1 expression, we first compared the transcriptomes of AML samples with lower and higher SPAG1 expression in the TCGA set. Up to 429 mRNAs and 13 miRNAs were found to be differentially expressed between two sets based on the following conditions: |log2 FC|> 1.5, FDR < 0.05 and P < 0.05 (Fig. 5a–c and Additional file 2: Table S4). Among these DEGs, 206 mRNAs and 7 miRNAs were found to be positively correlated with SPAG1 expression, whereas 223 genes and 6 miRNAs were negatively correlated with SPAG1 expression. Positively correlated genes such as MECOM were reported to have pro-leukemia effects [16] and were associated with prognosis in AML. Negatively correlated genes such as RUNX1T1 and LEP were reported to have anti-leukemia effects and were also informative for AML prognosis [13]. Moreover, GSEA revealed that SPAG1 might participate in HOXA9 dysregulation associated with AML (Fig. 5d).

Molecular signatures associated with SPAG1 in AML. a Expression heatmap of differentially expressed genes between AML patients with lower and higher SPAG1 expression groups among TCGA datasets (FDR < 0.05, P < 0.05 and |log2 FC|> 1.5). b Volcano plot of differentially expressed genes between AML patients with lower and higher SPAG1 expression (FDR < 0.05, P < 0.05, and |log2 FC|> 1.5). c Expression heatmap of differentially expressed microRNAs between AML patients with lower and higher SPAG1 expression (FDR < 0.05, P < 0.05, and |log2 FC|> 1.5). d GSEA analysis of SPAG1 expression associated with HOXA9 dysregulation oin AML (NOM P < 0.05 and FDR Q < 0.05)
Validation of SPAG1 expression and its clinical significance in AML in our research cohort
To verify the expression pattern and clinical significance of SPAG1 expression in AML, we further investigated SPAG1 mRNA expression in BMMNC samples from 86 AML patients at diagnosis, 45 AML patients in the CR period and 15 healthy donors collected in our hospital. As expected, SPAG1 expression was significantly increased in newly diagnosed AML patients compared with healthy controls and AML patients in CR (both P < 0.001, Fig. 6a). Moreover, ROC analysis revealed that SPAG1 expression may serve as a quantifiable biomarker for distinguishing AML from controls, presenting an AUC of 0.857 (95% CI: 0.783–0.93) (P < 0.001, Fig. 6b). Significantly, AML patients who did not achieve CR after 1–2 courses of induction therapy exhibited markedly higher SPAG1 expression levels at diagnosis than those who achieved CR after 1–2 courses of induction therapy (P = 0.020, Fig. 6c). According to the set point of 1.0198 determined by ROC analysis (sensitivity of 66.3% and specificity of 100%), we grouped AML patients into two sets to analyze the prognostic significance of SPAG1 expression. Kaplan–Meier analysis demonstrated a marked tendency of shorter OS time in AML patients with high SPAG1 expression than in those with low SPAG1 expression (P = 0.034, Fig. 6d).

Validation of SPAG1 expression and its clinical significance in AML. a SPAG1 expression in 15 controls, 86 AML patients at diagnosis time, and 45 AML patients who achieved complete remission. b ROC curve analysis of SPAG1 expression in distinguishing AML from controls. c SPAG1 expression at diagnosis time in AML patients who did and did not achieve CR after 1–2 course induction therapy. d Kaplan–Meier survival curves of overall survival regarding SPAG1 expression in whole-cohort AML from our hospital
Discussion
Recent evidence has characterized SPAG family member expression together with its functional roles in cancer development. For example, SPAG1 expression could be enrolled in the early spread and adverse outcome of pancreatic adenocarcinoma and prostate cancer [17, 18]. SPAG2/UAP1 has been shown to be a promising therapeutic target for bladder cancer as well as lung adenocarcinoma [19, 20]. SPAG4 could act as a potential biomarker of glioblastoma progression and prognosis, as well as in renal cell carcinoma and lung carcinoma [21,22,23]. Moreover, SPAG5 hyperexpression was connected to poor disease-free survival in breast cancer patients, and fueled breast cancer cell proliferation [24]. Interestingly, reduced expression of SPAG6, which is transcriptionally regulated by tumor specific DNA methylation, has been revealed in non-small-cell lung cancer [25]. A direct role for aberrant SPAG9 was identified in diverse human cancers such as Kaposi’s sarcoma, gastric cancer, prostate cancer, thyroid carcinoma, liver cancer, and bladder transitional cell carcinoma [26,27,28,29,30,31,32]. Notably, accumulating studies have shown that SPAG6 expression is correlated with the pathogenesis of myelodysplastic syndrome (MDS) and Burkett lymphoma (BL) [33,34,35,36,37]. Consequently, SPAG proteins serve as a novel type of CT antigen with contributions to cancer formation and are likely to be novel targets for tumor targeted therapies.
This study was the first to reveal SPAG1 expression as uniquely associated with poor prognosis in AML among all SPAG family members through both analysis of public data and validation in a research cohort. It was demonstrated that SPAG1 expression could be a promising prognostic biomarker and could be used to optimize the choice of therapy between chemotherapy and HSCT in AML. Unlike SPAG6, SPAG1 expression has rarely been studied in hematological malignances. Conversely, several reports have examined at the relationships between SPAG1 and solid tumors. Shamsara et al. demonstrated that the amplification of SPAG1 was associated with decreased survival in patients with prostate cancer [18]. Moreover, SPAG1 is an early expressed gene in pancreatic tumorigenesis and can promote the activity of cancer cells [17]. Lin et al. showed that SPAG1 expression was a crucial variable related to many clinicopathological features and to RFS in breast cancer [38]. Functionally, SPAG1 acts as an inhibitor of breast cancer cell proliferation and colony formation during breast cancer pathogenesis and development [38]. Since there was no deep insight into SPAG1 in AML, further mechanistic studies are essential for investigating the possible role of SPAG1 in leukemogenesis and AML development.
The current study also identified a significant association between SPAG1 expression and genetic (both cytogenetic and molecular) abnormalities in AML. We first found the associations of SPAG1 expression with FAB-M4/M5 disease, suggesting that SPAG1 expression may play a role in monocyte differentiation and monocyte leukemogenesis. In terms of cytogenetics, SPAG1 expression was positively correlated with +8 but negatively associated with t(8;21) (q22;q22). Since the SPAG1 gene is located in 8q22.2, it is not surprising that aberrant SPAG1 expression was associated with these chromosome abnormalities. Notably, further studies are needed to determine whether the functions of these chromosomal abnormalities during leukemogenesis occur through aberrant SPAG1 expression. Regarding gene mutations, SPAG1 expression was associated with FLT3 and DNMT3A mutations in AML, but the exact relationship between SPAG1 expression and these gene mutations still unclear. Importantly, there is no evidence showing the association of FLT3 and DNMT3A mutations with the above chromosome abnormalities. Consequently, we need to obtain deeper insight into the underlying mechanism of SPAG1 expression in leukemogenesis caused by FLT3 and/or DNMT3A mutations.
In general, our discoveries suggested that SPAG1 hyperexpression may function independently as a prognostic biomarker and assist treatment selection between HSCT and chemotherapy in AML.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–52.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, Levine RL, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz M, Sierra J, Tallman MS, Tien HF, Wei AH, Löwenberg B, Bloomfield CD. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Arber VJW, DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M, Vardiman JW,. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Burd A, Levine RL, Ruppert AS, Mims AS, Borate U, Stein EM, Patel P, Baer MR, Stock W, Deininger M, Blum W, Schiller G, Olin R, Litzow M, Foran J, Lin TL, Ball B, Boyiadzis M, Traer E, Odenike O, Arellano M, Walker A, Duong VH, Kovacsovics T, Collins R, Shoben AB, Heerema NA, Foster MC, Vergilio JA, Brennan T, Vietz C, Severson E, Miller M, Rosenberg L, Marcus S, Yocum A, Chen T, Stefanos M, Druker B, Byrd JC. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: feasibility and preliminary efficacy of the Beat AML Master Trial. Nat Med. 2020;26(12):1852–8.
Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5(8):615–25.
Siliņa K, Zayakin P, Kalniņa Z, Ivanova L, Meistere I, Endzeliņš E, Abols A, Stengrēvics A, Leja M, Ducena K, Kozirovskis V, Linē A. Sperm-associated antigens as targets for cancer immunotherapy: expression pattern and humoral immune response in cancer patients. J Immunother. 2011;34(1):28–44.
Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, Baty JD, Fulton LL, Fulton R, Heath SE, Kalicki-Veizer J, Kandoth C, Klco JM, Koboldt DC, Kanchi KL, Kulkarni S, Lamprecht TL, Larson DE, Lin L, Lu C, McLellan MD, McMichael JF, Payton J, Schmidt H, Spencer DH, Tomasson MH, Wallis JW, Wartman LD, Watson MA, Welch J, Wendl MC, Ally A, Balasundaram M, Birol I, Butterfield Y, Chiu R, Chu A, Chuah E, Chun HJ, Corbett R, Dhalla N, Guin R, He A, Hirst C, Hirst M, Holt RA, Jones S, Karsan A, Lee D, Li HI, Marra MA, Mayo M, Moore RA, Mungall K, Parker J, Pleasance E, Plettner P, Schein J, Stoll D, Swanson L, Tam A, Thiessen N, Varhol R, Wye N, Zhao Y, Gabriel S, Getz G, Sougnez C, Zou L, Leiserson MD, Vandin F, Wu HT, Applebaum F, Baylin SB, Akbani R, Broom BM, Chen K, Motter TC, Nguyen K, Weinstein JN, Zhang N, Ferguson ML, Adams C, Black A, Bowen J, Gastier-Foster J, Grossman T, Lichtenberg T, Wise L, Davidsen T, Demchok JA, Shaw KR, Sheth M, Sofia HJ, Yang L, Downing JR, Eley G. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, Maharry K, Paschka P, Larson RA, Berdel WE, Büchner T, Wörmann B, Mansmann U, Hiddemann W, Bohlander SK, Buske C; Cancer and Leukemia Group B; German AML Cooperative Group. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112:4193–201.
Kassambara A, Rème T, Jourdan M, Fest T, Hose D, Tarte K, Klein B. GenomicScape: an easy-to-use web tool for gene expression data analysis. Application to investigate the molecular events in the differentiation of B cells into plasma cells. PLoS Comput Biol. 2015;11:e1004077.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposed revised criteria for the classification of acute myeloid leukaemia. A report of the French-American-British Cooperative Group. Ann Intern Med. 1985;103(4):620–5.
Zhang TJ, Xu ZJ, Gu Y, Wen XM, Ma JC, Zhang W, Deng ZQ, Leng JY, Qian J, Lin J, Zhou JD. Identification and validation of prognosis-related DLX5 methylation as an epigenetic driver in myeloid neoplasms. Clin Transl Med. 2020;10(2):e29.
Zhou JD, Zhang TJ, Xu ZJ, Deng ZQ, Gu Y, Ma JC, Wen XM, Leng JY, Lin J, Chen SN, Qian J. Genome-wide methylation sequencing identifies progression-related epigenetic drivers in myelodysplastic syndromes. Cell Death Dis. 2020;11(11):997.
Zhang TJ, Xu ZJ, Gu Y, Ma JC, Wen XM, Zhang W, Deng ZQ, Qian J, Lin J, Zhou JD. Identification and validation of obesity-related gene LEP methylation as a prognostic indicator in patients with acute myeloid leukemia. Clin Epigenetics. 2021;13(1):16.
Chu MQ, Zhang TJ, Xu ZJ, Gu Y, Ma JC, Zhang W, Wen XM, Lin J, Qian J, Zhou JD. EZH2 dysregulation: potential biomarkers predicting prognosis and guiding treatment choice in acute myeloid leukaemia. J Cell Mol Med. 2020;24(2):1640–9.
Xu ZJ, Ma JC, Zhou JD, Wen XM, Yao DM, Zhang W, Ji RB, Wu DH, Tang LJ, Deng ZQ, Qian J, Lin J. Reduced protocadherin17 expression in leukemia stem cells: the clinical and biological effect in acute myeloid leukemia. J Transl Med. 2019;17(1):102.
Hinai AA, Valk PJ. Review: Aberrant EVI1 expression in acute myeloid leukaemia. Br J Haematol. 2016;172(6):870–8.
Neesse A, Gangeswaran R, Luettges J, Feakins R, Weeks ME, Lemoine NR, Crnogorac-Jurcevic T. Sperm-associated antigen 1 is expressed early in pancreatic tumorigenesis and promotes motility of cancer cells. Oncogene. 2007;26(11):1533–45.
Shamsara E, Shamsara J. Bioinformatics analysis of the genes involved in the extension of prostate cancer to adjacent lymph nodes by supervised and unsupervised machine learning methods: the role of SPAG1 and PLEKHF2. Genomics. 2020;112(6):3871–82.
Puttamallesh VN, Deb B, Gondkar K, Jain A, Nair B, Pandey A, Chatterjee A, Gowda H, Kumar P. Quantitative proteomics of urinary bladder cancer cell lines identify UAP1 as a potential therapeutic target. Genes. 2020;11(7):763.
Wang X, Chen X, Liu H. Expression and bioinformatics-based functional analysis of UAP1 in lung adenocarcinoma. Cancer Manag Res. 2020;12:12111–21.
Zhao J, Liu B, Yang JA, Tang D, Wang X, Chen Q. Human sperm-associated antigen 4 as a potential biomarker of glioblastoma progression and prognosis. NeuroReport. 2019;30(6):446–51.
Ji Y, Jiang J, Huang L, Feng W, Zhang Z, Jin L, Xing X. Sperm-associated antigen 4 (SPAG4) as a new cancer marker interacts with Nesprin3 to regulate cell migration in lung carcinoma. Oncol Rep. 2018;40(2):783–92.
Shoji K, Murayama T, Mimura I, Wada T, Kume H, Goto A, Ohse T, Tanaka T, Inagi R, van der Hoorn FA, Manabe I, Homma Y, Fukayama M, Sakurai T, Hasegawa T, Aburatani H, Kodama T, Nangaku M. Sperm-associated antigen 4, a novel hypoxia-inducible factor 1 target, regulates cytokinesis, and its expression correlates with the prognosis of renal cell carcinoma. Am J Pathol. 2013;182(6):2191–203.
Canu V, Donzelli S, Sacconi A, Lo Sardo F, Pulito C, Bossel N, Di Benedetto A, Muti P, Botti C, Domany E, Bicciato S, Strano S, Yarden Y, Blandino G. Aberrant transcriptional and post-transcriptional regulation of SPAG5, a YAP-TAZ-TEAD downstream effector, fuels breast cancer cell proliferation. Cell Death Differ. 2021;28(5):1493–511.
Li W, Wang F, Shi J, Feng Q, Chen Y, Qi X, Wang C, Lu H, Lu Z, Jia X, Yan Q, Gao SJ, Lu C. Sperm associated antigen 9 promotes oncogenic KSHV-encoded interferon regulatory factor-induced cellular transformation and angiogenesis by activating the JNK/VEGFA pathway. PLoS Pathog. 2020;16(8):e1008730.
Altenberger C, Heller G, Ziegler B, Tomasich E, Marhold M, Topakian T, Müllauer L, Heffeter P, Lang G, End-Pfützenreuter A, Döme B, Arns BM, Klepetko W, Zielinski CC, Zöchbauer-Müller S. SPAG6 and L1TD1 are transcriptionally regulated by DNA methylation in non-small cell lung cancers. Mol Cancer. 2017;16(1):1.
Sun QH, Yin ZX, Li Z, Tian SB, Wang HC, Zhang FX, Li LP, Zheng CN, Kong S. miR-874 inhibits gastric cancer cell proliferation by targeting SPAG9. BMC Cancer. 2020;20(1):522.
Xiao C, Li M, Huang Q, Si-Tu J. SPAG9 promotes prostate cancer proliferation and metastasis via MAPK signaling pathway. Am J Transl Res. 2019;11(8):5249–60.
Yan P, Su Z, Zhang Z, Gao T. LncRNA NEAT1 enhances the resistance of anaplastic thyroid carcinoma cells to cisplatin by sponging miR-9-5p and regulating SPAG9 expression. Int J Oncol. 2019;55(5):988–1002.
Luo S, Ren B, Zou G, Liu J, Chen W, Huang Y, Chen X, Fu Y. SPAG9/MKK3/p38 axis is a novel therapeutic target for liver cancer. Oncol Rep. 2019;41(4):2329–36.
Li X, Jiang F, Wang X, Gu X. SPAG9 regulates HEF1 expression and drives EMT in bladder transitional cell carcinoma via rac1 signaling pathway. Am J Cancer Res. 2018;8(12):2467–80.
Pan J, Yu H, Guo Z, Liu Q, Ding M, Xu K, Mao L. Emerging role of sperm-associated antigen 9 in tumorigenesis. Biomed Pharmacother. 2018;103:1212–6.
Zhang R, Zhu H, Yuan Y, Wang Y, Tian Z. SPAG6 promotes cell proliferation and inhibits apoptosis through the PTEN/PI3K/AKT pathway in Burkitt lymphoma. Oncol Rep. 2020;44(5):2021–30.
Zhang M, Luo J, Luo X, Liu L. SPAG6 silencing induces autophagic cell death in SKM-1 cells via the AMPK/mTOR/ULK1 signaling pathway. Oncol Lett. 2020;20(1):551–60.
Jiang M, Chen Y, Deng L, Luo X, Wang L, Liu L. Upregulation of SPAG6 in myelodysplastic syndrome: knockdown inhibits cell proliferation via AKT/FOXO signaling pathway. DNA Cell Biol. 2019;38(5):476–84.
Yin J, Li X, Zhang Z, Luo X, Wang L, Liu L. SPAG6 silencing induces apoptosis in the myelodysplastic syndrome cell line SKM-1 via the PTEN/PI3K/AKT signaling pathway in vitro and in vivo. Int J Oncol. 2018;53(1):297–306.
Yang B, Wang L, Luo X, Chen L, Yang Z, Liu L. SPAG6 silencing inhibits the growth of the malignant myeloid cell lines SKM-1 and K562 via activating p53 and caspase activation-dependent apoptosis. Int J Oncol. 2015;46(2):649–56.
Lin S, Lv Y, Zheng L, Mao G, Peng F. Expression and prognosis of sperm-associated antigen 1 in human breast cancer. Onco Targets Ther. 2021;14:2689–98.
Acknowledgements
None.
Funding
The work was supported by National Natural Science Foundation of China (81900166), Zhenjiang Clinical Research Center of Hematology (SS2018009), Social Development Foundation of Zhenjiang (SH2020055), Medical Field of Zhenjiang “Jin Shan Ying Cai” Project, Scientific Research Foundation of Affiliated People's Hospital of Jiangsu University for PhD (KFB202002), Key Project of Medical Education Collaborative Innovation Fund of Jiangsu University.
Author information
Authors and Affiliations
Contributions
JZ, TZ and JL conceived and designed the experiments; YG and MC performed the experiments; JZ and ZX analyzed the data; QY collected the clinical data; JL provided the technical support; JZ wrote the paper; all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
The present study approved by the Ethics Committee and Institutional Review Board of the Affiliated People’s Hospital of Jiangsu University, in compliance with the Declaration of Helsinki. Written informed consents were obtained from all enrolled individuals prior to their participation.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Clinic-pathologic characteristics of AML patients in our research cohort and Cox regression univariate and multivariate analysis of variables for leukemia free survival in AML patients in TCGA dataset.
Additional file 2:
Different expressed genes correlated with SPAG1 expression.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gu, Y., Chu, Mq., Xu, Zj. et al. Comprehensive analysis of SPAG1 expression as a prognostic and predictive biomarker in acute myeloid leukemia by integrative bioinformatics and clinical validation. BMC Med Genomics 15, 38 (2022). https://doi.org/10.1186/s12920-022-01193-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01193-0