
Abstract
Background
Relative haplotype dosage (RHDO) approach has been applied in noninvasive prenatal diagnosis (NIPD) of Duchenne muscular dystrophy (DMD). However, the RHDO procedure is relatively complicated and the parental haplotypes need to be constructed. Furthermore, it is not suitable for the diagnosis of de novo mutations or mosaicism in germ cells. Here, we investigated NIPD of DMD using a relative mutation dosage (RMD)-based approach—cell-free DNA Barcode-Enabled Single-Molecule Test (cfBEST), which has not previously been applied in the diagnosis of exon deletion.
Methods
Five DMD families caused by DMD gene point mutations or exon deletion were recruited for this study. After the breakpoints of exon deletion were precisely mapped with multiple PCR, the genotypes of the fetuses from the five DMD families were inferred using cfBEST, and were further validated by invasive prenatal diagnosis.
Results
The cfBEST results of the five families indicated that one fetus was female and did not carry the familial molecular alteration, three fetuses were carriers and one was male without the familial mutation. The invasive prenatal diagnosis results were consistent with those of the cfBEST procedure.
Conclusion
This is the first report of NIPD of DMD using the RMD-based approach. We extended the application of cfBEST from point mutation to exon deletion mutation. The results showed that cfBEST would be suitable for NIPD of DMD caused by different kinds of mutation types.
Similar content being viewed by others

Background
Duchenne/Becker muscular dystrophy (DMD/BMD) are X-linked recessive diseases that are caused by defects in the DMD gene [1]. It is one of the most common severe, untreatable neuromuscular disease, that presents as a progressive muscle disorder. DMD is a lethal inherited muscle disease mostly in young boys, with an incidence at birth of 1/3800–1/6300 [2]. BMD is a milder form with onset later in childhood and a low incidence of 1 in 20,000–30,000 males [3]. As no curative therapy is currently available, the importance of prevention is paramount. Traditional invasive procedures including chorion villus sampling and amniocentesis could raise anxiety in pregnant women and carry the risk of miscarriage or injury to the fetuses and the mothers [4]. Noninvasive procedures would be more easily accepted by pregnant women and their families [5].
The discovery of cell free fetal DNA (cffDNA) in the maternal plasma and the next generation sequencing (NGS) technology made the non-invasive prenatal testing (NIPT) possible [6, 7]. NIPT is already widely used in screening for aneuploidies [8,9,10,11]. Entire fetal genomes are represented in the maternal plasma at a constant relative proportion, which makes it possible to diagnose fetal genetic disorders prenatally in a noninvasive way [12]. To date, different noninvasive prenatal diagnosis strategies have been presented including the relative mutation dosage (RMD) approach and the relative haplotype dosage analysis (RHDO) [13,14,15,16]. The RHDO approach has been successfully used in DMD [17,18,19,20,21], but the procedures are relatively complicated [17, 22].
A new technology called cell-free DNA barcode-enabled single-molecule test (cfBEST) is able to detect monogenetic disease noninvasively with a high sensitivity and specificity [19]. With the strategy of cfBEST, it is possible to count the original and real molecules accurately by reducing the influence of random sequencing errors, unbalanced PCR amplification, and different PCR amplification efficiency between fetal and maternal cell free DNA (cfDNA). Here, we report on the RMD-based NIPD for DMD caused by point mutations or small insertions/deletions by target-position sequencing of the maternal plasma using cfBEST. Moreover, after precisely mapping the deletion breakpoints of a sample with one exon deletion, we also inferred the genotype of the fetus using cfBEST. All of the results were confirmed with subsequent invasive procedures.
Materials and methods
Study design
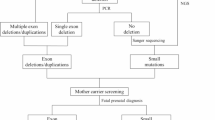
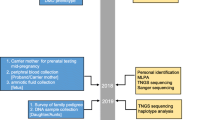
The study workflow is illustrated in Fig. 1. This clinical research study was conducted at the Prenatal Diagnosis Center of the First Affiliated Hospital of Zhengzhou University. DMD families that had a previous child affected with DMD from their first pregnancy were recruited to the study. After genetic counselling, all the probands and the mothers elected to undergo molecular diagnosis to identify the causative pathogenic DMD mutations. For clinical management of second pregnancies, NIPDs were blindly performed in parallel with invasive prenatal diagnosis (IPD). NIPD was performed through cfBEST after mapping the breakpoints. IPD was performed through chorionic villus sampling (CVS) followed by Sanger sequencing or Multiplex-ligation dependent probe amplification (MLPA). Finally, we assessed cfBEST for DMD through comparing the results of NIPD with IPD.

The workflow of this study. First, we collected samples from DMD families and identified the mutation genotypes of each case. Then, noninvasive prenatal diagnosis and invasive prenatal diagnosis were blindly conducted by two independent groups. Finally, we assessed the performance of cfBEST for DMD
Sample collection and DNA extraction
All subjects gave their informed consent for inclusion before they participated in the study. Samples were collected with consent from subjects at the First Affiliated Hospital of Zhengzhou University. A cohort of five DMD families were recruited. The characteristics of the families are summarized in Table 1. A healthy man with no DMD mutation was chosen to be the control. Each family cohort consisted of a proband and his mother with a second new singleton pregnancy. Blood samples (2 mL) from the probands and the control were collected. In addition, 10 mL blood samples were collected from pregnant women and 5 mg of chorionic villus from the fetuses were collected via CVS. DNA in white blood cells of the proband and the normal healthy control sample were extracted using the DNeasy Blood Tissue Kit (Qiagen, Dusseldorf, Germany). Genomic DNA was extracted from fetal cells using the Lab-Aid Nucleic Acid (DNA) Isolation Kit (Zeesan, Xiamen, China) according to the manufacturer’s instructions. Blood samples (10 mL) from the pregnant women were centrifuged twice to collect the plasma, which was used to extract cfDNA using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Dusseldorf, Germany).
Mapping breakpoints of the proband with exon 12 deletion of the DMD gene
We designed 19 pairs of primers (Additional file 1: Table S1) between exon 11 and exon 13 of the DMD gene (GenBank NG_012232) at contiguous intervals of approximately 1.25 kb (Fig. 2). This enabled division of the region between exon 11 and exon 13 into 19 parts (P1-P19) (Fig. 2), with each part being amplified by its relevant primers. PCR was conducted using the primers designed above in the proband and the healthy normal control sample. The presence/absence of these amplified products were verified by agarose gel electrophoresis. If the parts could not be amplified for the proband, the primers would be in the deletion region, as the primers could not bind to the templates. It was then possible to narrow down the region that included the breakpoints. Finally, genomic regions spanning the breakpoints were amplified and sequenced using the new primers (Additional file 1: Table S2) designed according to the results of the above agarose gel electrophoresis.

Localization of the 19 parts (P1-P19) between exon 11 and exon 13 of the DMD gene divided by the relevant primers. F: forward primer, R: reverse primer. E11-E13: exon 11-exon 13 of DMD gene
Primers design for the DMD cfBEST assay
Four primers were designed for each DMD mutation site based on the principle of cfBEST [19] (Additional file 1: Table S3). Furthermore, cfBEST primers for chromosome Y had been designed to detect fetal gender (Additional file 1: Table S4).
DNA sequencing library preparation and sequencing
cfBEST tag adaptors were ligated to cfDNA after A-tailing was added to cfDNA using the KAPA Hyper Prep Kit (Kapa Biosystems, Boston, USA). The pre-library amplified from cfDNA with an index primer and a universal primer was split into two parts [named “F” and “R”, Additional file 2: Fig. S1 shows only the “F” part)] to be amplified separately with two rounds of PCR. The two PCR products were pooled to execute a third PCR with universal primers U1 and U2 (Additional file 2: Fig. S1). Finally, the sequencing libraries from the three rounds of PCR were subjected to massively parallel sequencing on the NextSeq CN500 (Illumina, San Diego, USA).
Bioinformatics analysis and genotyping by cfBEST
The noise sequences were eliminated using the bioinformatic filtering step. After preprocessing of the raw sequences with barcode trimming, adapter trimming, and primer recognition, the preprocessed reads were aligned to hg19 and further filtered. After calling the consensus sequence, the allele would be counted. The allele count was modeled in a mixed binomial distribution, and the fetal cfDNA fraction was deduced using an EM algorithm with the 109 SNP [19]. Fetal gender was determined via the results of allele count on the Y chromosome, as there would be almost no count for female fetuses. The expected ratio of mutant alleles in plasma would be deduced according to the condition of the pregnant woman and the fetus (Additional file 1: Table S5). The final genotyping results would be deduced using an in-house R-script.
Invasive prenatal diagnosis
Fetal cells were retrieved by CVS at around 12 gestational weeks. The mutations were tested by PCR and Sanger sequencing or MLPA using standard protocols in the laboratory. Maternal contamination was excluded via quantitative fluorescent polymerase chain reaction (QF-PCR) using the GoldeneyeTM DNA ID System 20A Kit (Peoplespot, Beijing, China) in accordance with the manufacturer’s instructions.
Results
Family cases
For Family D1, the causative mutation was identified as insertion A between physical position 32,361,292–32,361,293. The causative mutations were nonsense mutations for Families D2-D4. Family D5 had an exon 12 deletion. All of the pregnant women from the DMD families except Family D4 were DMD carriers with a gestational age of about 12 weeks. For Family D4, the mutation of the proband may have been de novo or the mother of the proband may have had germline mosaicism. The age of the pregnant women ranged from 26 to 32 years of age. More details are shown in Table 1.
Map of the breakpoints
The specific primers in Additional file 1: Table S1 were used to amplify the genomic DNA of the proband from Family D5 and a normal control sample, respectively. The results showed that there was no signal in parts P11–P15 of the proband of Family D5, which indicated that the breakpoints of this sample may be localized in these parts (Fig. 3). In order to accurately locate the breakpoints, two more reverse primers were designed in the primers R14–F16 region (Additional file 1: Table S2). The forward primer of P11 was paired with the two new reverse primers respectively to amplify the genomic DNA (gDNA) of the proband from Family D5 and the control (Fig. 3). There was no signal in either 2P1 or 2P2 in the control sample, with the PCR products being about 10 kb and too long to be amplified, as the PCR conditions were suitable for PCR products with about of around 1.25 kb in size. There was no signal in the region 2P1 in the proband from Family D5, which indicated that this region was deleted. The signal of 2P2 in the proband from Family D5 indicated that the breakpoints were localized in this region. Later, the PCR products of the part 2P2 of the proband from Family D5 was sequenced and aligned with the DMD gene to map the breakpoints. Finally, the breakpoints of this sample were localized at 32,625,751 and 32,635,984 on chromosome X (g.32625752_32635983del) (Table 2).

The results of PCR using the same primers with different templates (normal control, and proband of Family D5). P11-P15 had no signal in proband of Family D5 compared with the normal control whose DMD gene was totally normal. 2P1 had no signal in both the control and proband of family D5. 2P2 had no signal in the normal control, but there was signal for proband of Family D5. All of these indicate that the breakpoints were localized in part 2P2
NIPD of DMD
NIPD was performed on plasma samples from the five second pregnancies at risk for DMD using the cfBEST assay targeting the five different DMD mutations. Plasma samples were sent to an independent laboratory for analysis without information on the fetal genotypes determined by IPD. The fetal fraction of the plasma tested was > 5%, except for the sample from Family D4, which was 3.00%. Finally, the NIPD results showed that the fetus was female and did not inherit the maternal mutation for Family D1. For families D2, D3 and D5, the fetuses were females and carried the same mutations as the proband. For family D4, the fetus was male and did not inherit the maternal mutation.
Invasive prenatal diagnosis through CVS
For Families D1-D4, genotyping of the fetuses took place via Sanger sequencing (Fig. 4), while, MLPA was used to genotype the fetus for Family D5 (Fig. 4). The IPD results were consistent with the deduced fetal genotypes determined for each of the five pregnancies by NIPD (Table 2).

The invasive prenatal diagnosis results of Families D1-D5 with Sanger sequencing (Families D1-D4) or MLPA (Family D5). For Families D1 and D4, the fetuses did not inherit the maternal mutation. For Families D2 and D3, the fetuses were female carriers. The fetus was a carrier with exon 12 heterozygous deletion for Family D5
Discussion
In the current study, both NIPD and IPD were performed on five pregnant women from DMD families caused by different mutation types to assess the performance of the cfBEST assay for quantification of different kinds of DMD mutation types in the cfDNA of pregnancy plasma. In all five cases with three different mutation types, including substitution mutation, small insertion, and exon deletion, the fetal genotypes were correctly inferred by the mutation percentage according to the cfBEST assay. The fetal genotypes determined by cfBEST were consistent with the IPD results. The results showed that cfBEST was suitable for different kinds of mutation types.
DMD is the second monogenic disease, after β-Thalassemia, that has been noninvasively detected using cfBEST [19]. This was the first application of cfBEST for exon deletion and X-linked diseases. Our study further indicated that cfBEST can be a universally applicable system for any other monogenic diseases with minor modifications.
Expanded noninvasive prenatal testing has already been used to screen chromosome aneuploidy and genome copy number variations regarding several mega base pairs [9]. Point mutations and small indels can be detected through the strategy of RHDO and RMD [23]. However, there are few effective methods to detect the copy number variation (CNV) with a size of around several dozens base to mega base using the RMD based strategy. We localized the breakpoints in the DMD gene using the proband’s genomic DNA, then cfBEST was used for noninvasive detection. The information of the breakpoints could be used for conventional prenatal diagnosis and preimplantation genetic diagnosis (PGD), providing a double guarantee for conventional methods. As reported previously [19], a minimum of 1000 unique reads were required for precisely calculating mutation ratios. In our study using the cfBEST approach, the total unique reads of Families D1 and D5 were 380 and 695, respectively, which were lower than the minimal unique reads. However, the fetal genotypes deduced by cfBEST were in accordance with the IPD results (Table 2). The high concentrations of the cfDNA from these two cases, 13.06% and 10.2% respectively, compensated the requirement of > 1000 total unique reads, enabling accurate diagnostic results from cfBEST. This indicated that the impact of the least unique reads on the accuracy of cfBEST also relied on the fetal fraction. The threshold of the parameters could be set according to the large dataset with comprehensive consideration of all of the influencing factors in the future.
With advances in molecular genetics, mosaicism in germ cells has been recognized as a relatively frequent cause of genetic disorders. Germline mosaicism has been found in 10% of the cases in a large series of sporadic patients with DMD [24]. Mothers with no detectable mutation in their lymphocytes may still have an elevated recurrence risk due to germline mosaicism [25]. In Family D4, the pregnant woman did not carry the mutation in her lymphocytes, indicating that the proband could be a de novo mutation or the woman could have germinal mosaicism, making it essential to perform a prenatal diagnosis in this case. The fetal DNA fraction of the pregnant woman from Family D4 was 3.00%, which was lower than the cutoff of 5% set previously [19]. However, the fetus was male, according to the Y chromosome signal from the sequencing results. If the fetus had been a patient, the mutation ratio would theoretically be at least 1.50%, which could be precisely detected via cfBEST, as this test shows excellent performance in detecting samples containing 0–0.5% mutations [19]. The mutation ratio of this case was 0.00% (Table 2), which indicated that the fetus was male and did not inherit the maternal mutation. Finally, the IPD result confirmed the fetal genotype given by cfBEST. Therefore, a low fetal fraction would not impact the accuracy of cfBEST results in such cases. The minimum fetal DNA fraction required in these cases should be determined and validated in extended DMD families.
Compared with these haplotype-based strategies used to detect DMD noninvasively [20, 26], cfBEST did not require construction of the haplotypes of the parents with no need for blood collected from the father, meaning that it would be less expensive and easy to perform [17, 27]. cfBEST could also be suitable for de novo mutations or mosaicism in germ cells [27], which are invalid for RHDO. Besides, if recombination events occur, the possibility of an incorrect fetal genotype classification for RHDO would increase [17, 20]. As for large segment deletion or duplication, we could map the breakpoints and conduct the detection targeting the breakpoints just as with the sample from Family D5 using cfBEST.
This is a preliminary study and additional improvements are necessary to ensure the highest clinical sensitivity and specificity of our NIPD strategy for DMD, despite the high performance of the current assay. Firstly, as this study only involved five DMD families, more cases should be recruited to further evaluate and optimize our assay in the future. Secondly, the minimum fetal fraction and unique reads required for our analyses will be determined according to the more data acquired in the future.Besides, enrichment of the fetal DNA would improve the fetal fraction to extend the application of our method to earlier gestational ages or to cases with a low fetal fraction due to other reasons.
Conclusion
In summary, our study demonstrated the feasibility of cfBEST for DMD caused by different kinds of mutation types including exon deletion. With further validation and innovation, our assay could become a better clinical application for noninvasive prenatal diagnosis of pregnancies at risk for DMD. Our study will provide a new approach for safely and non-invasively examining fetuses at a high risk of DMD.
Availability of data and materials
As public access was not consented for by the subjects in our study, the raw datasets are not publicly available. However, they are available from the corresponding author upon reasonable request.
Abbreviations
- DMD:
-
Duchenne muscular dystrophy
- NIPD:
-
Noninvasive prenatal diagnosis
- RMD:
-
Relative mutation dosage
- cfBEST:
-
Cell-free DNA Barcode-Enabled Single-Molecule Test
- BMD:
-
Becker muscular dystrophy
- cffDNA:
-
Cell free fetal DNA
- NGS:
-
Next generation sequencing
- NIPT:
-
Non-invasive prenatal testing
- RHDO:
-
Relative haplotype dosage analysis
- cfDNA:
-
Cell free DNA
- MLPA:
-
Multiplex ligation-dependent probe amplification
- CVS:
-
Chorion villus sampling
- Het:
-
Heterozygous
- N:
-
Normal wild-type allele
- IPD:
-
Invasive prenatal diagnosis
References
Guo R, Zhu G, Zhu H, Ma R, Peng Y, Liang D, et al. DMD mutation spectrum analysis in 613 Chinese patients with dystrophinopathy. J Hum Genet. 2015;60:435–42.
Landfeldt E, Lindgren P, Bell CF, Schmitt C, Guglieri M, Straub V, et al. The burden of Duchenne muscular dystrophy: an international, cross-sectional study. Neurology. 2014;83:529–36.
Bushby KMD, Thambyayah M, Gardner-Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet. 1991;337:1022–4.
Mujezinovic F, Alfirevic Z. Procedure-related complications of amniocentesis and chorionic villous sampling: a systematic review. Obstet Gynecol. 2007;110:687–94.
Scotchman E, Chandler NJ, Mellis R, Chitty LS. Noninvasive prenatal diagnosis of single-gene diseases: the next frontier. Clin Chem. 2019;66:53–60.
Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485–7.
Chiu RW, Chan KC, Gao Y, Lau VY, Zheng W, Leung TY, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci USA. 2008;105:20458–63.
Allyse MA, Wick MJ. Noninvasive prenatal genetic screening using cell-free DNA. JAMA. 2018;320:591–2.
Liang D, Cram DS, Tan H, Linpeng S, Liu Y, Sun H, et al. Clinical utility of noninvasive prenatal screening for expanded chromosome disease syndromes. Genet Med. 2019;21:1998–2006.
Xu L, Huang H, Lin N, Wang Y, He D, Zhang M, et al. Non-invasive cell-free fetal DNA testing for aneuploidy: multicenter study of 31 515 singleton pregnancies in southeastern China. Ultrasound Obstet Gynecol. 2020;55:242–7.
Bianchi DW, Parker RL, Wentworth J, Madankumar R, Saffer C, Das AF, et al. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med. 2014;370:799–808.
Lo YM, Chan KC, Sun H, Chen EZ, Jiang P, Lun FM, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2:61ra91.
Lun FM, Tsui NB, Chan KC, Leung TY, Lau TK, Charoenkwan P, et al. Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc Natl Acad Sci USA. 2008;105:19920–5.
Lv W, Wei X, Guo R, Liu Q, Zheng Y, Chang J, et al. Noninvasive prenatal testing for Wilson disease by use of circulating single-molecule amplification and resequencing technology (cSMART). Clin Chem. 2015;61:172–81.
Papasavva T, Kalikas I, Kyrri A, Kleanthous M. Arrayed primer extension for the noninvasive prenatal diagnosis of beta-thalassemia based on detection of single nucleotide polymorphisms. Ann N Y Acad Sci. 2008;1137:302–8.
Tsui NB, Kadir RA, Chan KC, Chi C, Mellars G, Tuddenham EG, et al. Noninvasive prenatal diagnosis of hemophilia by microfluidics digital PCR analysis of maternal plasma DNA. Blood. 2011;117:3684–91.
Jang SS, Lim BC, Yoo SK, Shin JY, Kim KJ, Seo JS, et al. Targeted linked-read sequencing for direct haplotype phasing of maternal DMD alleles: a practical and reliable method for noninvasive prenatal diagnosis. Sci Rep. 2018;8:8678.
Yoo SK, Lim BC, Byeun J, Hwang H, Kim KJ, Hwang YS, et al. Noninvasive prenatal diagnosis of duchenne muscular dystrophy: comprehensive genetic diagnosis in carrier, proband, and fetus. Clin Chem. 2015;61:829–37.
Yang X, Zhou Q, Zhou W, Zhong M, Guo X, Wang X, et al. A cell-free DNA barcode-enabled single-molecule test for noninvasive prenatal diagnosis of monogenic disorders: application to β-Thalassemia. Adv Sci (Weinh). 2019;6:1802332.
Xu Y, Li X, Ge HJ, Xiao B, Zhang YY, Ying XM, et al. Haplotype-based approach for noninvasive prenatal tests of Duchenne muscular dystrophy using cell-free fetal DNA in maternal plasma. Genet Med. 2015;17:889–96.
Chen M, Chen C, Huang X, Sun J, Jiang L, Li Y, et al. Noninvasive prenatal diagnosis for duchenne muscular dystrophy based on the direct haplotype phasing. Prenat Diagn. 2020;40:918–24.
Verhoef TI, Hill M, Drury S, Mason S, Jenkins L, Morris S, et al. Non-invasive prenatal diagnosis (NIPD) for single gene disorders: cost analysis of NIPD and invasive testing pathways. Prenat Diagn. 2016;36:636–42.
Allen S, Young E, Bowns B. Noninvasive prenatal diagnosis for single gene disorders. Curr Opin Obstet Gynecol. 2017;29:73–9.
Horn D, Delaunoy JP, Kunze J. Prenatal diagnosis in Coffin-Lowry syndrome demonstrates germinal mosaicism confirmed by mutation analysis. Prenat Diagn. 2001;21:881–4.
Bermudez-Lopez C, Garcia-de Teresa B, Gonzalez-del Angel A, Alcantara-Ortigoza MA. Germinal mosaicism in a sample of families with Duchenne/Becker muscular dystrophy with partial deletions in the DMD gene. Genet Test Mol Biomarkers. 2014;18:93–7.
Chen M, Chen C, Li Y, Yuan Y, Lai Z, Guo F, et al. Haplotype-Based noninvasive prenatal diagnosis for duchenne muscular dystrophy: a pilot study in South China. Eur J Obstet Gynecol Reprod Biol. 2019;240:15–22.
Chiu EKL, Hui WWI, Chiu RWK. cfDNA screening and diagnosis of monogenic disorders - where are we heading? Prenat Diagn. 2018;38:52–8.
Acknowledgements
We would like to thank the families for their participation in the study. We would like to thank Dr. Wenting Zhao and Dr. Weigang Lv for editing the manuscript.
Funding
The study was supported by a grant from the Science and Technology Research Program of Henan Province (Grant Number 202102310391), Key Scientific Research Projects in Colleges and Universities of Henan Province (Grant Number 22A320075), Science and Technology Huimin Project of Zhengzhou (2021KJHM0003), the National Key R&D Program of China (Grant Number 2018YFC1002206), and the Youth Innovation Fund of the First Affiliated Hospital of Zhengzhou University. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
XK designed and directed the study. GZ, XW, LL and PD performed the experiments and bioinformatic analysis. GZ and XK contributed to the writing and revision of the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University (2019-KY-286). All the participants provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Table S1. Primer sequences to map the breakpoints of the proband of Family D5; Table S2. Primer sequences to amplify parts 2P1 and 2P2; Table S3. Primers used for DMD in cfBEST. Table S4. Primers used for Y chromosome in cfBEST. Table S5. The expected mutant ratio of the plasma of the pregnant woman.
Additional file 2
. Fig. S1. Schematic representation of the cfBEST method. Red dot: the site of interest; UMI: Unique Molecular identifiers; Index: sample index; Primer F1: Target-specific primer in 1st PCR; Primer F2: Target-specific primer in 2nd PCR, which is close to the site of interest; Primer U1: a universal primer of P7; Primer U2: a universal tail part of P5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, G., Wang, X., Liu, L. et al. Noninvasive prenatal diagnosis of duchenne muscular dystrophy in five Chinese families based on relative mutation dosage approach. BMC Med Genomics 14, 275 (2021). https://doi.org/10.1186/s12920-021-01128-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-01128-1