
Abstract
Background
Copy number variations (CNVs) account for a substantial proportion of inter-individual genomic variation. However, a majority of genomic variation studies have focused on single-nucleotide variations (SNVs), with limited genome-wide analysis of CNVs in large cohorts, especially in populations that are under-represented in genetic studies including people of African descent.
Methods
We carried out a genome-wide copy number analysis in > 3400 healthy Bantu Africans from Tanzania. Signal intensity data from high density (> 2.5 million probes) genotyping arrays were used for CNV calling with three algorithms including PennCNV, DNAcopy and VanillaICE. Stringent quality metrics and filtering criteria were applied to obtain high confidence CNVs.
Results
We identified over 400,000 CNVs larger than 1 kilobase (kb), for an average of 120 CNVs (SE = 2.57) per individual. We detected 866 large CNVs (≥ 300 kb), some of which overlapped genomic regions previously associated with multiple congenital anomaly syndromes, including Prader-Willi/Angelman syndrome (Type1) and 22q11.2 deletion syndrome. Furthermore, several of the common CNVs seen in our cohort (≥ 5%) overlap genes previously associated with developmental disorders.
Conclusions
These findings may help refine the phenotypic outcomes and penetrance of variations affecting genes and genomic regions previously implicated in diseases. Our study provides one of the largest datasets of CNVs from individuals of African ancestry, enabling improved clinical evaluation and disease association of CNVs observed in research and clinical studies in African populations.
Similar content being viewed by others

Background
Copy number variations (CNVs) are a class of structural variation resulting from loss or gain of genomic fragments ≥ 1 kilobase (kb). CNVs can arise from genomic rearrangements such as deletions, duplications, insertions, inversions, or translocations [1,2,3] and have been implicated in the etiology of Mendelian disorders as well as complex traits [4]. Several pediatric disorders resulting from CNVs such as the 22q11 deletion syndrome, the Williams-Beuren syndrome, resulting from a microdeletion in 7q11.23, and the 15q13.3 microdeletion syndromes are characterized by the occurrence of multiple congenital anomalies, including intellectual and developmental disabilities, congenital heart defects, craniofacial dysmorphisms, or abnormalities in the development of other tissues and organs [5,6,7,8,9,10]. These types of CNVs can alter copy number of dosage-sensitive genes or disrupt regulatory elements, which result in pathogenic outcomes observed in patients [11]. For instance, 22q11.2 microdeletion region overlaps with genes essential for cortical circuit formation, and aberrations in cortical anatomy are two of the phenotypes observed in individuals with 22q11.2 deletion syndrome [12]. CNVs may also play a role in the etiology of common, complex diseases and traits including, diabetes, asthma, HIV susceptibility, cancer, and phenotypes in immune and environmental responses [13,14,15,16,17].
In addition to their role in disease, CNVs account for a high level of variation between healthy individuals, both within and between populations [1,2,3, 18, 19]. The 1000 Genomes Project was initiated to identify genetic variation in the human genome across diverse populations, and it has been instrumental in generating the largest catalog of genomic variants, including CNVs [20,21,22,23]. Nevertheless, CNVs remain largely understudied compared to single-nucleotide variations (SNVs) and are not commonly genotyped in a microarray-based analysis of genome-wide variation and association to disease phenotypes [24]. In 2015, Zarrei and colleagues compiled a CNV map of the human genome and estimated that 4.8–9.5% of the human genome contributes to CNV [25]. Furthermore, they identified approximately 100 genes whose loss is not associated with any severe consequences [25]. However, the vast majority of CNV data derive from individuals of European descent residing in Western countries, which might cause incorrect clinical interpretation of genomic variants [26,27,28]. Recently, resources such as the Genome Aggregation Database (gnomAD) have reported structural variations, including CNVs, in large cohorts of individuals of both European and non-European ancestries [29]. Regardless, knowledge of the genomic landscape of CNVs remains incomplete, especially in understudied populations such as Africans.
Based on the significant role of CNVs in health and disease, it is critical to have a set of reference CNVs observed in individuals from diverse populations. These population-specific reference datasets will greatly improve clinical interpretation and can help to refine a genomic region associated with diseases [30]. A recent study by Kessler and colleagues [31] demonstrated how lack of African ancestry individuals in variant databases may have resulted in the mischaracterization of variants in the ClinVar and Human Gene Mutation Databases.
In this study, we have detected CNVs in > 3400 healthy Bantu African children from Tanzania, using data from high-density (> 2.5 million probes) genotyping microarrays. We present a high-resolution map of CNVs ranging in size from 1 kb—3 Mb (million bases), providing a useful resource of CNV genetic variation for individuals of African ancestry. Additionally, we observe large CNVs in genomic regions previously implicated in syndromes and developmental disorders.
Methods
Sample description – populations
Our study was conducted using a previously collected cohort which included 3631 Bantu African children aged 3–21 living in Mwanza, Tanzania, a region with a population that is both genetically and environmentally relatively homogeneous [32]. The original study was aimed at studying the genetics of facial shape in children and adolescents aged 3–21 to minimize the potential and accumulating impact of the environment. Additionally, the majority of the sample were between the ages of 7 and 12 to also minimize the effects of puberty. Other parameters collected for individuals in the study included height, weight and BMI (Additional file 1). Individuals with a birth defect or having a relative with orofacial cleft were excluded [32]. The subjects were previously genotyped at the Center for Inherited Disease Research (CIDR) as part of the NIDCR FaceBase1 initiative. Genotyping using the Illumina HumanOmni2.5Exome-8v1_A (also referred to as Infinium Omni2.5–8) beadchip array and quality control (QC) was described previously [32, 33]. We obtained deidentified signal intensity data (*.idat) files for all the subjects in order to carry out copy number variation detection and analysis as described below.
CNV detection and analysis
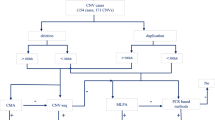
Signal intensity data (*.idat) files were processed and normalized using Illumina GenomeStudio software. The FinalReport files were used as the raw data to perform CNV calling with three CNV calling algorithms: PennCNV (version 1.0.1) [34], DNAcopy (version 1.46.0), [35] and VanillaICE (version 1.32.2), [36]. Both PennCNV and VanillaICE implement Hidden Markov Models (HMM), whereas DNAcopy implements a Circular Binary Segmentation (CBS) algorithm. GC correction was performed for PennCNV using the built-in function, and the R/Bioconductor package ArrayTV (version 1.8.0) [37] was used to perform GC correction for DNAcopy and VanillaICE. Codes used to run the algorithms are available at GitHub [38]. Individuals with a total number of CNVs ≥ 3 standard deviations above the cohort mean were removed from further analysis based on previously established criteria [39]. In all, 168 individuals were excluded from further analysis: 70 duplicate samples, 97 individuals with a total number of CNVs ≥ 3 standard deviation of the cohort mean, and one individual who had 0 CNVs after applying analysis pipeline thresholds described in Fig. 1. All subsequent analyses were performed on the remaining 3463 individuals and all CNV coordinates are based on NCBI build37/hg19.

CNV analysis and filtering pipeline. Workflow showing the various filtering steps applied to detected CNVs in order to obtain a set of high confidence CNVs used for further analysis
CNV calls with fewer than five probes and < 1000 bases in size were removed, followed by those with DNAcopy log-ratio between -0.1 and 0.1 (a threshold determined by a plateau plot in the DNAcopy R package that shows the copy number across the genome), and PennCNV calls with confidence score < 10 (recommended threshold by the developers of PennCNV) (Fig. 1). We used the intersect function in BEDTools v2.25 [40] to determine the proportion of overlap between CNV coordinates and genomic elements. CNV calls from two or more algorithms that overlap by 50% or more were considered concordant and included for further analyses. Next, CNV calls overlapping the centromere, telomere, or ≥ 50% with segmental duplications were removed.
PennCNV calls with copy numbers of 0 and 1 were annotated as copy number loss, 2 as diploid copy number, and 3, 4, 5 and 6 as copy number gain; VanillaICE calls with copy numbers of 1 and 2 were annotated as copy number loss, 3 and 4 as diploid copy number, and 5 and 6 as copy number gain; DNAcopy segments with log-ratio ≥ 0.1 were annotated as copy number gain, and log-ratio ≤ -0.1 as copy number loss.
CNV calling with PennCNV from genotype data using high-density SNP arrays often results in the artificial splitting of larger CNVs (i.e. > 500 kb) into multiple smaller CNVs [34]. Therefore, we merged adjacent CNVs of the same type (i.e., loss or gain) in the same individual using an approach described previously [34]. Briefly, for three adjacent genomic regions A, B, and C, where A and C represent two CNVs of the same type separated by a region B, the length of B was divided by the total length of all three segments (A + B + C). If this fraction was ≤ 15%, then three regions were merged into one CNV. This approach was used to generate a list of CNVs that passed quality metrics and filtering criteria in individual samples from the Bantu cohort (Additional file 2).
In silico quality assessment of CNVs
To assess the quality of CNV calls in the Bantu population, we compared the overlap of CNVs in the Bantu population with the Database of Genomic Variants (DGV) Gold Standard (GS) variants [41]. DGV GS variants are a curated set of variants from a select number of studies with high resolution and high quality, which were evaluated for accuracy and sensitivity. Therefore, an overlap with DGV GS variants indicates that our CNV calls are likely true positives. To assess whether the overlap was more than expected by chance, we permuted the genomic locations (n = 1000) using the shuffle function in BEDTools v2.25 [40]. Permutation tests were performed within each chromosome with the same number and size distribution of CNVs observed in the Bantu population as recommended for genomic elements that are unevenly distributed across the genome [42].
CNV regions (CNVRs)
CNV regions (CNVRs) were generated by merging all overlapping CNVs of the same type (i.e. loss or gain) from multiple individuals in our cohort, using the merge function in BEDTools v2.25 [40]. This resulted in a list of loss-only and gain-only CNVRs, which were further merged into overlapping CNVRs of all types (Additional file 3).
Comparison to other CNV datasets
We compared Bantu CNVRs to variants obtained from DGV (release date 2020–02-15) [41], the Genome Aggregation Database (gnomAD v2.1) [29, 43], African CNVR [44] and CNVs identified in low-mappability regions [45]. DGV CNVs dataset were downloaded from DGV website [46]. gnomAD SV 2.1 sites BED file was downloaded from Broad Institute website [47], which were filtered by SV Type and SV Filter, and only “DEL”, “DUP”, “CN” SV types, and SVs with “PASS” SV Filter were included. The CNV dataset for low-mappability regions obtained from Monlong and colleagues’ publication additional material Sect. [45]. CNVs obtained from tumor samples were excluded. CNVRs were generated using a similar approach as described above, and we then compared to the list of Bantu CNVRs to identify overlap.
CNV blocks
We generated a list of ‘CNV blocks’ from a set of unrelated individuals in our cohort (the description of unrelated individuals is explained in Ref. 32) to obtain a more accurate count of the number of times any given CNV was observed. First, all overlapping CNVs localizing to a given genomic region were aligned as shown (Fig. 2a,b). The largest region encompassed by these overlapping CNVs (A-D in Fig. 2) was segmented by start and end coordinates of individual CNV calls (A-K in Fig. 2), which resulted into multiple CNV blocks (A-E, E-C, C-J in Fig. 2, Additional file 4). An example for CNV blocks is represented in Fig. 2b. We then counted the number of times each CNV block was observed in unrelated individuals in our cohort. Based on these counts, CNV blocks were categorized into four groups: CNV blocks observed in ≥ 5% (common CNV blocks), ≥ 1 and < 5% (low frequency CNV blocks), ≥ 0.1 and < 1% (rare CNV blocks), and ≤ 0.1% (very rare CNV blocks).

CNV blocks. a A schematic demonstrating the delineation of CNV blocks followed by determination of total count within the Bantu cohort and categorization based on frequency. Black and gray rectangles, a-h and j, k represent five overlapping CNVs observed in different individuals [1,2,3,4,5]. a–k represent the start and end coordinates of the CNVs. Blue rectangles represent CNV blocks. b Represents an actual example of CNV block delineation from our CNV dataset
CNVs in regions associated with disease
To assess which CNVs from our cohort overlap genes associated with developmental disorders, we identified overlap (at least 1 bp) of our common (≥ 5%), low frequency (≥ 1—< 5%), and rare (≥ 0.1—< 1%) CNV blocks with genes catalogued in the Developmental Disorders Genotype–Phenotype Database (Additional file 5) (DDG2P, [48]), compiled based on known implication in disease etiology. The following “STATUS” categories were included in the analysis: Confirmed developmental disorder (DD) Gene, Probable DD Gene, Possible DD Gene, and Both DD and IF (incidental finding). We determined the degree of overlap between using a bi-directional approach; first we calculated how much of the CNV block overlapped with gene (CNVvsGeneOverlap% in Additional file 6) and then how much of the gene overlapped with the CNV block (GenevsCNVOverlap% in Additional file 6).
To assess whether large CNVs from our cohort overlap loci associated with genomic disorders, we first generated a list of 866 large CNVs (≥ 300 kb) observed in our cohort (Additional file 7). We then determined the proportion overlap of these CNVs with known CNVs previously implicated in the etiology of syndromes and genomic disorders catalogued in The DatabasE of genomiC variation and Phenotype in Humans using Ensembl Resources [49, 50] (Additional file 8). DECIPHER is an expert-curated database of microdeletion and microduplication syndromes in developmental disorders.
Results
CNV detection and analysis
We identified 448,337 CNVs in the genomes of 3463 Bantu African children (Fig. 1). Adjacent CNVs of the same type within a given individual were merged, resulted in a total of 416,877 CNVs across all autosomes, including 355,027 losses and 61,850 gains (Table 1, Additional file 2). Of these, 72,205 (17.3%) CNVs were concordantly called by all three CNV calling algorithms used. The average number of CNVs per subject was 120 (min = 27, max = 1569, mean = 120.38, stdev = 151.04, IQR = 45) with a median length of 7558 nucleotides (nt) and an average length of 18,145 nt (min = 1,001 nt, max = 2,929,312 nt). We further categorized CNVs based on their genomic size, as shown (Table 1). The vast majority of detected CNVs were smaller, with 247,314 (59.3%) that were 1–10 kb and 158,190 (38.0%) that were 10–100 kb. However, a sizable proportion were ≥ 100 kb with over a thousand that were ≥ 300 kb. Our CNV calls were significantly enriched for the Database of Genomic Variants (DGV) Gold Standard (GS) variants compared to randomly selected CNV regions (permuted p-value < 0.001), indicating that CNV calls detected in this study are likely true positives.
We next assembled copy number variation regions (CNVRs) by merging overlapping CNVs of the same type (loss or gain) detected in multiple individuals in the Bantu cohort (Additional file 3). These CNVRs were further divided into 13,738 loss only, 1100 gain only and 2656 with both gain and loss, for a total of 17,494 CNVRs (Additional file 3). The assembly into CNVRs further allowed us to determine that CNVs observed in our cohort covered a total of approximately 600 million nucleotides, about 20% of the genome. The distribution of CNVRs across the genome suggested that the number of CNVRs was not proportional to the size of the chromosome (Fig. 3), consistent with previous reports [25].

Genomic Map of CNVRs. CNVRs detected in our cohort are shown as colored density plots across individual chromosomes represented by ideograms. The genome was divided into 1 million equal sized windows and the number of CNVRs within each window were counted and plotted on the density plot. Color key—red: loss CNVRs, blue: gain CNVRs, green: loss and gain CNVRs. Density was calculated by dividing the genome in equal sized windows (n = 1,000,000) and counting the number of CNVRs overlapping each of the windows
Comparison to other CNV datasets
To determine overlap with existing CNV datasets, we compared the CNVRs observed in our cohort with existing CNV databases including DGV (40,418 CNVRs) [41], gnomAD (54,851 CNVRs) [29, 43], and current studies that focus on CNVs in different African populations (7608 CNVRs) [44] and low-mappability regions (12,242 CNVRs) [45]. This comparison identified 1952 (11.16%) CNVRs in our cohort overlapping all four and 10,046 (57.46%) overlapping any three datasets, while a majority overlapped CNVRs in only one, two, or three of the databases (Table 2).
Additionally, we observed 48 CNVRs in our cohort that did not overlap with any CNV datasets mentioned above (Fig. 4, Additional file 9). These 48 CNVRs encompass a total of 209,951 nt with three (very rare frequency CNVRs) overlapping genes reported to be associated with developmental disorders in the Developmental Disorders Genotype–Phenotype Database (DDG2P) (Additional file 5).

Novel CNVRs. The chromosomal locations of CNVRs detected in the Bantu cohort, which did not overlap with known CNV datasets included in our comparison analysis. Vertical, colored lines represent individual CNVRs. Color key—red: loss blue: gain, green: loss and gain
CNVs in regions associated with disease
We next wanted to determine whether CNVs observed in the Bantu cohort overlapped genes and genomic regions previously associated with disease phenotypes. Using CNVs from 2696 unrelated subjects in our cohort, we identified 121,334 CNV blocks from 323,667 CNV calls (Additional file 4). We further classified CNV blocks into four categories based on how often they were observed in these 2696 unrelated individuals: a) 6913 CNV blocks observed in ≥ 5% of unrelated subjects were categorized as common; b) 24,908 CNV blocks observed in 1–5% were categorized as low frequency; c) 44,910 CNV blocks observed in 0.1–1% were categorized as rare; and d) 44,603 CNV blocks were observed in ≤ 0.1% and were categorized as very rare; most of the very rare CNV blocks were singletons.
We then determined the overlap between common (≥ 5%), low frequency (1–5%), and rare (0.1–1%) CNV blocks and genes reported to be associated with developmental disorders in the DDG2P Database (Additional file 5). We identified 11,835 CNV blocks that overlapped 1627 DDG2P genes (Additional file 6). We used reciprocal approach to identify ≥ 50% overlap between DDG2P genes and CNV blocks, which identified 125 CNV blocks (83 loss, 21 gain, 21 loss and gain) which overlapped with 125 DDG2P genes with reciprocal overlap percentage of ≥ 50%.
Additionally, we identified 866 relatively large CNVs (≥ 300 kb) (Additional file 7) in unrelated individuals within our cohort. We investigated whether any of these large CNVs overlap (≥ 1 bp) CNVs previously implicated in syndromes or genomic disorders catalogued in DECIPHER (DatabasE of genomiC variation and Phenotype in Humans using Ensembl Resources; Additional file 8) [49]. We identified 83 large CNVs, including 62 gain CNVs ranging in size from ~ 300–2740 kb and 21 loss CNVs ranging in size from ~ 309–1532 kb that overlap CNVs implicated in the etiology of 24 known syndromes and genomic disorders (Additional file 10). Fourteen individuals had CNVs, including 1 loss (~ 442 kb) and 13 gains (~ 414–537 kb), that overlap with the genomic region implicated in Prader-Willi /Angelman syndromes (Type 1), which is caused by a ~ 5.69 mb deletion on chromosome 15. Thirty-two individuals had CNVs, including 7 losses and 25 gains, ranging in size from ~ 300–485 kb that overlapped with the region implicated in ATR-16 syndrome, which is caused by a 775 kb deletion on chromosome 16.
Discussion
The vast majority of existing genetic variation analyses have been performed on individuals of European descent [26,27,28]. These types of analyses have resulted in an incomplete view of the genetic variation across populations and hindered the understanding and discovery of associations between diseases and genetic variations in non-European populations. To better catalog the full extent of genetic variation across human populations, targeted analyses of genetic variation in under-represented populations are needed. Several recent studies have undertaken such analyses, including of single-nucleotide variations (SNVs), small insertion-deletions (indels), and copy number variations (CNVs) in under-represented populations including people of African, Asian, Latinx and Native American ancestry [29, 51,52,53,54,55,56,57,58,59]. Here, we present a catalog of genome-wide copy number variations in a large cohort of healthy individuals of African ancestry.
One of the earliest studies reporting CNVs in a population of African descent was an analysis of 385 individuals of African American ancestry, which identified 1362 total CNVs [51]. Compared to the results we show here, this study used a lower resolution array platform that contained fewer probes, which resulted in a relatively small number of CNVs being identified [51]. Over the years, additional studies of individuals from diverse populations, including of African descent as part of 1000 Genomes Project, reported an increasing number of CNVs (> 50,000) [20,21,22,23]. Most recently, CNVs and other structural variants (> 400,000) in 4937 individuals of African and African American ancestry were reported as part of the Genome Aggregation Database (gnomAD) [29, 43], and novel CNVRs were identified by Nyangiri and colleagues [44]. In our study, we identified 48 CNVRs which may represent CNVRs that are either specific to the Bantu African population or that may be very rare in populations currently represented in existing CNV datasets.
One of the limitations of our study is that the genotyping array platforms are limited to detecting copy number differences of sequences present in the human genome reference assembly used to design probes [60, 61]. This suggests that the current reference genome, which is mostly derived from people of European descent, may not be adequate for population-based analysis of human genome variation. A recent study showed that there is an unprecedented variation on highly repetitive 22q11.2 segmental duplication regions within individuals and populations [62] which might be missed by genotyping platforms. Furthermore, there is a high level of variation between human genome assemblies hg19 (GRCh37) and hg38 (GRCh38), which is mainly due to gaps associated with complex genomic regions, missing sequences, sequencing errors and representation of centromeres and telomeres in individual assemblies [63]. In the array used in our study, the probes were selected based on human genome reference assembly hg19 (GRCh37), which is likely missing DNA that exists in people of African ancestry. Another limitation is the ability to detect CNVs which varies between platforms, as SNP-based array platforms are more likely to underestimate gain CNVs than are array CGH platforms [64, 65]. Therefore, the number of detected losses is usually higher than the number of detected gains. CNVRs observed in our dataset, but not in other existing databases are likely to be either specific to Africans or rare in other populations, underscoring the importance of genetic reference datasets derived from diverse ancestral populations.
We observed a considerable overlap between genes within common CNV blocks and genes previously implicated in developmental disorders curated within the DDG2P Database. These observations raise the possibility that dosage alteration of these genesis either not pathogenic or incompletely penetrant in people of African ancestry. Additionally, of the 866 large CNVs (≥ 300 kb) we identified, 87 overlap with CNVs previously implicated in syndromes catalogued in DECIPHER [49]. Thirty of these (34%) are in the same direction (loss or gain) as observed in these known syndromes but are smaller than the pathologic CNVs. One potential explanation for this could be that the region responsible for the clinical outcomes observed in syndromic patients is smaller and our data may allow further refinement of the critical region for these syndromes. Alternatively, these results may also point to variable expressivity and/or reduced penetrance of CNVs in these regions in Africans. These findings underscore the need for population specific CNV datasets for comparison in order to determine the impact of CNVs on clinical outcomes observed in patients [66, 67].
A recent study [68] showed that the African “pan-genome”, built using sequence data from 910 individuals of African descent, contained ~ 10% more DNA not present in hg38 (GRCh38), suggesting that the current reference genome may not fully represent genomic variation in diverse human populations. This suggests the need for de novo sequencing of a large number of genomes from African and other under-represented populations, in order to comprehensively assess genomic variation within and between diverse populations.
Conclusion
The increasing number of African samples being analyzed as part of the 1000 Genomes Project, gnomAD, and several other projects continues to improve our understanding of genetic diversity in this population. More importantly, our results suggests that the determination of the clinical impact and phenotypic outcomes of CNVs, in diverse populations, will require appropriate datasets from healthy individuals from the same population for comparison. The data we present contribute to this effort by providing a rich dataset of CNVs observed in a large cohort of Bantu Africans. However, based on the level of genomic diversity that exists within African subpopulations, we suggest that additional, larger datasets will be required in order to capture all the existing genomic variation within the African population [69,70,71,72,73].
Availability of data and materials
The CNV data presented in this article has been deposited in the FaceBase Consortium Database (https://www.facebase.org/) available at https://doi.org/10.25550/1-7330 and included within the article (Additional file 2). The genotype data used for CNV detection were previously deposited in the Database of Genotypes and Phenotypes (dbGaP: http://www.ncbi.nlm.nih.gov/gap; dbGaP study accession: phs000622.v1.p1).
Abbreviations
- CNV:
-
Copy number variation
- CNVR:
-
Copy number variant region
- SNV:
-
Single nucleotide variation
- DGV:
-
Database of Genomic Variants
- GS:
-
Gold Standard
- DDG2P:
-
Developmental Disorders Genotype–Phenotype Database
- gnomAD:
-
Genome Aggregation Database
- SE:
-
Standard Error
- IQR:
-
Interquartile
References
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–54.
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36(9):949–51.
Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, et al. Large-scale copy number polymorphism in the human genome. Science (80-). 2004;305(5683):525–8.
Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61(1):437–55.
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43(9):838–46.
Watson CT, Tomas M-B, Sharp AJ, Mefford HC. The genetics of microdeletion and microduplication syndromes: an update. Annu Rev Genomics Hum Genet. 2014;15(1):215–44.
Harel T, Lupski JR. Genomic disorders 20 years on—mechanisms for clinical manifestations. Clin Genet. 2018;93(3):439–49.
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JAS, et al. 22Q11.2 Deletion syndrome. Nat Rev Dis Prim. 2015;1(11).
Carvalho CMB, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016;17(4):224–38.
Yilmaz F, Shaikh TH, Emanuel BS. Segmental duplications and genetic disease. eLS. 2017;23:1–8.
Rice AM, McLysaght A. Dosage sensitivity is a major determinant of human copy number variant pathogenicity. Nat Commun [Internet]. 2017;8:1–11.
Meechan DW, Maynard TM, Tucker ES, Fernandez A, Karpinski BA, Rothblat LA, et al. Modeling a model: Mouse genetics, 22q112 deletion syndrome, and disorders of cortical circuit development. Prog Neurobiol [Internet]. 2015;130:1–28.
Henrichsen CN, Chaignat E, Reymond A. Copy number variants, diseases and gene expression. Hum Mol Genet. 2009;18(R1):1–8.
Lee JA, Lupski JR. Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron. 2006;52(1):103–21.
Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41(7):849–53.
Girirajan S, Campbell CD, Eichler EE. Human copy number variation and complex genetic disease. Annu Rev Genet. 2011;45(1):203–26.
Seifert M, Friedrich B, Beyer A. Importance of rare gene copy number alterations for personalized tumor characterization and survival analysis. Genome Biol. 2016;17(1):1–25.
Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2008;84(2):148–61.
McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40(10):1166–74.
Altshuler DL, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73.
Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65.
Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81.
Lauer S, Gresham D. An evolving view of copy number variants. Curr Genet. 2019;65(6):1287–95.
Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16(3):172–83.
Bentley AR, Callier S, Rotimi CN. Diversity and inclusion in genomic research: why the uneven progress? J Community Genet. 2017;8(4):255–66.
Manolio TA. Using the data we have: improving diversity in genomic research. Am J Hum Genet [Internet]. 2019;105(2):233–6.
Landry LG, Ali N, Williams DR, Rehm HL, Bonham VL. Lack of diversity in genomic databases is a barrier to translating precision medicine research into practice. Health Aff. 2018;37(5):780–5.
Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, et al. A structural variation reference for medical and population genetics. Nature. 2020;581(7809):444–51.
Wright CF, West B, Tuke M, Jones SE, Patel K, Laver TW, et al. Assessing the pathogenicity, penetrance, and expressivity of putative disease-causing variants in a population setting. Am J Hum Genet. 2019;104(2):275–86.
Kessler MD, Yerges-Armstrong L, Taub MA, Shetty AC, Maloney K, Jeng LJB, et al. Challenges and disparities in the application of personalized genomic medicine to populations with African ancestry. Nat Commun. 2016;7.
Cole JB, Manyama M, Kimwaga E, Mathayo J, Larson JR, Liberton DK, et al. Genomewide association study of african children identifies association of SCHIP1 and PDE8A with facial size and shape. PLoS Genet. 2016;12(8):1–19.
Laurie CC, Doheny KF, Mirel DB, Pugh EW, Bierut LJ, Bhangale T, et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet Epidemiol. 2010;34(6):591–602.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SFA, et al. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17(11):1665–74.
Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23(6):657–63.
Scharpf RB, Parmigiani G, Pevsner J, Ruczinski I. Hidden Markov models for the assessment of chromosomal alterations using high-throughput SNP arrays. Ann Appl Stat. 2008;2(2):687–713.
Halper-stromberg AE. Package ‘ ArrayTV .’ 2019;
Codes used to run CNV calling algorithms. https://github.com/dpastling/facebase_cnv
Gai X, Perin JC, Murphy K, O’Hara R, D’arcy M, Wenocur A, et al. CNV Workshop: An integrated platform for high-throughput copy number variation discovery and clinical diagnostics. BMC Bioinformatics. 2010;11:1–9.
Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2.
MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42(D1):986–92.
De S, Pedersen BS, Kechris K. The dilemma of choosing the ideal permutation strategy while estimating statistical significance of genome-wide enrichment. Brief Bioinform. 2013;15(6):919–28.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv. 2020;581(May):531210.
Nyangiri OA, Noyes H, Mulindwa J, Ilboudo H, Kabore JW, Ahouty B, et al. Copy number variation in human genomes from three major ethno-linguistic groups in Africa. BMC Genom. 2020;21(1):1–15.
Monlong J, Cossette P, Meloche C, Rouleau G, Girard SL, Bourque G. Human copy number variants are enriched in regions of low mappability. Nucleic Acids Res. 2018;46(14):7236–49.
Database of Genomic Variants. http://dgv.tcag.ca/dgv/app/downloads?ref=GRCh37/hg19. Accessed on 2 March 2020.
Genome Aggregation Database v2.1. https://gnomad.broadinstitute.org/downloads. Accessed on 30 June 2020.
Developmental Disorders Genotype-Phenotype database. https://decipher.sanger.ac.uk/info/ddg2p. Accessed on 29 July 2020.
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet. 2009;84(4):524–33.
DECIPHER CNV syndromes. https://decipher.sanger.ac.uk/disorders #syndromes/overview DECIPHER. Accessed on 29 July 2020.
McElroy JP, Nelson MR, Caillier SJ, Oksenberg JR. Copy number variation in African Americans. BMC Genet. 2009;10:15.
Ku CS, Pawitan Y, Sim X, Ong RTH, Seielstad M, Lee EJD, et al. Genomic copy number variations in three southeast Asian populations. Hum Mutat. 2010;31(7):851–7.
Suktitipat B, Naktang C, Mhuantong W, Tularak T, Artiwet P, Pasomsap E, et al. Copy number variation in Thai population. PLoS One. 2014;9(8).
Vidal EA, Moyano TC, Bustos BI, Pérez-Palma E, Moraga C, Riveras E, et al. Whole genome sequence, variant discovery and annotation in Mapuche-Huilliche native South Americans. Sci Rep. 2019;9(1):1–11.
Lindo J, Rogers M, Mallott EK, Petzelt B, Mitchell J, Archer D, et al. Patterns of genetic coding variation in a Native American population before and after European contact. Am J Hum Genet. 2018;102(5):806–15.
Lin CH, Lin YC, Wu JY, Pan WH, Chen YT, Fann CSJ. A genome-wide survey of copy number variations in Han Chinese residing in Taiwan. Genomics. 2009;94(4):241–6.
Lou H, Li S, Jin W, Fu R, Lu D, Pan X, et al. Copy number variations and genetic admixtures in three Xinjiang ethnic minority groups. Eur J Hum Genet. 2015;23(4):536–42.
Narang A, Jha P, Kumar D, Kutum R, Mondal AK, Dash D, et al. Extensive copy number variations in admixed Indian population of African ancestry: Potential involvement in adaptation. Genome Biol Evol. 2014;6(12):3171–81.
Fu R, Mokhtar SS, Phipps ME, Hoh BP, Xu S. A genome-wide characterization of copy number variations in native populations of Peninsular Malaysia. Eur J Hum Genet. 2018;26(6):886–97.
Kidd JM, Sampas N, Antonacci F, Graves T, Fulton R, Hayden HS, et al. Characterization of missing human genome sequences and copy-number polymorphic insertions. Nat Methods. 2010;7(5):365–71.
Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet [Internet]. 2011;12(5):363–76.
Demaerel W, Mostovoy Y, Yilmaz F, Vervoort L, Pastor S, Hestand MS, et al. The 22q11 low copy repeats are characterized by unprecedented size and structural variability. Genome Res. 2019;29(9):1389–401.
Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017;27(5):849–64.
Pinto D, Darvishi K, Shi X, Rajan D, Rigler D, Fitzgerald T, et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol. 2011;29(6):512–20.
Pang AWC, MacDonald JR, Yuen RKC, Hayes VM, Scherer SW. Performance of high-throughput sequencing for the discovery of genetic variation across the complete size spectrum. G3 Genes, Genomes, Genet. 2014;4(1):63–5.
Buchanan JA, Scherer SW. Contemplating effects of genomic structural variation. Genet Med. 2008;10(9):639–47.
Nowakowska B. Clinical interpretation of copy number variants in the human genome. J Appl Genet. 2017;58(4):449–57.
Sherman RM, Forman J, Antonescu V, Puiu D, Daya M, Rafaels N, et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat Genet. 2019;51(1):30–5.
Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, et al. Genetic structure of human populations. Science (80-). 2002;298(5602):2381–5.
Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, et al. The genetic structure and history of Africans and African Americans. Science (80-). 2009;324(5930):1035–44.
Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K, et al. The African genome variation project shapes medical genetics in Africa. Nature. 2015;517(7534):327–32.
Rotimi CN, Tekola-Ayele F, Baker JL, Shriner D. The African diaspora: history, adaptation and health. Curr Opin Genet Dev. 2016;41:77–84.
Gurdasani D, Carstensen T, Fatumo S, Chen G, Franklin CS, Prado-Martinez J, et al. Uganda genome resource enables insights into population history and genomic discovery in Africa. Cell. 2019;179(4):984-1002.e36.
Acknowledgements
We would like to thank the FaceBase Consortium (https://www.facebase.org/) for providing the genotyping data used in this study. University of Colorado Anschutz Medical Campus Department of Biochemistry and Molecular Genetics’ research cluster was used to perform analyses. We would like to thank the DECIPHER community (http://decipher.sanger.ac.uk), including all the centres who contributed to the generation of the data used in this study.
Funding
This work was supported in part by grant # DE025363 from the National Institutes of Health to T.H.S. DECIPHER (http://decipher.sanger.ac.uk) is funded by the Wellcome Trust. The funding body had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
T.H.S and A.E.H conceived the study. F.Y., D.A. and H-C. Y. performed the copy number analysis and data interpretation. R.A.S, B.H. and M.M. led the original study that collected samples and conducted the genotyping study of the Bantu cohort. J.C, S.A.S and R.A.S provided the deidentified data and other relevant information on the sample used in this study. F.Y., M.N., A.E.H. and T.H.S. drafted the manuscript which was read and critically revised by all authors. Final approval of the version to be published was given by F.Y., M.N., D.A., H-C.Y., J. C., S. A. S., B.H., M.M., R. A. S., A. E. H., and T.H.S. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol (#09–0731) and participating investigators were reviewed and approved by the Colorado Multiple Institutional Review Board (USA) as the official Institutional Review Board of Record. The study protocol and investigators were additionally reviewed and approved by the Institutional Review Boards of the Catholic University of Health and Allied Sciences (Mwanza, Tanzania), the University of Calgary (Canada), Florida State University (USA), and the National Institute for Medical Research (Tanzania). The study conformed to the tenets of the Declaration of Helsinki. Subjects were recruited and the study explained by the local investigator in Swahili. All study subjects were aged 3–21. The legal age of majority in Tanzania is 18; accordingly, as required, written informed consent to participate was obtained from all subjects aged 18–21 or from the parents of subjects aged 3–17, using a consent form in either English or Swahili, as per the choice of the person providing consent.
Consent for publication
Not applicable.
Competing interests
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Bantu Samples Demographic Info. Description: The list of Bantu samples with demographic information.
Additional file 2.
The list of CNVs. Description: The list of CNVs detected in our study.
Additional file 3.
The list of CNVRs. Description: The list of CNVRs identified in our study.
Additional file 4.
The list of CNV blocks. Description: The list of Bantu CNV blocks identified in our study.
Additional file 5.
The list of DDG2P genes used in our analysis. Description: The list of DDG2P genes used in our analysis to detect genes overlapping with Bantu CNV blocks.
Additional file 6.
CNV blocks overlapped with DDG2P genes. Description: The list of Bantu CNV blocks overlapped with DDG2P genes.
Additional file 7.
The list of large (>300kb) CNVs observed in unrelated individuals in our cohort. Description: The list of large CNVs identified in our study.
Additional file 8.
The list of CNVs associated with DECIPHER Syndromes used in our analysis: Description: The list of DECIPHER CNV syndromes used in our study.
Additional file 9.
Novel CNVs observed in our cohort. Description: The list of novel CNVs detected in our study.
Additional file 10
. CNVs associated with DECIPHER syndromes overlapping large CNVs observed in our cohort. Description: The list of DECIPHER CNV syndromes overlapped with DECIPHER CNV syndromes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yilmaz, F., Null, M., Astling, D. et al. Genome-wide copy number variations in a large cohort of bantu African children. BMC Med Genomics 14, 129 (2021). https://doi.org/10.1186/s12920-021-00978-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-00978-z