
Abstract
Background
Competitive endogenous RNA (ceRNA) networks have revealed a new mechanism of interaction between RNAs, and play crucial roles in multiple biological processes and development of neoplasms. They might serve as diagnostic and prognosis markers as well as therapeutic targets.
Methods
In this work, we identified differentially expressed mRNAs (DEGs), lncRNAs (DELs) and miRNAs (DEMs) in sarcomas by comparing the gene expression profiles between sarcoma and normal muscle samples in Gene Expression Omnibus (GEO) datasets. Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analyses were applied to investigate the primary functions of the overlapped DEGs. Then, lncRNA-miRNA and miRNA-mRNA interactions were predicted, and the ceRNA regulatory network was constructed using Cytoscape software. In addition, the protein–protein interaction (PPI) network and survival analysis were performed.
Results
A total of 1296 DEGs were identified in sarcoma samples by combining the GO and KEGG enrichment analyses, 338 DELs were discovered after the probes were reannotated, and 36 DEMs were ascertained through intersecting two different expression miRNAs sets. Further, through target gene prediction, a lncRNA–miRNA–mRNA ceRNA network that contained 113 mRNAs, 69 lncRNAs and 29 miRNAs was constructed. The PPI network identified the six most significant hub proteins. Survival analysis revealed that seven mRNAs, four miRNAs and one lncRNA were associated with overall survival of sarcoma patients.
Conclusions
Overall, we constructed a ceRNA network in sarcomas, which might provide insights for further research on the molecular mechanism and potential prognosis biomarkers.
Similar content being viewed by others

Background
Sarcomas are rare types of malignant neoplasms of mesenchymal origin and often spread to other tissues in the body [1]. Sarcomas can be classified into more than 100 distinct subtypes, which are typically divided into two heterogeneous groups, including bone sarcomas and soft tissue sarcomas [2, 3]. Despite the use of extensive surgical resection, intensive and multiagent chemotherapy, radiotherapy, and emerging treatment strategies consisting of molecular targeting agents, immune checkpoint inhibitors, and adoptive T-cell therapy, the overall 5-year survival rate of sarcomas has not been significantly improved over the past few decades [2, 4, 5]. Unfortunately, sarcoma patients accompanied with metastatic spread show a lower 5-year overall survival rate [6]. Further investigations into the pathogenesis of sarcomas and identification of the novel prognostic biomarkers that facilitate the improvement of therapy and prognosis of sarcomas are urgently needed.
Noncoding RNAs (ncRNAs), such as long noncoding RNAs (lncRNAs) and microRNAs (miRNAs), function as key regulators of gene expression, their involvement in various human diseases is being gradually revealed, and the multilayered regulatory networks formed by cross-linked ncRNAs and mRNAs seemly provide new insights into their regulatory mechanism with regards to both physiology and pathology [7, 8]. The competing endogenous RNAs (ceRNAs) have been suggested to be involved in essential biological processes and play crucial roles in the initiation and development of neoplasms, and they potentially serve as diagnostic and prognosis markers or therapeutic targets [9]. miRNAs are small RNAs of 19–25 nucleotides in length that can guide the posttranscriptional repression of protein-coding genes by binding to their mRNAs [10]. The lncRNAs that harbor a miRNA response element can compete with other RNA transcripts and thus theoretically function as ceRNAs [11]. A single miRNA can regulate multiple target RNAs that contain the specific miRNA response element and these RNAs can be regulated by multiple miRNAs lay the foundation for the construction of a ceRNA network [12]. Recent studies have shown novel roles of ceRNA network in lung cancer, breast cancer, gastric cancer, esophageal adenocarcinoma, esophageal squamous cell carcinoma, cholangiocarcinoma, ovarian cancer, and uterine corpus endometrial carcinoma [13,14,15,16,17,18,19,20,21]. In addition, a previous study has constructed a ceRNA network and predicted the prognosis of soft tissue sarcoma recurrence [22]. However, the ceRNA network based on sarcoma and normal tissue samples has not been reported.
In the present study, the genes expression profiles of sarcomas in the Gene Expression Omnibus (GEO) database were integrated, and the differentially expressed genes (DEGs), lncRNAs (DELs) and miRNAs (DEMs) were identified. Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analyses were performed to screen out significant functional groups of DEGs. Thereafter, a ceRNA network consisting of lncRNA–miRNA–mRNA was constructed based on miRNA-mRNA and lncRNA-miRNA interactions. In addition, protein–protein interaction (PPI) network of the DEGs involved in the ceRNA network and prognostic analysis based on survival information of sarcoma patients were conducted to explore the effects of these transcripts on sarcomas.
Methods
Acquisition of available datasets and lncRNA annotation
Microarray data including high-throughput genes or miRNA expression profiling of sarcoma patients and normal samples were acquired from the GEO database (www.ncbi.nlm.nih.gov/geo/). The Affymetrix Human Genome U133 Plus 2.0 Array was reannotated according to the latest release of the updated gene database to filter lncRNA probes [23]. The process of lncRNA annotation was as follows: transcription sequences beginning with NM and XM in the RefSeq database were selected as a mRNA database, and ncRNAs with a length greater than 200 nucleotides that had gene transcription sequences beginning with NR and XR were selected from RefSeq, Ensembl and NONCODE2016 as a lncRNA database. Sequence alignments between 54,675 probes that were downloaded from the annotation file of the Affymetrix Human Genome U133 Plus 2.0 Array and the mRNA database were performed by using BLAST, and the E value was set as 10–5. Probes that match the respective gene were included in the mRNA probe set, and others were compared with the lncRNA database. The E value was set as 10–5, and probes that match the respective gene were included in the lncRNA probe set, which was use for the following analysis.
Identification of DEGs, DELs and DEMs
DEGs, DELs and DEMs between sarcoma and normal muscle samples were determined by using the limma package in the R software (version 3.3.2) [24, 25]. P < 0.05, the false discovery rate (FDR) < 0.05 and fold change (FC) > 3 was used as the screening criteria for DEGs and DELs, while P < 0.05, FDR < 0.05 and FC > 2 was used to screen DEMs. Hierarchical clustering was performed using EPCLUST to represent the relationships among the samples based upon the expression data matrix of DEGs, DELs and DEMs, respectively.
GO and KEGG pathway enrichment analysis of DEGs
GO enrichment analysis of DEGs was performed to identify the potential functional genes according to the GO database, which can organize genes into hierarchical categories and uncover the gene regulatory network on the basis of biological processes and molecular function [26]. KEGG pathway analysis of DEGs was carried out to ascertain the potential functions of these genes that participated in the pathways based on the KEGG database [27,28,29]. Fisher’s exact test was used to classify the GO category and identify the significant genes through the P-value and FDR [30].
Target genes and lncRNAs prediction of DEMs
Three algorithms, miRanda (http://www.microrna.org/), Targetscan (http://www.targetscan.org/) and miRWalk (http://129.206.7.150/) were used for miRNA-mRNA target gene prediction. The target genes that overlapped with genes that were identified by GO and KEGG pathway enrichment analyses were considered as the predicted results. MiRanda and PITA (https://genie.weizmann.ac.il/pubs/mir07/mir07_exe.html) were used to predict target lncRNAs, and those that overlapped with DELs between sarcomas and normal muscle samples were used for the construction of the ceRNA network.
Construction of the ceRNA network
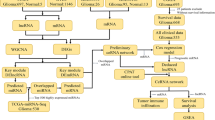
Based on the ceRNA theory that lncRNA can served as an endogenous “sponge” to regulate the expression of mRNA by sinking miRNA [31], upregulated or downregulated miRNAs, and lncRNAs or mRNAs that are inverse relationships with miRNAs in the miRNA-mRNA and lncRNA-miRNA interaction pairs were chose to construct the lncRNA–miRNA–mRNA ceRNA network [32]. The ceRNA network was constructed and visualized using Cytoscape software (version 2.8.2) [33]. The flow chart, shown in Fig. 1, represents the overall process of ceRNA network construction.

Flow chart of constructing the ceRNA network
PPI network analysis
The PPI network of the DEGs that were included in the ceRNA network was constituted by using the STRING database version 11.0 (https://string-db.org/). Cytoscape software (version 2.8.2) was used to visualize the PPI network with a combined score of protein pairs > 0.4 as the cut-off value [34].
Survival analysis
High-throughput experimental data with survival profiles of sarcoma patients was extracted from the TCGA database. The associations between biomarkers in the ceRNA network and overall survival of sarcoma patients were evaluated by using log rank test, with P-values less than 0.05 recognized as statistically significant.
Results
Available datasets of sarcomas
There were four datasets (GSE55625, GSE31045, GSE17674 and GSE18546) obtained from GEO, whereas the GSE55625 and GSE31045 datasets, with multiple zero and negative expression values, were excluded. The GSE17674 dataset based on the array platforms of Affymetrix Human Genome U133 Plus 2.0 Array contains 32 Ewing sarcoma and 18 normal muscle samples, from which we extracted probes for lncRNA annotation. The GSE18546 dataset includes 10 synovial sarcoma, 5 Ewing sarcoma and 5 normal muscle samples.
Identification of DEGs, DELs and DEMs in sarcomas
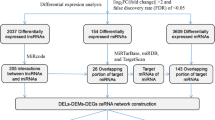
DEGs and DELs were identified in 32 Ewing sarcoma samples compared with 18 normal muscle samples in the GSE17674 dataset. DEMs were identified in 10 synovial sarcoma and 5 Ewing sarcoma samples compared with normal muscle samples in the GSE18546 dataset. A total of 3415 mRNAs (2554 upregulated and 861 downregulated genes) and 338 lncRNAs (234 upregulated and 104 downregulated lncRNAs) were deferentially expressed in sarcoma patients of the GSE17674 dataset (Additional file 2: Table S1; Additional file 3: Table S2).
There were two sarcoma subtypes, Ewing sarcoma and synovial sarcoma, in the GSE18546 dataset, and two different expression miRNAs sets were identified. Fifty-two miRNAs (39 upregulated and 13 downregulated miRNAs) were differentially expressed in Ewing sarcoma and normal muscle samples. In addition, there were 145 (109 upregulated and 36 downregulated miRNAs) DEMs between synovial sarcoma and normal muscle samples. We created a Venn diagram intersecting the two DEM sets, and 26 upregulated DEM s and 10 downregulated DEMs were identified (Additional file 1: Fig. S1; Additional file 4: Table S3). Hierarchical clustering of the identified DEGs, DELs and DEMs was displayed as a heatmap (Fig. 2).

a The heatmap of DEGs between sarcoma and normal muscle samples. b The heatmap of DELs between sarcoma and normal muscle samples. c The heatmap of DEMs between synovial sarcoma and normal muscle samples. d The heatmap of DEMs between Ewing sarcoma and normal muscle samples. Blue represents a low expression level and red represents a high level
GO and KEGG enrichment analysis of DEGs
GO enrichment analysis and KEGG pathways analysis of 3415 DEGs were performed to identify the potential functional genes. The upregulated and down-regulated genes were analyzed, respectively. The top 25 significantly enriched upregulated DEGs are presented in Fig. 3a, including transcription, DNA-templated (GO:0006351; P = 2.64 × 10–104), cell division (GO:0051301; P = 2.57 × 10–82) and regulation of transcription, DNA-templated (GO:0006355; P = 7.99 × 10–75). The most significantly enriched downregulated DEGs are presented in Fig. 3b, including muscle contraction (GO:0006936; P = 1.89 × 10–57), muscle filament sliding (GO:0030049; P = 5.78 × 10–46) and sarcomere organization (GO:0045214; P = 8.40 × 10–38). The main pathways for both the upregulated genes and the downregulated genes are metabolic pathways (Fig. 3c, d). The significant GO terms and pathways of upregulated and downregulated DEGs are presented in Additional file 5: Table S4 and Additional file 6: Table S5, respectively.

Top 25 enriched DEGs from the GO and KEGG pathway analyses. The bar plot shows the enrichment scores of GO and KEGG pathways. Different colors of bars represent different P values
Finally, 1296 intersecting DEGs were extracted from the significantly enriched genes in GO and KEGG pathway analyses, which involved the upregulated and downregulated genes (Additional file 7: Table S6).
Target genes and lncRNAs of DEMs
In this study, we have identified 36 DEMs, and we focused on whether these miRNAs would target the 1296 DEGs and 338 DELs. Based on the predicted targets of DEMs, 448 miRNA-mRNA interactor pairs (including 34 miRNAs and 269 mRNAs) and 454 lncRNA-miRNA interaction pairs (including 36 miRNAs and 117 lncRNAs) were obtained.
ceRNA network
According the ceRNA theory, we used the shared miRNA as a junction, that is, upregulated miRNAs, accompany with downregulated lncRNAs and mRNAs, and downregulated miRNAs, accompany with upregulated lncRNAs and mRNAs in the miRNA-mRNA and miRNA-lncRNA interaction pairs, to constructed a lncRNA–miRNA–mRNA ceRNA network. Finally, the ceRNA network was constructed with 1440 interactions, including 29 miRNAs, 69 lncRNAs and 113 mRNAs (Fig. 4; Additional file 8: Table S7).

The lncRNA–miRNA–mRNA ceRNA network. Rectangles, circles and triangles represent miRNAs, mRNAs and lncRNAs, respectively. Red and green color represent upregulated and downregulated biomarkers in sarcomas. Gray lines indicate interactions between miRNA-mRNA and miRNA-lncRNA
PPI network analysis
To further explore the most significant clusters of DEGs in the ceRNA network, we conducted PPI network analysis using the STRING database version 11.0 and visualization by Cytoscape. The most significant hub upregulated proteins were IGF1, PRKCB and GNAI3, and the most significant hub downregulated proteins were AR, CYCS and PPP1CB in the PPI network (Fig. 5; Additional file 9: Table S8).

PPI network of DEGs created by STRING. Red and green nodes represent upregulated and downregulated proteins, respectively. The edge between nodes represents the interaction between two connected proteins
Survival analysis
Furthermore, we performed the survival analysis based on the lncRNAs, miRNAs and mRNAs that were involved in the ceRNA network. The results show that seven mRNAs, four miRNAs and one lncRNA were significantly associated with the overall survival of sarcoma patients (P < 0.05). Among them, high expression levels of SMARCC1, SRSF10, PRPF38A, JARID2, GNAI3, miR-301a-3p, miR-106b-5p, miRNA-130b-3p, miR-423-3p and LINC01296 and low expression levels of ARF3 and PRKCB were associated with shorter overall survival in sarcomas (Fig. 6).

Kaplan–Meier survival curves of lncRNAs, miRNAs and mRNAs associated with overall survival of sarcomas patients
Discussion
Increasing evidence shows that lncRNAs and miRNAs were differentially expressed and implicated in series of molecular processes, including differentiation, proliferation, metastasis, and transcriptional regulation, in sarcomas [35, 36]. However, the entire regulatory network that links the functions of coding and noncoding RNAs has not been extensively discussed. In the present study, bioinformatics analysis was utilized to integrate available sequencing datasets of sarcomas, and 1296 DEGs were identified in sarcoma samples by combining the GO and KEGG pathway enrichment analyses. A total of 338 DELs were discovered after the probes were reannotated, and 36 DEMs were ascertained through intersecting two different expression miRNAs sets. Further, 448 miRNA–mRNA interactions and 454 lncRNA–miRNA interactions were obtained through target gene prediction, and then, we constructed a lncRNA–miRNA–mRNA ceRNA network that contained 29 miRNAs, 69 lncRNAs and 113 mRNAs.
The ceRNA network identified in our study provided useful clues for further study. According to the DEGs in the ceRNA network, we constructed a PPI network, which showed the upregulated hub nodes, including IGF1, PRKCB and GNAI3, and the downregulated hub nodes, including AR, CYCS and PPP1CB. In addition, there were twelve RNAs in the ceRNA network associated with the prognosis of sarcomas based on the TCGA database. Among the seven survival-associated mRNAs, the high expression levels of SMARCC1, SRSF10, PRPF38A, JARID2 and GNAI3 were significantly associated with shorter overall survival in sarcomas (P = 0.0018, P = 0.037, P = 0.0058, P = 0.0093, and P = 0.0234, respectively). However, the high expressions of ARF3 and PRKCB were significantly associated with longer overall survival (P = 0.0018 and P = 0.0162), indicating that ARF3 and PRKCB overexpression could be positive prognostic factors in sarcoma patients. In these overall survival-associated mRNAs, GNAI3 and PRKCB, with the highest degree of connectivity, were hubs and tended to be essential [37]. Moreover, the PPI network showed that GNAI3 and PRKCB interacted with each other. The protein encoded by PRKCB is one of the PKC family members that has been reported to be involved in many different cellular functions. Surdez et al.’s study showed PRKCB is strongly overexpressed in Ewing sarcoma. PRKCB inhibition significantly increased apoptosis in Ewing sarcoma cells and prevented tumor growth in vivo [38]. GNAI3 encodes an alpha subunit of guanine nucleotide-binding proteins and is involved in various transmembrane signaling pathways. In addition, in the ceRNA network, both GNAI3 and ARF3 can be regulated by miR-133b and miR-133a-3p, which are at the center of the regulatory network and interact with lncRNAs including NONHSAT001973.2, NONHSAT203034.1, SNHG12 and ZNF37BP. Recent research has demonstrated that SNHG12 was significantly overexpressed in osteosarcoma, and high expression of SNHG12 tended to lead to a poor prognosis of osteosarcoma patients. Researchers further confirmed that SNHG12 promoted tumorigenesis and metastasis by activating the Notch signaling pathway, wherein it functioned as a ceRNA, modulating the expression of Notch2 by competing with miR-195-5p [39]. Therefore, the interaction of SNHG12, miR-133a-3p/miR-133b and GNAI3 in sarcoma was worthwhile of exploration. Additionally, ARF3 could also be regulated by miR-378a-3p and miR-422a, and among them, miR-378a-3p targeted LINC01296. It has been revealed that the high expressions were significantly associated with shorter overall survival of sarcoma patients (P = 0.0297) through our survival analysis.
The high expressions of miRNA-301a-3p, miRNA-106b-5p, miRNA-130b-3p, and miRNA-423-3p were significantly associated with a shorter overall survival of sarcoma patients (P < 0.0001, P = 0.0046, P = 0.0128, and P = 0.0147, respectively). The overexpression of miR-301a was revealed in Ewing sarcoma cell lines. Additionally, the transfection of anti-miR-301a inhibited the proliferation and cell cycle progression of Ewing sarcoma cells [40]. In the ceRNA network, the NEAT1/miR-301a-3p, XIST/miR-301a-3p, miR-301a-3p/NR3C2, and miR-301a-3p/AR interactions were identified. XIST was significantly upregulated in osteosarcoma tissues and cell lines, and increased XIST expression was associated with the poor overall survival of patients [41]. Nakatani et al. have defined miR-130b as an independent predictor of risk for disease progression and survival by microarray analysis [42]. Until now, there is no study that reports the association of miRNA-423-3p and miRNA-106b-5p with sarcomas. The NEAT1/miR-301a-3p/AR, XIST/miR-301a-3p/AR, NEAT1/miR-301a-3p/NR3C2, and XIST/miR-301a-3p/AR axes predicted by the construction of the ceRNA network may provide a more precise research direction for exploring biological mechanisms extending from this ceRNA network.
There were several limitations in the present study. Sarcomas contain multiple distinct subtypes, and only two subtypes were involved in our study. More RNA sequencing datasets of all subtypes of sarcomas are required for the construction of a ceRNA network of sarcomas or a particular subtype. Furthermore, regulatory pathways of the ceRNA network are very complex, and ceRNA activity is influenced by multiple factors, such as the abundance and subcellular location of ceRNA components, miRNA/ceRNA affinity, RNA editing and RBPs [9]. This study only investigated the putative interactions of lncRNAs, miRNAs and genes, which requires further validation.
Conclusion
In conclusion, our work successfully constructed a ceRNA network by bioinformatics analysis based on the GEO database, providing a comprehensive resource for investigating the ceRNA regulation in sarcomas. Importantly, we screened out candidate prognostic biomarkers that are involved in the ceRNA network, which may exhibit important roles in the therapeutic target and prognosis analysis in sarcoma patients.
Availability of data and materials
The dataset supporting this article’s conclusions is available in the NCBI GEO repository, with accession number GSE17674 in https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE17674 and GSE18546 in https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE18546. The survival data of sarcomas patients were obtained from https://gtexportal.org/home/datasets.
Abbreviations
- GEO:
-
Gene expression omnibus
- DEGs:
-
Differentially expressed genes
- DELs:
-
Differentially expressed lncRNAs
- DEMs:
-
Differentially expressed miRNAs
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PPI:
-
Protein–protein interaction
- FDR:
-
False discovery rate
- TCGA:
-
The cancer genome atlas
References
Ressing M, Wardelmann E, Hohenberger P, Jakob J, Kasper B, Emrich K, Eberle A, Blettner M, Zeissig SR. Strengthening health data on a rare and heterogeneous disease: sarcoma incidence and histological subtypes in Germany. BMC Public Health. 2018;18(1):235.
Alavi SN, Florou V, Tinoco G, Trent JC, Wilky BA. A precision medicine approach in sarcoma: identification of patients who may benefit from early use of pazopanib. Discov Med. 2018;25(137):131–44.
Doyle LA. Sarcoma classification: an update based on the 2013 World Health Organization Classification of Tumors of Soft Tissue and Bone. Cancer. 2014;120(12):1763–74.
Miwa S, Yamamoto N, Hayashi K, Takeuchi A, Igarashi K, Tsuchiya H. Therapeutic targets for bone and soft-tissue sarcomas. Int J Mol Sci. 2019;20(1):170.
Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, Kovar H, Grimer R, Whelan J, Claude L, et al. Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol. 2015;33(27):3036–46.
Meyer WH. Improving outcomes for patients with pulmonary metastatic ewing sarcoma. J Clin Oncol. 2019;2019:1902522.
Cava C, Bertoli G. Portrait of tissue-specific coexpression networks of noncoding RNAs (miRNA and lncRNA) and mRNAs in normal tissues. Comput Math Methods Med. 2019;2019:9029351.
Zhu KP, Zhang CL, Ma XL, Hu JP, Cai T, Zhang L. Analyzing the interactions of mRNAs and ncRNAs to predict competing endogenous RNA networks in osteosarcoma chemo-resistance. Mol Ther. 2019;27(3):518–30.
Qi X, Zhang DH, Wu N, Xiao JH, Wang X, Ma W. ceRNA in cancer: possible functions and clinical implications. J Med Genet. 2015;52(10):710–8.
Li L, Peng M, Xue W, Fan Z, Wang T, Lian J, Zhai Y, Lian W, Qin D, Zhao J. Integrated analysis of dysregulated long non-coding RNAs/microRNAs/mRNAs in metastasis of lung adenocarcinoma. J Transl Med. 2018;16(1):372.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–8.
Plotnikova O, Baranova A, Skoblov M. Comprehensive analysis of human microRNA-mRNA interactome. Front Genet. 2019;10:933.
Wang XW, Guo QQ, Wei Y, Ren KM, Zheng FS, Tang J, Zhang HY, Zhao JG. Construction of a competing endogenous RNA network using differentially expressed lncRNAs, miRNAs and mRNAs in nonsmall cell lung cancer. Oncol Rep. 2019;42(6):2402–15.
Wang JJ, Huang YQ, Song W, Li YF, Wang H, Wang WJ, Huang M. Comprehensive analysis of the lncRNA-associated competing endogenous RNA network in breast cancer. Oncol Rep. 2019;42(6):2572–82.
Le K, Guo H, Zhang Q, Huang X, Xu M, Huang Z, Yi P. Gene and lncRNA co-expression network analysis reveals novel ceRNA network for triple-negative breast cancer. Scientific reports. 2019;9(1):15122.
Pan H, Guo C, Pan J, Guo D, Song S, Zhou Y, Xu D. Construction of a competitive endogenous RNA network and identification of potential regulatory axis in gastric cancer. Front Oncol. 2019;9:912.
Yu Y, Chen X, Cang S. Cancer-related long noncoding RNAs show aberrant expression profiles and competing endogenous RNA potential in esophageal adenocarcinoma. Oncol Lett. 2019;18(5):4798–808.
Li CY, Zhang WW, Xiang JL, Wang XH, Wang JL, Li J. Integrated analysis highlights multiple long non-coding RNAs and their potential roles in the progression of human esophageal squamous cell carcinoma. Oncol Rep. 2019;42(6):2583–99.
Xu F, Zhao Y, Qin G, Huan Y, Li L, Gao W. Comprehensive analysis of competing endogenous RNA networks associated with cholangiocarcinoma. Exp Ther Med. 2019;18(5):4103–12.
Li G, Han L, Ren F, Zhang R, Qin G. Prognostic value of the tumor-specific ceRNA network in epithelial ovarian cancer. J Cell Physiol. 2019;234(12):22071–81.
Ouyang D, Li R, Li Y, Zhu X. Construction of a competitive endogenous RNA network in uterine corpus endometrial carcinoma. Med Sci Monit. 2019;25:7998–8010.
Huang R, Meng T, Chen R, Yan P, Zhang J, Hu P, Zhu X, Yin H, Song D, Huang Z. The construction and analysis of tumor-infiltrating immune cell and ceRNA networks in recurrent soft tissue sarcoma. Aging. 2019;11(22):10116–43.
Chalifa-Caspi V, Yanai I, Ophir R, Rosen N, Shmoish M, Benjamin-Rodrig H, Shklar M, Stein TI, Shmueli O, Safran M, et al. GeneAnnot: comprehensive two-way linking between oligonucleotide array probesets and GeneCards genes. Bioinformatics (Oxford). 2004;20(9):1457–8.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl Acids Res. 2015;43(7):e47.
Zou JB, Chai HB, Zhang XF, Guo DY, Tai J, Wang Y, Liang YL, Wang F, Cheng JX, Wang J, et al. Reconstruction of the lncRNA-miRNA–mRNA network based on competitive endogenous RNA reveal functional lncRNAs in Cerebral Infarction. Sci Rep. 2019;9(1):12176.
The Gene Ontology (GO) project in 2006. Nucleic acids research 2006, 34(Database issue):D322–326.
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucl Acids Res. 2004;32(Database issue):D277-280.
Yi M, Horton JD, Cohen JC, Hobbs HH, Stephens RM. WholePathwayScope: a comprehensive pathway-based analysis tool for high-throughput data. BMC Bioinform. 2006;7:30.
Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, Georgescu C, Romero R. A systems biology approach for pathway level analysis. Genome Res. 2007;17(10):1537–45.
Wei D. A multigene support vector machine predictor for metastasis of cutaneous melanoma. Mol Med Rep. 2018;17(2):2907–14.
Guo LL, Song CH, Wang P, Dai LP, Zhang JY, Wang KJ. Competing endogenous RNA networks and gastric cancer. World J Gastroenterol. 2015;21(41):11680–7.
Xu Y, Chen J, Yang Z, Xu L. Identification of RNA Expression Profiles in Thyroid Cancer to Construct a Competing Endogenous RNA (ceRNA) Network of mRNAs, Long Noncoding RNAs (lncRNAs), and microRNAs (miRNAs). Med Sci Monit. 2019;25:1140–54.
Demchak B, Hull T, Reich M, Liefeld T, Smoot M, Ideker T, Mesirov JP. Cytoscape: the network visualization tool for GenomeSpace workflows. F1000Research. 2014;3:151.
Bhushan R, Rani A, Ali A, Singh VK, Dubey PK. Bioinformatics enrichment analysis of genes and pathways related to maternal type 1 diabetes associated with adverse fetal outcomes. J Diabetes Complic. 2020;34(5):107556.
Ma L, Sun X, Kuai W, Hu J, Yuan Y, Feng W, Lu X. LncRNA SOX2 overlapping transcript acts as a miRNA sponge to promote the proliferation and invasion of Ewing’s sarcoma. Am J Transl Res. 2019;11(6):3841–9.
Li M, Chen H, Zhao Y, Gao S, Cheng C. H19 Functions as a ceRNA in promoting metastasis through decreasing miR-200s activity in osteosarcoma. DNA Cell Biol. 2016;35(5):235–40.
He X, Zhang J. Why do hubs tend to be essential in protein networks? PLoS Genet. 2006;2(6):e88.
Surdez D, Benetkiewicz M, Perrin V, Han ZY, Pierron G, Ballet S, Lamoureux F, Redini F, Decouvelaere AV, Daudigeos-Dubus E, et al. Targeting the EWSR1-FLI1 oncogene-induced protein kinase PKC-beta abolishes ewing sarcoma growth. Can Res. 2012;72(17):4494–503.
Zhou S, Yu L, Xiong M, Dai G. LncRNA SNHG12 promotes tumorigenesis and metastasis in osteosarcoma by upregulating Notch2 by sponging miR-195-5p. Biochem Biophys Res Commun. 2018;495(2):1822–32.
Kawano M, Tanaka K, Itonaga I, Iwasaki T, Tsumura H. MicroRNA-301a promotes cell proliferation via PTEN targeting in Ewing’s sarcoma cells. Int J Oncol. 2016;48(4):1531–40.
Li GL, Wu YX, Li YM, Li J. High expression of long non-coding RNA XIST in osteosarcoma is associated with cell proliferation and poor prognosis. Eur Rev Med Pharmacol Sci. 2017;21(12):2829–34.
Nakatani F, Ferracin M, Manara MC, Ventura S, Del Monaco V, Ferrari S, Alberghini M, Grilli A, Knuutila S, Schaefer KL, et al. miR-34a predicts survival of Ewing’s sarcoma patients and directly influences cell chemo-sensitivity and malignancy. J Pathol. 2012;226(5):796–805.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Science and Technology Program of Sichuan Province (2018JY0165). We thank all of the participants in this study for their contributions. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
LG and LZ conceived and designed the study. LG and YZ wrote the manuscript. LG, YZ and XM were responsible for the data visualization, and analysis. XM and LZ reviewed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Fig. S1
: Common DEMs of Ewing sarcoma and synovial sarcoma comparisons in the expression profile of GSE18546.
Additional file 2. Table S1
: DEGs between sarcoma samples and normal muscle samples.
Additional file 3. Table S2
: DELs between sarcoma and normal muscle samples.
Additional file 4. Table S3
: The intersection of significantly DEMs of Ewing sarcoma and synovial sarcoma comparisons.
Additional file 5. Table S4
: Significantly enriched GO terms of DEGs.
Additional file 6. Table S5
: Significantly enriched pathway terms of DEGs.
Additional file 7. Table S6
: The intersecting DEGs of GO and KEGG pathway enrichment.
Additional file 8. Table S7
: The lncRNA–miRNA–mRNA ceRNA network of sarcomas.
Additional file 9. Table S8
: The PPI nodes of DEGs involved in the ceRNA network.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gao, L., Zhao, Y., Ma, X. et al. Integrated analysis of lncRNA–miRNA–mRNA ceRNA network and the potential prognosis indicators in sarcomas. BMC Med Genomics 14, 67 (2021). https://doi.org/10.1186/s12920-021-00918-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-00918-x