
Abstract
Background
Congenital hearing loss is one of the most common birth defects. Early identification and management play a crucial role in improving patients’ communication and language acquisition. Previous studies demonstrated that genetic screening complements newborn hearing screening in clinical settings.
Methods
We developed a multiplex PCR amplicon sequencing assay to sequence the full coding region of the GJB2 gene, the most pathogenic variants of the SLC26A4 gene, and hotspot variants in the MT-RNR1 gene. The sensitivity, specificity, and reliability were validated via samples with known genotypes. Finally, a pilot study was performed on 300 anonymous dried blood samples.
Results
Of 103 samples with known genotypes, the multiplex PCR amplicon sequencing assay accurately identified all the variants, demonstrating a 100% sensitivity and specificity. The consistency is high in the analysis of the test–retest reliability and internal consistency reliability. In the pilot study, 12.3% (37/300) of the newborns were found to carry at least one pathogenic variant, including 24, 10, and 3 from the GJB2, SLC26A4, and MT-RNR1 gene, respectively. With an allele frequency of 2.2%, the NM_004004.6(GJB2):c.109G>A was the most prevalent variant in the study population.
Conclusion
The multiplex PCR amplicon sequencing assay is an accurate and reliable test to detect hearing loss variants in the GJB2, SLC26A4, and MT-RNR1 genes. It can be used to screen genetic hearing loss in newborns.
Similar content being viewed by others

Background
Congenital hearing loss is one of the most common birth defects in children. Around 30,000 newborns are estimated to born with congenital hearing loss every year in China [1]. Genetic factors and congenital cytomegalovirus infections accounted for approximately 60% and 21% of congenital hearing loss, respectively [2]. Early detection of congenital hearing loss is proven to be beneficial for language acquisition and academic performance [3].
Universal newborn hearing screening is performed in the nursery before discharge to discern the deaf and hard of hearing infants [4]. The methods include otoacoustic emission test and automated auditory brainstem response test [5]. Universal newborn hearing screening has been implemented in China for more than 20 years. It has made significant contributions to the early detection, diagnosis, and interventions of hearing loss [6], leading to improved language development [7]. However, it has limitations in identifying cytomegalovirus-induced hearing loss [8], aminoglycoside-antibiotic-induced ototoxicity [9], and delayed-onset prelingual hearing loss [10]. Additionally, the universal newborn hearing screening has a low positive predictive value, which may cause parental anxiety [11] and result in unnecessary follow-up tests [12]. These challenges urged the need for genetic screens to integrate into the universal newborn hearing screening program [13].
Genetic screens for hearing loss were initially proposed in 2006 [14]. Considering the highly genetic heterogeneity [15], a test covering hundreds of deafness-related genes was costly and hard to implement as a screening test in clinical settings. Limited genetic screens, targeting a limited number of variants, are feasible because hotspot variants existed and the ten most frequently encountered causative variants accounted for 30.4% of genetic diagnoses [5]. Large epidemiological studies demonstrated that GJB2, SLC26A4, and MT-RNR1 are the most common disease-causing genes in the Chinese population [16, 17]. Recently, the clinical benefits of limited genetic screens of variants in these three genes were well studied [18,19,20].
Several techniques were developed to detect a limited number of variants. Wan et al. described a multiplex genetic screening system called the SNPscan assay technique to screen for 115 deafness-related variants [21]. Wang et al. developed a multicolor melting curve analysis-based assay to detect 12 deafness-related variants simultaneously [22]. Li et al. developed a PCR-reverse dot blot assay to screen 20 variants in hearing loss genes [23].
In this study, we developed a multiplex PCR amplicon sequencing assay to sequence the full coding region of the GJB2 gene, the most pathogenic variants of the SLC26A4 gene, and hotspot variants in the MT-RNR1 gene. We then validated and piloted the genetic test in a newborn population.
Material and methods
Multiplex PCR
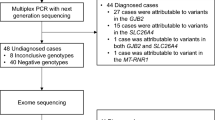
The workflow of the multiplex PCR amplicon sequencing assay is shown in Fig. 1. DNA was extracted from peripheral blood or dried blood spots by DNA Extraction Kit (BGI Biotech, Wuhan, China). Twenty-six pairs of indexed primers were used to amplify targeted regions by multiplex PCR (Additional file 1: Table S1). The amplification was conducted in a reaction mixture with a final volume of 25 µl containing 2 × KAPA 2G Fast Multiple PCR Mix (KAPA BIOSYSTEMS, Wilmington, MA, USA), 0.2uM Primer Mix, and 1–5 ng DNA templet. The PCR cycling program was as follows: 95 °C for 5 min; followed by ten cycles of denaturation at 95 °C for 30 s, annealing at 65 °C for 50 s (− 1 °C/cycle); then 25 cycles of denaturation at 95 °C for 30 s, annealing at 58 °C for 30 s; and a final extension at 72 °C for 15 s; 72 °C for the 60 s and holding at 12 °C.

The workflow of the multiplex PCR amplicon sequencing assay
Library construction, sequencing, and analysis
PCR products of 48 samples (tag1-48) were pooled together. The pooled amplicons were purified and used for library construction. Briefly, 200 ng purified products were taken for end repair and A-tailing before adapter ligation. And then, the adapter-ligated-products were amplified by four-cycle PCR. Finally, after single-stranded circle formation and DNA nano balls preparation, the libraries were sequenced by MGISEQ-2000 sequencer (MGI, Shenzhen, China) in pair-end 100.
The cleaned reads were grouped to each sample based on index primers and then were mapped to the human reference genome (hg19) using Bowtie and SAMtools to create BAM and index files. Alignment data were next subjected to a strategic procedure for variant calling by GATK [24]. All hotspots are checked the mutation rate in bams and add the variants with a high mutation rate (> 0.1) and high depth (> 30X) as a complement to GATK detection. The genotype call is expressed as homozygous (allele fraction ≥ 0.8), heterozygous (0.1 ≤ allele fraction < 0.8), wildtype (allele fraction < 0.1).
Validation and pilot of the assay
To validate the assay, we employed 103 dried blood samples with known genotypes to analyze the sensitivity and specificity, including 93 samples with positive genotypes and 11 samples with negative genotypes, respectively. Then, we performed experiments to access the test–retest reliability and internal consistency reliability. We used 14 samples and 31 samples with known genotypes in the analysis of the test–retest reliability and internal consistency reliability, respectively.
To test the performance of the assay, we randomly selected 300 anonymous dried blood samples. The detected genotypes were confirmed by sanger sequencing. This study was approved by the Institutional Review Board of BGI.
Results
Design and establishment of the assay
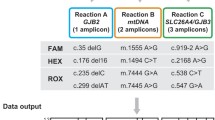
The workflow of the multiplex PCR amplicon sequencing assay is displayed in Fig. 1. To full sequence the coding region of the GJB2 gene, the seven GJB2 primers were divided into two groups for PCR reactions in parallel. The sixteen SLC26A4 primers were divided to minimize the interaction between primers. As a result, well 1 included four primers in the GJB2 gene, eight primers in the SLC26A4 gene, three primers in the MT-RNR1 gene; well 2 included three primers in the GJB2 gene and eight primers in the SLC26A4 gene (Fig. 2). The amplicons were then mixed into a single tube for library construction and referred to as sequencing.

A schematic diagram showing the location of primers and hotspot variants. White boxes indicate exons, and the black boxes indicate the coding region. The lines above the coding regions indicate primers. The red lines present the primers in well 1, blue lines present the primers in well 2. The variants were pathogenic variants with an allele frequency greater than 1/10,000 in the gnomAD database
Finally, the coding region of the GJB2 gene was completely covered (Fig. 2a). Although only 63% of the coding region of the SLC26A4 gene was covered, the known pathogenic variants in the ClinVar database with an allele frequency greater than 1/10,000 were all included (Fig. 2b). Additionally, three mitochondrial variants (m.1095T>C, m.1494C>T, and m.1555A>G) were included. The m.1494C>T and m.1555A>G are prevalent in 0.23% of the Chinese population [19]. The prevalence of m.1095T>C was 0.61% in a large cohort of 1642 Han Chinese pediatric subjects with aminoglycoside-induced and nonsyndromic hearing loss [25].
Analytical studies
To verify the sensitivity and specificity of the multiplex PCR amplicon sequencing assay, we selected 11 negative and 92 positive samples with known genotypes (101 variants in total), including 65 heterozygous and 1 homozygous states in the GJB2 gene; 30 heterozygous states in the SLC26A4 gene; and 5 mitochondrial variants (4 homoplasmy and 1 heteroplasmy) in the MT-RNR1 gene. The variants were all confirmed by Sanger sequencing. Our assay accurately identified all the 101 variants from the positive samples, indicating a 100% sensitivity (Table 1). No pathogenic variants were identified in the targeted region from negative samples, indicating a 100% specificity.
To validate the assay, we analyzed the test–retest reliability and internal consistency reliability. In the test–retest reliability, we performed the assay on 31 samples with known genotypes three independent times. In the internal consistency reliability, 14 samples were repeatedly tested in an internal test. All the variants in a heterozygous state were identified in a ratio between 0.1 and 0.8 and the variants in a homozygous state were identified in a ratio greater than 0.8 (Fig. 3, Additional file 2: Table S2, Additional file 3: Table S3). There was no significant difference in the ratio for any of the samples. These results demonstrated the multiplex PCR amplicon sequencing assay has a high consistency in detecting variants in targeted regions.

The reliability of the multiplex PCR amplicon sequencing assay. In the analysis of test–retest reliability (a), 31 samples with known genotypes were tested three independent times. In the analysis of internal consistency reliability (b), 14 samples with known genotypes were repeatedly tested in an internal test
Pilot evaluation
Of 300 anonymous dried blood spots, 37 (12.3%) were found to carry at least one pathogenic variant. One sample was identified in a homozygous and three samples were identified in homoplasmy. All other samples were in a heterozygous state (Table 2). NM_004004.6:c.109G>A in the GJB2 gene was the most prevalent variant in the cohort, accounting for an allele frequency of 2.2%. The complete sequence of the coding region in the GJB2 gene revealed a carrier rate of 8% (24/300), contributed to c.109G>A (4.3%), c.235delC (2.7%), c.176_191del (0.7%), c.299_300delAT(0.3%). Additionally, four variants in the SLC26A4 genes were identified; they are c.919-2A>G (1.3%), c.1975G>C (1.0%), c.589G>A (0.7%) and c.1173C>T (0.3%). The carrier rate of m.1095T>C in the MT-RNR1 gene was 1.0%.
Discussion
Hearing loss is the most common neurosensory disorder in humans, with an incidence of one in 1000 worldwide [26]. The most common pathogenic variants were from three genes (GJB2, SLC26A4, and MT-RNR1) in the Chinese population [1, 27,28,29]. Previous studies primarily focused on a number of limited hotspot variants, such as NM_004004.6:c.235delC in the GJB2 gene, NM_000441.2:c.919-2A>G in the SLC26A4 gene, and m.1555A>G and m.1494C>T in the MT-RNR1 gene [20]. In this study, we developed a multiplex PCR amplicon sequencing assay, covering the full coding region of the GJB2 gene, the most pathogenic variants in the SLC26A4 gene, and the three hotspot variants in the MT-RNR1 gene.
In this study, the multiplex PCR amplicon sequencing assay was designed to cover the entire coding region of the GJB2 gene. The implement of GJB2 screening in newborns is considered necessary and feasible for the following reasons. First, GJB2 is the most common gene causing congenital hearing loss [30]. It is estimated that the single gene contributed to 21% of the congenital hearing loss and 15% of the childhood hearing loss at 4 years [14]. Second, the coding region of the GJB2 gene is short (681 bp) enough to achieve a complete sequence at a low cost. Third, previous studies mainly focused on several hotspot variants including NM_004004.6:c.235delC, NM_004004.6:c.299_ 300delAT, NM_004004.6:c.176del16, and NM_004004.6:c.35delG, et al. These studies identified some hearing loss patients with inconclusive genotypes (a heterozygous variant in a single gene) [19, 20]. The second variant might be identified by a complete sequence of the coding region in the GJB2 gene.
Our study recovered pathogenic variants in the GJB2 gene had a carrier rate (8%) in the newborn population. The high rate is attributable to the inclusion of NM_004004.6:c.109G>A (4.3%) in our assay. NM_004004.6:c.109G>A is the most prevalent variant in Eastern Asia [31]. Although the penetrance is low, it was proven to have strong associations with mild or moderate hearing loss [32]. Recently, the ClinGen Hearing Loss Expert Panel reached a consensus interpretation of this variant as a pathogenic variant [33]. The documentation of patients’ longitudinal auditory features with NM_004004.6(GJB2):c.109G>A supports including it in a screening panel [34].
The SLC26A4 gene is another common cause of hearing loss. It is attributed to only 3% of the congenital hearing loss, but the proportion significantly increased to 12% at the age of four [14]. This is because that the pathogenic variants in the SLC26A4 gene cause enlarged vestibular aqueduct, which is late-onset. More importantly, the late-onset hearing loss cannot be identified by conventional newborn hearing screens because the hearing status was normal at birth when the tests were performed. In this scenario, genetic screens are proven to be a complementary test to conventional newborn hearing screens [20]. In our study, the full coding region of the SCL26A4 gene was not fully covered because it has over 20 coding regions. Still, we covered all the hotspot variants in the Chinese population (Fig. 2b), including c.919-2A>G, c.2168A>G, c.1975G>C, which accounts for 13.39% of the cases contributed to the SLC26A4 gene in a cohort of 864 Chinese patients [35].
MT-RNR1 is a mitochondrial gene associated with aminoglycoside-induced hearing loss [36]. Individuals carrying such variants have normal hearing functions until aminoglycoside exposure. The drug susceptibility is unable to be identified by conventional physiologic hearing screens but detectable by genetic screens [20]. This genetic information is not only useful for the newborns who received the genetic screens, but also beneficial for the maternal relatives because the mitochondrial variants are transmitted in maternal inheritance. In this study, 1% of newborns were identified to carry mitochondrial variants, higher than the proportion (0.24%) from the previous studies [18]. This might be attributable to the small sample size.
The genetic hearing loss test was proposed to incorporate into the newborn physiologic hearing screening program before hospital discharge [5]. The results of the genetic test are valuable for the choice of following-up tests. Considering a high risk of hearing loss, audiologic evaluations should be arranged directly if a homozygous or compound heterozygous status was identified [20]. This strategy can identify newborns with hearing loss missed by physiologic screens, whom may benefit from a prompt intervention and management [18,19,20]. If a newborn with a heterozygous variant failed the physiologic screens, a following-up physiologic re-test or a comprehensive genetic test should be suggested. Asymptomatic newborns with MT-RNR1 variants and the maternal family members should be informed of their predisposition for aminoglycoside toxicity [37].
We employed a multiplex PCR amplicon sequencing assay to analyze variants in deaf-related genes for two reasons. First, multiplex PCR allows the simultaneous detection of multiple targets of interest in an easy and efficient way [38]. Second, this assay relied on the next-generation sequencing techniques, facilitating high throughput processing of a large number of samples in a short time. These characters allow the test to implement in a large population as a screening test.
Conclusion
In conclusion, we developed and validated a multiplex PCR amplicon sequencing assay to detect variants in the GJB2, SLC26A4, and MT-RNR1 genes. Our results demonstrated that the assay is an accurate and reliable test and can be used to screen genetic hearing loss in newborns.
Availability of data and materials
The data used and/or analyzed during the current study are available from the corresponding author on reasonable request. The data are not publicly available due to privacy or ethical restrictions.
Abbreviations
- PCR:
-
Polymerase chain reaction
- DNA:
-
Deoxyribonucleic acid
References
Wang QJ, Zhao YL, Rao SQ, Guo YF, He Y, Lan L, Yang WY, Zheng QY, Ruben RJ, Han DY, et al. Newborn hearing concurrent gene screening can improve care for hearing loss: a study on 14,913 Chinese newborns. Int J Pediatr Otorhinolaryngol. 2011;75(4):535–42.
Cynthia C, Morton PD, Walter E, Nance MD. Newborn hearing screening—a silent revolution. N Engl J Med. 2006;354:2151–64.
Yoshinaga-Itano C, Sedey AL, Coulter DK, Mehl AL. Language of early- and later-identified children with hearing loss. Pediatrics. 1998;102(5):1161–71.
Mason JA, Herrmann KR. Universal infant hearing screening by automated auditory brainstem response measurement. Pediatrics. 1998;101(2):221–8.
Shearer AE, Shen J, Amr S, Morton CC, Smith RJ. Newborn hearing screening working group of the national coordinating center for the regional genetics N: a proposal for comprehensive newborn hearing screening to improve identification of deaf and hard-of-hearing children. Genet Med. 2019;21(11):2614–30.
Huang LH, Zhang L, Tobe RY, Qi FH, Sun L, Teng Y, Ke QL, Mai F, Zhang XF, Zhang M, et al. Cost-effectiveness analysis of neonatal hearing screening program in China: should universal screening be prioritized? BMC Health Serv Res. 2012;12:97.
Kennedy CR, McCann DC, Campbell MJ, Law CM, Mullee M, Petrou S, Watkin P, Worsfold S, Yuen HM, Stevenson J. Language ability after early detection of permanent childhood hearing impairment. N Engl J Med. 2006;354(20):2131–41.
Fowler KB, Dahle AJ, Boppana SB, Pass RF. Newborn hearing screening: will children with hearing loss caused by congenital cytomegalovirus infection be missed? J Pediatr. 1999;135(1):60–4.
Zhang J, Wang P, Han B, Ding Y, Pan L, Zou J, Liu H, Pang X, Liu E, Wang H, et al. Newborn hearing concurrent genetic screening for hearing impairment—a clinical practice in 58,397 neonates in Tianjin, China. Int J Pediatr Otorhinolaryngol. 2013;77(12):1929–35.
Kim BG, Shin JW, Park HJ, Kim JM, Kim UK, Choi JY. Limitations of hearing screening in newborns with PDS mutations. Int J Pediatr Otorhinolaryngol. 2013;77(5):833–7.
Mohd Khairi MD, Rafidah KN, Affizal A, Normastura AR, Suzana M, Normani ZM. Anxiety of the mothers with referred baby during universal newborn hearing screening. Int J Pediatr Otorhinolaryngol. 2011;75(4):513–7.
Kennedy CR. Controlled trial of universal neonatal screening for early identification of permanent childhood hearing impairment: coverage, positive predictive value, effect on mothers and incremental yield. Wessex Universal Neonatal Screening Trial Group. Acta Paediatr Suppl. 1999;88(432):73–5.
Howell RR. We must now put in place an updated, comprehensive newborn screening program for deaf and hard-of-hearing infants. Genet Med. 2019;21:2439–41.
Morton CC, Nance WE. Newborn hearing screening—a silent revolution. N Engl J Med. 2006;354(20):2151–64.
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT, Campbell CA, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135(4):441–50.
Dai P, Yu F, Han B, Liu X, Wang G, Li Q, Yuan Y, Liu X, Huang D, Kang D, et al. GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J Transl Med. 2009;7:26.
Dai P, Liu X, Han D, Qian Y, Huang D, Yuan H, Li W, Yu F, Zhang R, Lin H, et al. Extremely low penetrance of deafness associated with the mitochondrial 12S rRNA mutation in 16 Chinese families: implication for early detection and prevention of deafness. Biochem Biophys Res Commun. 2006;340(1):194–9.
Dai P, Huang LH, Wang GJ, Gao X, Qu CY, Chen XW, Ma FR, Zhang J, Xing WL, Xi SY, et al. Concurrent hearing and genetic screening of 180,469 neonates with follow-up in Beijing, China. Am J Hum Genet. 2019;105(4):803–12.
Wang Q, Xiang J, Sun J, Yang Y, Guan J, Wang D, Song C, Guo L, Wang H, Chen Y, et al. Nationwide population genetic screening improves outcomes of newborn screening for hearing loss in China. Genet Med. 2019;21(10):2231–8.
Guo L, Xiang J, Sun L, Yan X, Yang J, Wu H, Guo K, Peng J, Xie X, Yin Y, et al. Concurrent hearing and genetic screening in a general newborn population. Hum Genet. 2020;139(4):521–30.
Du W, Cheng J, Ding H, Jiang Z, Guo Y, Yuan H. A rapid method for simultaneous multi-gene mutation screening in children with nonsyndromic hearing loss. Genomics. 2014;104(4):264–70.
Wang X, Hong Y, Cai P, Tang N, Chen Y, Yan T, Liu Y, Huang Q, Li Q. Rapid and reliable detection of nonsyndromic hearing loss mutations by multicolor melting curve analysis. Sci Rep. 2017;7:42894.
Li S, Peng Q, Liao S, Li W, Ma Q, Lu X. A reverse dot blot assay for the screening of twenty mutations in four genes associated with NSHL in a Chinese population. PLoS ONE. 2017;12(5):e0177196.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–8.
Lu J, Li Z, Zhu Y, Yang A, Li R, Zheng J, Cai Q, Peng G, Zheng W, Tang X, et al. Mitochondrial 12S rRNA variants in 1642 Han Chinese pediatric subjects with aminoglycoside-induced and nonsyndromic hearing loss. Mitochondrion. 2010;10(4):380–90.
Dai P, Yu F, Han B, Yuan Y, Li Q, Wang G, Liu X, He J, Huang D, Kang D, et al. The prevalence of the 235delC GJB2 mutation in a Chinese deaf population. Genet Med. 2007;9(5):283–9.
Yu H, Liu D, Yang J, Wu Z. Prevalence of mutations in the GJB2, SLC26A4, GJB3, and MT-RNR1 genes in 103 children with sensorineural hearing loss in Shaoxing, China. Ear Nose Throat J. 2018;97(6):E33-e38.
Liu Y, Hu C, Liu C, Liu D, Mei L, He C, Jiang L, Wu H, Chen H, Feng Y. A rapid improved multiplex ligation detection reaction method for the identification of gene mutations in hereditary hearing loss. PLoS ONE. 2019;14(4):e0215212.
Ouyang XM, Yan D, Yuan HJ, Pu D, Du LL, Han DY, Liu XZ. The genetic bases for non-syndromic hearing loss among Chinese. J Hum Genet. 2009;54(3):131–40.
Guan Q, Balciuniene J, Cao K, Fan Z, Biswas S, Wilkens A, Gallo DJ, Bedoukian E, Tarpinian J, Jayaraman P, et al. AUDIOME: a tiered exome sequencing-based comprehensive gene panel for the diagnosis of heterogeneous nonsyndromic sensorineural hearing loss. Genet Med. 2018;20(12):1600–8.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Huang S, Huang B, Wang G, Yuan Y, Dai P. The Relationship between the p.V37I mutation in GJB2 and hearing phenotypes in Chinese individuals. PLoS ONE. 2015;10(6):e0129662.
Shen J, Oza AM, Del Castillo I, Duzkale H, Matsunaga T, Pandya A, Kang HP, Mar-Heyming R, Guha S, Moyer K, et al. Consensus interpretation of the p.Met34Thr and p.Val37Ile variants in GJB2 by the ClinGen hearing loss expert panel. Genet Med. 2019;21(11):2442–52.
Wu CC, Tsai CH, Hung CC, Lin YH, Lin YH, Huang FL, Tsao PN, Su YN, Lee YL, Hsieh WS, et al. Newborn genetic screening for hearing impairment: a population-based longitudinal study. Genet Med. 2017;19(1):6–12.
Yuan Y, Li Q, Su Y, Lin Q, Gao X, Liu H, Huang S, Kang D, Todd NW, Mattox D, et al. Comprehensive genetic testing of Chinese SNHL patients and variants interpretation using ACMG guidelines and ethnically matched normal controls. Eur J Hum Genet. 2019;28:231–43.
Van Camp G, Smith RJ. Maternally inherited hearing impairment. Clin Genet. 2000;57(6):409–14.
Barbarino JM, McGregor TL, Altman RB, Klein TE. PharmGKB summary: very important pharmacogene information for MT-RNR1. Pharmacogenet Genomics. 2016;26(12):558–67.
Onda Y, Takahagi K, Shimizu M, Inoue K, Mochida K. Multiplex PCR targeted amplicon sequencing (MTA-Seq): simple, flexible, and versatile SNP genotyping by highly multiplexed PCR amplicon sequencing. Front Plant Sci. 2018;9:201.
Acknowledgements
We express our gratitude to the participants for their kind permission to contribute their data for anonymous research use.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
HY wrote the first and final draft of the paper. HL, GZ, JZ provided input on data analysis and critical comments on the paper. JX and ZP coordinated the study and commented on the drafts and the final paper. JX designed and supervised the conduct of the study, supervised the data analysis, interpreted the data, and reviewed and commented on all drafts and the final paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Institutional Review Board of the BGI approved this study (No. BGI-IRB 20072-T1). All patients, parents or legal guardians signed an informed consent for sample collection.
Consent for publication
Not applicable.
Competing interests
HL, GZ, JZ, ZP, and JX were employed at BGI at the time of submission.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Primers of GJB2, SLC24A4 and MT-RNR1 gene.
Additional file 2: Table S2.
The test–retest reliability on 31 samples with known genotypes. Table S3: The internal consistency reliability on 14 samples with known genotypes.
Additional file 3: Table S3.
The internal consistency reliability on 14 samples with known genotypes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, H., Luo, H., Zhang, G. et al. A multiplex PCR amplicon sequencing assay to screen genetic hearing loss variants in newborns. BMC Med Genomics 14, 61 (2021). https://doi.org/10.1186/s12920-021-00906-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-00906-1