
Abstract
Background
Mutations in lysyl-tRNA synthetase (KARS1), an enzyme that charges tRNA with the amino acid lysine in both the cytoplasm and mitochondria, have been associated thus far with autosomal recessive Charcot–Marie–Tooth type CMTRIB, hearing loss type DFNB89, and mitochondrial encephalohepatopathy (MEH) featuring neurodevelopmental disorders with microcephaly, white matter changes, and cardiac and hepatic failure in less than 30 patients.
Case presentation
We report the clinical, biochemical and molecular findings of a 14-month-old girl with severe MEH compatible clinical features, profound sensorineural hearing loss, leopard spot retinopathy, pancytopenia, and advanced liver disease with portal hypertension leading to death at the age of 30 months.
Conclusions
Whole exome sequencing identified two rare variants in KARS1 gene. Our report expands the allelic and clinical features of tRNA synthase disorders. Moreover, with our report we confirm the usefulness of WES as first tier diagnostic method in infants with complex multisystem phenotypes.
Similar content being viewed by others

Background
Aminoacyl tRNA synthetase (ARS) proteins are fundamentally known as the first enzymes of translation that catalyze amino acid attachment to their cognate tRNA. This catalytic process, called tRNA charging, is necessary for the translation of genetic sequences into polypeptide chains [1]. The KARS1 gene codes for both the mitochondrial and cytoplasmic isoforms of the t-RNA synthase of Lysine, a ubiquitous enzyme responsible for the link between the amino acid Lysine and the cognate RNA transfer [2]. To date, sixteen mutations in the KARS1 gene have been associated with autosomal recessive Charcot–Marie–Tooth type CMTRIB [3], hearing loss type DFNB89 [4], and mitochondrial encephalohepatopathy (MEH) featuring neurodevelopmental disorders with microcephaly, white matter changes, and cardiac and hepatic failure in 26 patients.
We report the clinical, biochemical, neuroradiological and molecular findings of an additional child. The clinical features observed largely recall the wide spectrum seen in previously reported patients harboring bi-allelic variants in KARS1 and satisfy the clinical criteria for mitochondrial encephalocardiohepatopathy [5] with limited muscular involvement of oxidative-phosphorylation. Ocular and hepatic findings which have never described up to now will be described.
Case presentation
This female child was the first baby born at 38 weeks of gestational age to healthy, unrelated parents from a complicated pregnancy due to intrauterine growth retardation detected at 32 weeks of gestational age. At birth weight was 2240gr (− 1.97 SD), length 48 cm (− 0.39 SD), and cranial circumference 31.5 cm (− 2 SD). First neonatal neurological examination highlighted severe global hypotonia, poor crying and sucking with feeding difficulties. At the age of 4 months, a first brain MRI showed white matter lesions and a major amount of periventricular calcifications. Moreover, MR-Spectroscopy with the voxel localized on left parietal white matter region showed reduced NAA/Cr ratio (1.47) and Cho/Cr ratio (1.40) and an elevated lactate peak (Fig. 1). Follow-up neuroimaging investigations showed progression of cortical and subcortical atrophy and increase of calcifications in subcortical regions and deep brain and the cerebellum.

Neuroradiological findings. a 1.5T Brain MRI at 4 months (T2-weighted axial section): diffuse and bilateral hyperintense alterations in nucleocapsular and peritrigonal regions, mild calcifications and thin corpus callosum; b 1.5T Brain MRI at 30 months (T2-weighted axial section): evolution of progressive damage involving diffusively the white matter leading to cortical, subcortical and cerebellar atrophy with more prominent amount of bilateral calcifications; c Computed Tomography (CT) scan at 30 months: generalized and massive cortical and subcortical atrophy with hydrocephalus; d MR-Spectroscopy (MRS) at 4 months, volume of interest in left parietal white matter region: reduced NAA/Cr ratio and Cho/Cr ratio and occurrence of a high lactate peak (at 1.3 ppm)
Neurological and physical examinations at 21 months showed apostural tetraparesis, severe developmental and growth delay, epileptic encephalopathy with spasms, myoclonic and focal seizures. She also showed hyposomatism with low weight 8900 gr (− 2.35 SD) and length 76 cm (− 1.79 SD), microcephaly with reduced head circumference of 39 cm (− 5 SD) at 14 months, and bilateral neurosensorial hearing loss. Echocardiogram revealed pulmonary valve stenosis.
Ophthalmological examination showed exotropia and poor fixation. Indirect fundus examination revealed the presence of a bilateral, hypoplasic appearance of the optic nerve head and bilateral, diffuse and mottled retinal pigmentation described as “leopard spot” retinopathy. A first spectral domain optical coherence tomography (SD-OCT) scan at age of 3 months revealed subnormal peripapillary retinal nerve fiber layer (pRNFL) in both eyes with overall preserved retinal architecture. A second SD-OCT scan performed at 14 months of age revealed a severe thinning of pRNFL and retinal thickness in both eyes suggestive of progressive retinal degeneration (Fig. 2).

SD-OCT scan findings. a Wide field retinal imaging showing ophthalmoscopic appearance of bilateral leopard spot retinopathy and mild hypoplasic appearance of optic nerve head; b SD-OCT scan showing normal foveal profile and retinal segmentation; c SD-OCT optic nerve scan showing severe and bilateral pRNFL thinning coherent with a neurodegenerative process and bilateral optic atrophy
Liver ultrasound at 5 months of age showed a nodular liver surface, round edge, hypoechoic nodules in the liver parenchyma and calcifications in the 6th hepatic segment. The portal vein was non-obstructed, and the spleen was normal (length 4.7 cm). Serum aminotransferases, gamma-glutamil-transpeptidase, albumin and international normalized ratio were normal. Total serum bile acids were slightly elevated (2 times the upper limit of normal). She was occasionally found to have portal hypertension at 5 months of age (grade 2 esophageal varices) and at 14 months of age she presented with acute hematemesis from esophageal varices and then started on propranolol (1–2 mg/kg/day). The child underwent a complete diagnostic work-up and the most common causes of cholestasis were excluded.
High levels of serum lactate (4.9 mM/L, reference range 0.63–2.44 mM/L) and normal pyruvate value were also detected.
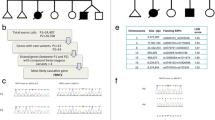
Shortly after, clinical manifestations evolved to a progressive pancytopenia with the observation of ring sideroblasts at a bone marrow biopsy suggestive of a Pearson marrow-pancreas-like syndrome in spite of normal metabolic investigations in blood and urine. Also, the minimal alterations of the respiratory chain complex enzyme activities in a skeletal muscle biopsy and lack of deletion/duplication or punctuate mutations in the mitochondrial genome did not support this possibility. The clinical situation of the baby worsens, and she died at 30 months of life. Array-CGH analysis was negative. Whole exome sequencing (WES) in the family trio (Additional file 1) revealed the paternal c.815T>G/p.[Phe272Cys] (rs138062606) and the maternal c.1570T>C/p.[Cys524Arg] (rs776736207) (Fig. 3a) variants in KARS1 (NM 001130089.1). Paternal mutation has already reported elsewhere [6], while the other variant has not yet been described. Both mutations are ultra-rare and in silico predictions using multiple prediction software programs indicate that the two variants are probably pathogenetic (Fig. 3b) and both alter highly conserved nucleotides in the various species (Fig. 3c). Western Blot (WB) analysis in muscle biopsy homogenate using a rabbit polyclonal anti-KARS antibody (Proteintech; dilution 1:500) showed a severe reduction of the gene product (Fig. 3d, Additional files 2, 3, 4). This implies an impairment of mitochondrial protein synthesis machinery, though this was not directly tested.

Molecular findings in this patient. a Pedigree and electropherograms; b in silico predictions using multiple prediction software programs; c multiple species alignment; d Western Blot analysis in muscle biopsy homogenate
Discussion and conclusions
Whole exome sequencing allows us to identify two rare genetic variants and functional studies (WB in muscle biopsy) showed a severe reduction of the gene product supporting the direct role of the KARS1 gene in this child’s disease.
The clinical features observed in our patient largely recall the wide spectrum seen in the 26 reported patients harboring bi-allelic variants in KARS1 (Table 1) and satisfy the clinical criteria for mitochondrial encephalocardiohepatopathy with limited muscular involvement of oxidative phosphorylation [5].
Our patient presents some new clinical findings never reported before. Presence of early-onset portal hypertension complicated by bleeding from esophageal varices is a new feature in KARS1-related syndromes, but it remains unclear how this occurs. In a previously reported child [6], hepatoportal sclerosis (HPS) was demonstrated on the liver biopsy. HPS is a rare case of non-cirrhotic portal hypertension characterized by sclerosis of the intrahepatic portal veins. HPS could present with normal or mildly abnormal liver function tests and, in advanced cases, with liver nodularity and atrophy producing an imaging appearance indistinguishable from that of cirrhosis [8]. It is tempting to speculate that energy failure due to low mitochondrial protein synthesis might be invoked in this case. Although the child described in the present report did not undergo a liver biopsy, her clinical presentation could be consistent with the diagnosis of HPS.
Our patient showed novel ocular findings. We observed early and severe signs of optic neuropathy and “leopard spot” retinopathy, a condition which has so far been associated with leukemia [9], uveal effusion syndrome [10], systemic argyrosis [11], pseudoxanthoma elasticum [12], Warburg syndrome [13], β thalassaemia [14] and neonatal adrenoleucodystrophy [15] but not with multisystem mitochondrial encephalopathies.
Whilst it was essential for the diagnosis of early optic neuropathy, SD-OCT scans failed to reveal alterations in choroidal and outer retinal layers corresponding to areas of retinal pigmentation, as described in adult KARS1 patients [16].
Our patient also showed the simultaneous presence of cerebral and cerebellar atrophies combined with white matter calcifications seen at brain MRI imaging and the pancytopenia that are rare findings also pointing to a metabolic condition [5, 7].
From the analysis of the literature (Table 1) it appears that cerebral calcifications are present in 44.4% of patients (12/27) while other neurological characteristics are less common: cerebellar calcifications are seen in 5 of 27 patients, and cerebellar and cerebral atrophies in 4 and 2 children, respectively. The coexistence of these neurological features, as in our patient, is an extremely rare condition seldom seen in previous patients. An additional novel feature is the presence of pancytopenia in our patient. Mutations in KARS1 can cause haematological alterations such as microcytic anemia, thrombocytopenia and hypogammaglobulinemia but pancytopenia occurred in a single child.
Due to the small number of patients, it is remains hard to draw genotype–phenotype correlations. An analysis of the literature (Table 1) shows that some features are shared by most of the patients, while others are rare and unrelated to type and site of mutation. It will be necessary to describe additional patients to improve our correlations.
In summary, our report expands the phenotype of new biallelic mutations in KARS1 further stressing the importance to propose WES studies as first tier diagnostic method in infants with complex multisystem phenotypes. We also highlight the key role of SD-OCT examination in the multidisciplinary assessment of children with mitochondrial disorders.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. The DNA sequencing data is available in NCBI BioProject under the accession number PRJNA673368. The direct web link to the NM_001130089.1 dataset and to the human reference hg19 genome that we used in this study are respectively https://www.ncbi.nlm.nih.gov/nuccore/NM_001130089.1 and https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13.
Abbreviations
- MEH:
-
Mitochondrial encephalohepatopathy
- ARS:
-
Aminoacyl tRNA synthetase
- SD-OCT:
-
Spectral domain optical coherence tomography
- pRNFL:
-
Peripapillary retinal nerve fiber layer
- WES:
-
Whole exome sequencing
- HPS:
-
Hepatoportal sclerosis
- WB:
-
Western Blot
References
Fujishima K, Kanai A. tRNA gene diversity in the three domains of life. Front Genet. 2014;5:142.
Tolkunova E, Park H, Xia J, King MP, Davidson E. The human lysyl-tRNA synthetase gene encodes both the cytoplasmic and mitochondrial enzymes by means of an unusual alternative splicing of the primary transcript. J Biol Chem. 2000;275:35063–9.
McLaughlin HM, Sakaguchi R, Liu C, Igarashi T, Pehlivan D, Chu K, et al. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet. 2010;87:560–6.
Santos-Cortez RLP, Lee K, Azeem Z, Antonellis PJ, Pollock LM, Khan S, et al. Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am J Hum Genet. 2013;93:132–40.
Walker UA, Collins S, Byrne E. Respiratory chain encephalomyopathies: a diagnostic classification. Eur Neurol. 1996;36:260–7.
Ardissone A, Tonduti D, Legati A, Lamantea E, Barone R, Dorboz I, et al. KARS-related diseases: progressive leukoencephalopathy with brainstem and spinal cord calcifications as new phenotype and a review of literature. Orphanet J Rare Dis. 2018;13:45.
Itoh M, Dai H, Horike S-I, Gonzalez J, Kitami Y, Meguro-Horike M, et al. Biallelic KARS pathogenic variants cause an early-onset progressive leukodystrophy. Brain J Neurol. 2019;142:560–73.
Schouten JN, Verheij J, Seijo S. Idiopathic non-cirrhotic portal hypertension: a review. Orphanet J Rare Dis. 2015;10:67.
Clayman HM, Flynn JT, Koch K, Israel C. Retinal pigment epithelial abnormalities in leukemic disease. Am J Ophthalmol. 1972;74:416–9.
Schepens CL, Brockhurst RJ. Uveal effusion. 1. Clinical picture. Arch Ophthalmol Chic Ill. 1963;70:189–201.
Cohen SY, Quentel G, Egasse D, Cadot M, Ingster-Moati I, Coscas GJ. The dark choroid in systemic argyrosis. Retina Phila Pa. 1993;13:312–6.
Zürcher M. Schipper I [Spot-like to reticular pigment displacement in a patient with pseudoxanthoma elasticum (Grönblad-Strandberg syndrome)]. Klin Monatsbl Augenheilkd. 1990;196:30–2.
Barth RA, Pagon RA, Bunt-Milam AH. “Leopard spot” retinopathy in Warburg syndrome. Ophthalmic Paediatr Genet. 1986;7:91–6.
Sorcinelli R, Sitzia A, Figus A, Lai ME. Ocular findings in beta-thalassemia. Metab Pediatr Syst Ophthalmol N Y N. 1985;1990(13):23–5.
Lyons CJ, Castano G, McCormick AQ, Applegarth D. Leopard spot retinal pigmentation in infancy indicating a peroxisomal disorder. Br J Ophthalmol. 2004;88:191–2.
Jabbarpoor Bonyadi MH, Ownagh V, Rahimy E, Soheilian M. Giraffe or leopard spot chorioretinopathy as an outstanding finding: case report and literature review. Int Ophthalmol. 2019;39:1405–12.
Acknowledgements
We want to acknowledge the family for their partecipation. We thank Elena Andreucci and Giacomo Maria Bacci for their help in drafting the manuscript, Laura Dosa, Matteo Della Monica and Roberto Caputo to follow up the patient.
Funding
None of the Authors have funding to declare.
Author information
Authors and Affiliations
Contributions
FP and VP drafted the initial manuscript, retrieved the pertinent literature and interpret the data, GI, FM have drafted the work, RP, EP and CN have made contributions to the conception and design of the work; RG, FS and SG have supervised and substantively reviewed the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was performed with parental written informed consent. Specific Institutional Review Board (IRB) approval had been obtained prior to this study.
Consent for publication
Written informed consent for publication of the children’s clinical information and molecular data were obtained from their parents.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Methods.
Additional file 2.
Full image of KARS Western blot.
Additional file 3.
Sequencing of the region flanking the mutations using muscle cDNA analysis served to assess “semiquantitatively” the presence and the abundance of the wild-type and mutant transcripts.
Additional file 4.
Full gel images of cDNA analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Peluso, F., Palazzo, V., Indolfi, G. et al. Leopard-like retinopathy and severe early-onset portal hypertension expand the phenotype of KARS1-related syndrome: a case report. BMC Med Genomics 14, 25 (2021). https://doi.org/10.1186/s12920-020-00863-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-00863-1