
Abstract
Background
Elevated triglycerides (TG) are associated with, and may be causal for, cardiovascular disease (CVD), and co-morbidities such as type II diabetes and metabolic syndrome. Pathogenic variants in APOA5 and APOC3 as well as risk SNVs in other genes [APOE (rs429358, rs7412), APOA1/C3/A4/A5 gene cluster (rs964184), INSR (rs7248104), CETP (rs7205804), GCKR (rs1260326)] have been shown to affect TG levels. Knowledge of genetic causes for elevated TG may lead to early intervention and targeted treatment for CVD. We previously identified linkage and association of a rare, highly conserved missense variant in SLC25A40, rs762174003, with hypertriglyceridemia (HTG) in a single large family, and replicated this association with rare, highly conserved missense variants in a European American and African American sample.
Methods
Here, we analyzed a longitudinal mixed-ancestry cohort (European, African and Asian ancestry, N = 8966) from the Electronic Medical Record and Genomics (eMERGE) Network. We tested associations between median TG and the genes of interest, using linear regression, adjusting for sex, median age, median BMI, and the first two principal components of ancestry.
Results
We replicated the association between TG and APOC3, APOA5, and risk variation at APOE, APOA1/C3/A4/A5 gene cluster, and GCKR. We failed to replicate the association between rare, highly conserved variation at SLC25A40 and TG, as well as for risk variation at INSR and CETP.
Conclusions
Analysis using data from electronic health records presents challenges that need to be overcome. Although large amounts of genotype data is becoming increasingly accessible, usable phenotype data can be challenging to obtain. We were able to replicate known, strong associations, but were unable to replicate moderate associations due to the limited sample size and missing drug information.
Similar content being viewed by others

Background
Triglyceride (TG) levels are a marker for health outcomes. High TG, (defined as 200 < TG < 500 mg/dL in adults), are associated with, and may cause, cardiovascular disease (CVD) [1,2,3,4]. High TG is also associated with type II diabetes and metabolic syndrome [5, 6]. TG levels are affected by environmental variables such as diet, alcohol consumption, smoking, and exercise [7,8,9,10,11]. Maintaining healthy levels of TGs may improve health outcomes [6, 12].
Both hypo- and hypertriglyceridemia (HTG) have major genetic components such as heterozygous variation at APOC3 and APOA5, respectively [13,14,15]. More recently, homozygous and compound heterozygous null variation at ANGPTL3 and ANGTPL4 have been shown to be associated with low TG [16, 17]. SNVs in the genes APOE, GCKR, APOA1/C3/A4/A5 gene cluster, INSR and CETP have smaller effects on TG [18,19,20].
We previously identified linkage and association of a rare, highly conserved missense SNV, rs762174003, in SLC25A40 with HTG in a large family, and replicated the association with rare missense SNVs at highly conserved sites in SLC25A40 in a European American (EA) and African American (AA) sample [21]. SLC25A40 encodes a mitochondrial transmembrane protein [22]. In drosophila, SLC25A40 loss-of-function was shown to contribute to mitochondrial damage due to excessive oxidation [23]. Here, we attempt to replicate the association between deleterious variation at SLC25A40, as well as other reported associations with variation at APOE, GCKR, APOA1/C3/A4/A5 gene cluster, INSR, and CETP, with TG in a multi-ancestry population from the Electronic Medical Records and Genomics (eMERGE) Network phase 3 study [24, 25].
Methods
Participants and phenotype data
Participants were ascertained from each of nine phase 3 eMERGE sites, a study tasked with investigating return of results from the sequencing of a panel of ~ 100 genes in a large (N = 25,000), diverse cohort of participants [24]. Participants ages 18 years and older were included, as the distribution for TG levels in children differs from that of adults. We ascertained relevant patient data from the clinical records, including TG level measured in mg/dL (N = 12,229 adult participants), age, sex, weight, height and body mass index (BMI), measured at the same clinical visit. We also ascertained relevant ICD9 and ICD10 codes for lipidemias, pancreatic cancer, and pancreatitis. Thirteen participants with chylomicronemia (ICD9 code 272.3, ICD10 code E78.3), 12 participants with Kaposi’s sarcoma (ICD9 code 176 and ICD10 code C46) and 2,539 participants with morbid obesity (ICD9 code 278.01 and ICD10 code E66.01) or bariatric surgery were removed as their TG values are likely associated with unaccounted for exposures. Quality control was performed within participants as well. Participants who had maximum TG (maxTG) < 40 and had more than one record verifying this low TG were retained. However, one participant who had a single record with TG = 19 mg/dL was removed. Records within 2 weeks prior or 4 weeks after a diagnosis of pancreatitis (ICD9 code 577.0, ICD10 code K85.9) for an individual were removed (382 participants), as high TG are correlated with pancreatitis and is likely due to an environmental exposure such as alcohol. Similarly, individual level records coincident and after a diagnosis of pancreatic cancer were removed (147 participants). Quality control of BMI was performed using height and weight data as described by Goodloe et. al [26]. For participants with only BMI data (i.e., no height or weight data), values more than 5 standard deviations from the participant specific median BMI were removed.
Genotype data
A subset of individuals (N = 24,956) in eMERGE were genotyped using a custom capture target containing sequence for 788 genes with partial to full exon coverage, including 58 of the 59 genes considered actionable by the American College of Medical Genetics (ACMG) [27, 28], and detects 62,051 SNVs. Sequencing was performed at two sites: Partners Healthcare Laboratory for Molecular Medicine (LMM) and Baylor. Samples from dropped out participants, that failed quality control or had sex discrepancy were removed (N = 229). After removing duplicate data for a single participant, sequence data for all participants was aligned to a merged target sequence map, using the Burrows-Wheeler Aligner version 0.7.10 [29]. Genotypes were jointly called on all participants using the Genome Analysis Toolkit version 3.5 [30]. 896 Variants with genotype missingness rate > 5% were removed. Individual level genotyping rate was > 98% for all participants. After cleaning the data, 8970 participants with both phenotype and genotype data remained in the analysis. Four additional participants were removed due to their influence on the residual model (see results).
Principal components (PC) of ancestry was calculated on a pruned set of 1571 SNVs (r2 < 0.7, MAF > 0.05). Self-reported ancestry aligned well with the first two PCs.
Exon sequence data was available for the relevant genes SLC25A40, APOC3, and APOA5. Two individuals with known pathogenic variants in APOA5 (rs201079485, rs147528707) were removed from analysis. Exon sequence data was also available for known TG risk SNVs rs429358 and rs7412 (which determine the APOE ε2/3/4 genotype), rs7248104 (INSR), rs1260326 (GCKR), rs7205804 (CETP), and rs964184 (APOA1/C3/A4/A5 gene cluster).
Analysis
The phenotype of interest, medTGres, was calculated as the residuals from the median log10 transformed TG for each individual, adjusted for median age, sex, median BMI, presence of hypertriglyceridemia (HTG, ICD9 code 272.1 and ICD10 code E78.1), presence of hyperlipidemia (HL, ICD9 codes 272.2, 272.4, 272.9 and ICD10 codes E78.2, E78.4, E78.9), APOE genotype, known risk SNVs, known APOC3 protective SNVs, the first two principal components (PCs) of ancestry, and site. The variable for median age was centered around age 50, as this is when TG levels tend to descend, and then squared, as the relationship between TG levels and age is parabolic. We log10 transformed TG as the distribution is highly skewed and the transformation makes it nearly Normal. We used the median transformed TG (medTG) value over all records for each individual because the median protects against outliers that could be due to non-fasting measurements. The resulting residuals from the above linear model (medTGres) were used as the primary phenotype. We also used a similar residual from the adjusted maximum log10 transformed TG (maxTGres), as using the maximum protects against errors introduced from treatment with Niacin, fibrates or binders for high TG. Given our more complete local data from the University of Washington and Kaiser Permanente Washington (UWKP) site, we expect about 3% of the participants to be receiving such treatment at some time point in their clinical records [31]. However, this data was not available to us study-wide.
In addition to assessing the significance of the known risk variation, we analyzed variants in SLC25A40 that are likely to affect TG levels [21]. This included rare (maximum population MAF < 0.005) variants that either caused an early termination in the first 90% of the coding region, or changed the amino acid sequence. Missense variants were further constrained to be evolutionarily conserved (Genomic Evolutionary Rate Profiling (GERP) score > 4.8) [32]. The maximum population MAF was derived from populations in the gnomAD database that have not experienced a bottleneck (i.e., ignored MAF from Ashkenazi Jewish or Finnish populations) [33, 34]. We performed four separate gene-wise tests for association with medTGres and maxTGres using 1. Genotype at single SNVs with more than 10 heterozygotes, 2. All other SNVs collapsed into a single indicator variable, 3. All other rare missense SNVs collapsed into a single indicator variable and 4. All other early terminations collapsed into a single indicator variable.
Results
Demographics
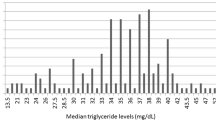
Demographics of the participants included in these analyses are given in Table 1. There were slightly more females than males at most sites. The distribution of age varied widely among the sites, with younger participants from Children’s Hospital of Philadelphia (CHOP) and Cincinnati Children’s Hospital Medical Center. Median BMI tended to be above normal (> 25), but there were some participants with low BMI (< 18.5) (Columbia, Mayo). There were 51 participants with very low median BMI < 11, all from Mayo, with at least 3 records supporting this measurement. The median TG ranged between 19 and 1371 mg/dL over all participants, but the majority of participants had median TG < 150 mg/dL, which is defined as normal (Fig. 1). The number of TG records available per participant varied widely, with a median of 6 overall. Most participants were of European ancestry (77%) followed by African ancestry (7%) and Asian ancestry (6%) (Table 2, Fig. 2). Most of the participants of Asian ancestry (82%) were from UWKP and most of the participants of African ancestry (62%) were from Northwestern and Cincinnati.

Distribution of median triglyceride by site. The horizontal line at 150 mg/dL represents the maximum normal level for an adult

First and second principal components of ancestry. AS Asian ancestry, EA European ancestry, AA African ancestry, UN unknown ancestry
RiskSNVs
Genotype counts for APOE and risk SNVs at APOC3, APOA5, APOA1/C3/A4/A5 gene cluster and GCKR are given in Tables 3 and 4. The distribution of APOE genotypes follows expectations, given known allele frequencies. Sixty-two participants were heterozygous for 1 of 4 protective APOC3 SNVs (rs76353203, rs147210663, rs138326449, rs140621530). Eighty-three participants had at least one of two risk alleles at APOA5 SNV rs2075291.
SLC25A40 genotype data
Twenty-one rare SNVs, consisting of 2 stop gains, 3 frame shifts and 16 evolutionarily conserved missense variants, were heterozygous in the data set for 32 participants (Table 5). There were 119 participants who were heterozygous at the missense rs724665 and this SNV is considered separately from the others. Each participant was heterozygous for at most one of the SNVs. Three participants were heterozygous at the frame shifts and 2 participants were heterozygous at the stop gains, limiting the power of the tests.
Residual model
The final residual model, containing only significant covariates is shown in Table 6. Although centered-median-age-squared was significantly associated with log10(medTG), its effect is near zero (β = − 4e−05). Males had higher log10(medTG) (β = 0.02). Increased BMI was associated with higher log10(medTG) (β = 0.01). Similarly, individuals with HTG or HL had higher log10(medTG) (β = 0.3 and 0.08, respectively) than other participants. SITE was also highly associated with log10(medTG) (F-test p < 2e−16). Neither the INSR or CETP SNVs were significantly associated with median or maximum TG (p > 0.16), and were not included in the model (data not shown). Four potentially influential outlying participants (as defined by their cook’s distance [35]) in either model (medTGres or maxTGres) were removed, leaving a total of 8,966 participants with complete phenotype and genotype data for analysis. None of the participants with low BMI (median BMI < 15) appeared to influence the models.
Association with known TG risk SNVs
Previously published risk SNVs at APOE, GCKR, APOA5 and APOC3 were associated with TG (Table 6). The minor alleles at the APOE SNVs rs429358 and rs7412 were associated with increased log10(medTG) (β = 0.01 and 0.04, respectively) and are jointly significantly associated with log10(medTG) (F-test p = 1.1e−11). Under an additive model, the minor alleles at GCKR and APOA1/C3/A4/A5 gene cluster were also significantly positively associated with log10(medTG) (GCKR: β = 0.03, p < 2e−16, APOA1/C3/A4/A5 gene cluster: β = 0.04, p < 2e−16). Under a dominant model, presence of either minor allele at APOA5 was positively associated with log10(medTG) (β = 0.09, p = 2.7e−05). Jointly, a single copy of any of the minor alleles at the four APOC3 SNVs were negatively associated with log10(medTG) (β = -0.23, p = 1.3e−21).
Primary analysis
None of the SLC25A40 SNVs were significantly associated with medTGres, either separately or in combination (p > 0.4, Table 7, Fig. 3). Furthermore, the estimated effect of the variants was negative in all situations (rs724665 β = − 0.01; all SNVs excluding rs724665 β = − 0.02; all rare missense β = − 0.01; rare early terminations β = − 0.07). Similar results were observed for maxTGres and for analysis including the EA-only subset.

Association between variation at SLC25A40 and medTGres. a rs724665; b all rare variation; c all rare missense variation; d all rare stop gains and frameshifts. Width of the boxplots is proportional to sample size
Discussion
We replicated association between TG and risk SNVs at APOE, GCKR, APOA5 and APOC3, with the respective directions of effect as expected. Association between variation at APOA5 and APOC3 with TG has been well documented, and our ability to detect these effects supports the reliability of these TG data. Similarly, replication of an association between APOE and TG lends further support to the association of APOE ε2/3/4 and lipid traits in general. The association between GCKR and TG further supports a biological mechanism between diabetes and TG [36]. However, we did not replicate the reported association between TG and risk SNVs at INSR and CETP. The association between INSR rs7248104 and TG was first reported by a large GWAS with a sample size of 176,000 subjects [18]. It is possible that the sample size here is too small to detect any effect. Similarly, the association with CETP rs7205804 was discovered in a cohort of 95,000 subjects [37], and therefore it is possible that the sample size here is too small to detect any effect with TG.
Additionally, we failed to replicate an association between putatively deleterious variation at SLC25A40 and TG. Previous studies relied on linkage in a large family and data from the Exome Sequencing Project (ESP). It is possible that the original signal found in the family is due to another variant in linkage with rs762174003, but unrelated to the function of SLC25A40. Further research into this locus may benefit from whole genome sequencing of selected individuals from this family. However, this would not explain why an association was detected in the relatively small sample from the ESP. Results for maximum TG also showed no association.
It is also possible that there is a relationship between SLC25A40 and TG, but we are not able to detect it in this sample. One major caveat is that we are missing data on treatment for high TG. In our local data set, UWKP, where treatment data were available, we discovered approximately 3% of individuals were being treated for high TG with niacin, fibrates or binders [31]. When we accounted for these treatments in the local data we found evidence supporting an association between TG and rare early termination and conserved missense variation at SLC25A40 [31]. As the number of participants who harbor such a variant in SLC25A40 is small (1.7% of our sample), an unaccounted for 3% treatment rate could obscure any signal. Although we used median TG to reduce the effect of unknown treatment, it may have been insufficient as we do not know when treatment would have begun. We did not use the mean as it can be influenced by outliers and TG have a right skewed distribution. Furthermore, this is secondary use of data, which is prone to a high error rate in the data. Although we attempted to clean the data using multiple strategies, it is possible that enough error remained in the data to obscure any true association with SLC25A40. In addition, the small numbers of heterozygotes that we observe in this study limit the power of our tests.
Conclusion
Although we replicated known, strong associations with TG, these data do not replicate the previously reported association between SL25A40 variation and TG. Larger datasets with more complete data on fasting and medication use may be required to further explore this association. Furthermore, use of secondary data, such as EHR data, needs extensive quality control and would benefit from more comprehensive data extraction methods.
Availability of data and materials
Data have been posted to dbGaP. Imputed SNV genotype data are available at Study Accession: phs001584.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001584.v1.p1). Custom capture sequence data are available at Study Accession: phs001616.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001616.v1.p1). Covariate data are available at Dataset Accession: pht009072.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/dataset.cgi?study_id=phs001584.v1.p1&pht=9072). Median and maximum TG is available upon request.
Abbreviations
- Colu:
-
Columbia University
- CVD:
-
Cardiovascular disease
- eMERGE:
-
Electronic Medical Records and Genomics
- Geis:
-
Geisinger
- GERP:
-
Genomic evolutionary rate profiling
- Harv:
-
Harvard
- HTG:
-
Hypertriglyceridemia
- maxTG:
-
Maximum triglycerides
- medTG:
-
Median triglycerides
- Nwes:
-
Northwestern University
- PC:
-
Prinicpal components
- TG:
-
Triglycerides
- UWKP:
-
University of Washington and Kaiser Permanente Washington
- Vand:
-
Vanderbilt
References
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–421.
Stampfer MJ, Krauss RM, Ma J, Blanche PJ, Holl LG, Sacks FM, Hennekens CH. A prospective study of triglyceride level, low-density lipoprotein particle diameter, and risk of myocardial infarction. Jama J Am Med Assoc. 1996;276:882–8.
Triglyceride Coronary Disease Genetics Consortium and Emerging Risk Factors Collaboration, Sarwar N, Sandhu MS, Ricketts SL, Butterworth AS, Di Angelantonio E, Boekholdt SM, Ouwehand W, Watkins H, Samani NJ, et al. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010;375:1634–9.
Hokanson JE, Austin MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk. 1996;3:213–9.
Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–5.
AbouRjaili G, Shtaynberg N, Wetz R, Costantino T, Abela GS. Current concepts in triglyceride metabolism, pathophysiology, and treatment. Metabolism. 2010;59:1210–20.
Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arterioscler Thromb. 1992;12:911–9.
Sopko G, Leon AS, Jacobs DR, Foster N, Moy J, Kuba K, Anderson JT, Casal D, McNally C, Frantz I. The effects of exercise and weight loss on plasma lipids in young obese men. Metabolism. 1985;34:227–36.
Pownall HJ, Ballantyne CM, Kimball KT, Simpson SL, Yeshurun D, Gotto AM. Effect of moderate alcohol consumption on hypertriglyceridemia: a study in the fasting state. Arch Intern Med. 1999;159:981–7.
Chait A, Mancini M, February AW, Lewis B. Clinical and metabolic study of alcoholic hyperlipidaemia. Lancet. 1972;2:62–4.
Willett W, Hennekens CH, Castelli W, Rosner B, Evans D, Taylor J, Kass EH. Effects of cigarette smoking on fasting triglyceride, total cholesterol, and HDL-cholesterol in women. Am Heart J. 1983;105:417–21.
Bruckert E, Labreuche J, Deplanque D, Touboul PJ, Amarenco P. Fibrates effect on cardiovascular risk is greater in patients with high triglyceride levels or atherogenic dyslipidemia profile: a systematic review and meta-analysis. J Cardiovasc Pharmacol. 2011;57:267–72.
Pennacchio LA, Olivier M, Hubacek JA, Krauss RM, Rubin EM, Cohen JC. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Hum Mol Genet. 2002;11:3031–8.
Talmud PJ, Hawe E, Martin S, Olivier M, Miller GJ, Rubin EM, Pennacchio LA, Humphries SE. Relative contribution of variation within the APOC3/A4/A5 gene cluster in determining plasma triglycerides. Hum Mol Genet. 2002;11:3039–46.
Johansen CT, Kathiresan S, Hegele RA. Genetic determinants of plasma triglycerides. J Lipid Res. 2011;52:189–206.
Romeo S, Yin W, Kozlitina J, Pennacchio LA, Boerwinkle E, Hobbs HH, Cohen JC. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119:70–9.
Pisciotta L, Favari E, Magnolo L, Simonelli S, Adorni MP, Sallo R, Fancello T, Zavaroni I, Ardigo D, Bernini F, et al. Characterization of three kindreds with familial combined hypolipidemia caused by loss-of-function mutations of ANGPTL3. Circ Cardiovasc Genet. 2012;5:42–50.
Global Lipids Genetics Consortium, Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83.
van de Woestijne AP, van der Graaf Y, de Bakker PI, Asselbergs FW, Spiering W, Visseren FL, SMART Study Group. Rs964184 (APOA5-A4-C3-A1) is related to elevated plasma triglyceride levels, but not to an increased risk for vascular events in patients with clinically manifest vascular disease. PLoS ONE. 2014;9:e101082.
Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–6.
Rosenthal EA, Ranchalis J, Crosslin DR, Burt A, Brunzell JD, Motulsky AG, Nickerson DA, NHLBI GO Exome Sequencing Project, Wijsman EM, Jarvik GP. Joint linkage and association analysis with exome sequence data implicates SLC25A40 in hypertriglyceridemia. Am J Hum Genet. 2013;93(1035):1045.
Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–23.
Slabbaert JR, Kuenen S, Swerts J, Maes I, Uytterhoeven V, Kasprowicz J, Fernandes AC, Blust R, Verstreken P. Shawn, the drosophila homolog of SLC25A39/40, Is a mitochondrial carrier that promotes neuronal survival. J Neurosci. 2016;36:1914–29.
Fossey R, Kochan D, Winkler E, Pacyna JE, Olson J, Thibodeau S, Connolly JJ, Harr M, Behr MA, Prows CA, et al. Ethical considerations related to return of results from genomic medicine projects: the eMERGE network (phase III) experience. J Pers Med. 2018. https://doi.org/10.3390/jpm8010002.
Kullo IJ, Haddad R, Prows CA, Holm I, Sanderson SC, Garrison NA, Sharp RR, Smith ME, Kuivaniemi H, Bottinger EP, et al. Return of results in the genomic medicine projects of the eMERGE network. Front Genet. 2014;5:50.
Goodloe R, Farber-Eger E, Boston J, Crawford DC, Bush WS. Reducing clinical noise for body mass index measures due to unit and transcription errors in the electronic health record. AMIA Jt Summits Transl Sci Proc. 2017;2017:102–11.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74.
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–55.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D., et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv (2017).
Rosenthal EA, Gordon AS, Stanaway IB, Crosslin DR, Albertson-Junkans L, Larson EB, Carrell D, Jarvik GP. Replication of association between triglyceride level and SLC25A40 in a multi-ancestry sample. Poster presented at the American Society of Human Genetics 68th Annual Meeting. San Diego, CA (2018).
Cooper GM, Stone EA, Asimenos G, NISC Comparative Sequencing Program, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Karczewski KJ, Francioli LC, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss of function intolerance across human protein coding genes. bioRxiv (2019).
Cook RD, Weisberg S. Residuals and influence in regression. New York: Chapman and Hall; 1982.
Beer NL, Tribble ND, McCulloch LJ, Roos C, Johnson PR, Orho-Melander M, Gloyn AL. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. 2009;18:4081–8.
Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13.
Acknowledgements
The authors would like to thank all participants in this study as well as individuals who contributed to the eMERGE network.
Funding
This phase of the eMERGE Network was initiated and funded by the National Human Genome Research Institute (NHGRI) through the following grants: U01HG8657 (Kaiser Permanente Washington/University of Washington); U01HG8685 (Brigham and Women’s Hospital); U01HG8672 (Vanderbilt University Medical Center); U01HG8666 (Cincinnati Children’s Hospital Medical Center); U01HG6379 (Mayo Clinic); U01HG8679 (Geisinger Clinic); U01HG8680 (Columbia University Health Sciences); U01HG8684 (Children’s Hospital of Philadelphia); U01HG8673 (Northwestern University); U01HG8701 (Vanderbilt University Medical Center serving as the Coordinating Center); U01HG8676 (Partners Healthcare/Broad Institute); and U01HG8664 (Baylor College of Medicine); U01HG004438 (CIDR) and U01HG004424 (the Broad Institute) serving as Genotyping Centers; U01HG004438 (CIDR) serving as a Sequencing Center. The funding supported overall planning, sequencing of participants, data collection, phenotype harmonization, coordination across sites, return of results to participants when relevant, and manuscript preparation.
Author information
Authors and Affiliations
Contributions
EAR conceived the analysis plan, performed all analyses and wrote the manuscript. DRC, DSC, JG acquired, maintained and analyzed the data used in this study. ASG, WQW, JCD, QPF, AS, ACS, MDR, JAP, LJR, JJC provided editorial feedback. IBS performed genotyping quality control and merged the genotype data. EBL obtained funding, acquired data and provided editorial feedback. HH provided editorial feedback and suggested some analysis. XF acquired, maintained and performed quality control on the data used in this study and provided editorial feedback. MS acquired and maintained the data used in this study and provided editorial feedback. IJK provided editorial feedback and obtained funding. GPJ supervised this work, obtained funding, acquired data and provided editorial feedback. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent was obtained at each site, under each site’s Institutional Review Board (IRB)-approved protocols [24]. Informed consent was obtained for all participants as either written or electronic. Some informed consents were carried out over the phone at Vanderbilt, as part of their collection of a prospective cohort, and this was approved by their IRB. Phone and verbal consents were approved at Geisinger in order to increase study participation, and this was approved by their IRB. The two sequencing centers had IRB-approved protocols that deferred consent to the participating sites. Protocol numbers are as follows: Partners Healthcare (2015P000929), Baylor College of Medicine (#H-40455). The federal-wide assurance number is FWA00002344. Ethics committees and reference numbers for each site are as follows: CHOP, The Committees for the Protection of Human Subjects, Federal Wide Assurance number FWA00000459, IRB Registration number IRB00000316; Cincinnati, Institution Review Board, Cincinnati Children’s Hospital Medical Center; Columbia, Human Research Protection Office and IRBs, AAAP7926, AAAQ9205; Geisinger, Geisinger Institutional Review Board, IRB Registration number IRB00008345; Harvard, Partners Human Research Committee, Protocol number 2015P000983/BWH; Mayo, Mayo Clinic Institutional Review Board – IRB-C, DHHS registration numbers: IRB00000020, IRB00003294, IRB00003295, and IRB00005256. Mayo Clinic holds Federal wide Assurance (FWA) number 00005001 covering Mayo Clinic in Rochester, Minnesota, Mayo Clinic in Arizona, Mayo Clinic in Florida, and Mayo Clinic Health Systems; Northwestern, Northwestern University's Institutional Review Board, IRN number STU00201971; UWKP, Kaiser Permanente Washington Research and Humans Subjects Review Office, IRB Registration number IRB00010902; Vanderbilt, Vanderbilt University Institutional Review Board, IRB number 151299. The network data does not include HIPAA identifiers. Only adult participants are included in this study. All data is shared under a consortia-wide data use agreement. No administrative permissions and/or licenses were acquired in order to access the clinical/personal patient data used in this research.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rosenthal, E.A., Crosslin, D.R., Gordon, A.S. et al. Association between triglycerides, known risk SNVs and conserved rare variation in SLC25A40 in a multi-ancestry cohort. BMC Med Genomics 14, 11 (2021). https://doi.org/10.1186/s12920-020-00854-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-00854-2