
Abstract
Background
Prognostic signatures are vital to precision medicine. However, development of somatic mutation prognostic signatures for cancers remains a challenge. In this study we developed a novel method for discovering somatic mutation based prognostic signatures.
Results
Somatic mutation and clinical data for lung adenocarcinoma (LUAD) and colorectal adenocarcinoma (COAD) from The Cancer Genome Atlas (TCGA) were randomly divided into training (n = 328 for LUAD and 286 for COAD) and validation (n = 167 for LUAD and 141 for COAD) datasets. A novel method of using the log2 ratio of the tumor mutation frequency to the paired normal mutation frequency is computed for each patient and missense mutation. The missense mutation ratios were mean aggregated into gene-level somatic mutation profiles. The somatic mutations were assessed using univariate Cox analysis on the LUAD and COAD training sets separately. Stepwise multivariate Cox analysis resulted in a final gene prognostic signature for LUAD and COAD. Performance was compared to gene prognostic signatures generated using the same pipeline but with different somatic mutation profile representations based on tumor mutation frequency, binary calls, and gene-gene network normalization. Signature high-risk LUAD and COAD cases had worse overall survival compared to the signature low-risk cases in the validation set (log-rank test p-value = 0.0101 for LUAD and 0.0314 for COAD) using mutation tumor frequency ratio (MFR) profiles, while all other methods, including gene-gene network normalization, have statistically insignificant stratification (log-rank test p-value ≥0.05). Most of the genes in the final gene signatures using MFR profiles are cancer-related based on network and literature analysis.
Conclusions
We demonstrated the robustness of MFR profiles and its potential to be a powerful prognostic tool in cancer. The results are robust according to validation testing and the selected genes are biologically relevant.
Similar content being viewed by others

Background
Lung and colon cancer are the leading cause of death over all cancers in the United States in 2017, with 155,870 and 50,260 deaths, respectively [1]. Prognostic signatures and risk stratification are vital to clinical decision making of treatment options in cancer precision medicine. As patient prognosis remains poor [2], researchers are seeking to develop improved prognostic signatures using molecular information, such as incorporating long non-coding RNA expression [3, 4].
However, incorporating somatic mutation profiles into prognostic signatures has remained a challenge and is often overlooked due to the sparse and binary nature of somatic mutation data [5]. The sparsity of the data arises from the observation that the vast majority of mutated genes are not shared among patients [6]. Save for a few frequently mutated driver genes, most somatically mutated genes are likely to be composed of only passenger mutations that do not provide growth advantage [7].
To investigate the prognostic value of somatic mutations, studies have chosen to tackle the challenge by confronting the sparsity problem. Le Morvan et al. [8] uses gene-gene networks as prior knowledge to de-sparsify the data. A patient’s binary somatic mutation profile is transformed by removing non-essential mutations and adding proxy mutations based on gene-gene network topology to normalize tumor mutational burden within a sample of patients. However, gene-gene networks vary from tissue to tissue and a single set of canonical gene-gene networks as prior knowledge may omit or overemphasize some interactions [9]. To address this issue, other studies have elected to use cancer-specific co-expression networks based on RNA expression data [10] or canonical pathways [11].
In this study, we confront the challenge of the binary nature of somatic mutation data rather than the sparsity problem. We propose the usage of the quantitative mutation frequency ratio of tumor vs. normal tissue from whole exome sequencing in building somatic mutation profiles. Using somatic mutation data for lung adenocarcinoma (LUAD) and colorectal adenocarcinoma (COAD) from The Cancer Genome Atlas (TCGA) [12, 13], we evaluate the risk stratification and prognostic performance of somatic mutation signatures generated by using two types of continuous somatic mutation profiles: mutation frequency ratio (MFR) profiles and tumor mutation frequency (TMF) profiles. We compare to two existing types of binary mutation profiles, raw binary mutation (BM) profiles and gene-gene network normalized profiles provided by NetNorM [8]. We show that the somatic mutation signatures generated by MFR profiles consistently provides statistically significant risk stratification while the other types of profiles do not.
Results
Identification of prognostic somatically mutated genes
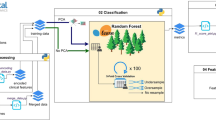
To identify and evaluate prognostic somatically mutated genes using different types of somatic mutation profiles, we used a pipeline (Fig. 1) adapted from Shukla et al.’s RNA-seq pipeline [3]. Clinical and controlled somatic mutation data for LUAD and COAD was gathered from TCGA [12, 13]. The data (Table 1) was partitioned randomly into training (n = 328 for LUAD and n = 286 for COAD) and validation (n = 167 for LUAD and n = 141 COAD) datasets and somatic mutation profiles generated.

Identification of prognostic somatic mutation gene signature. DNA-seq prognostic analysis and signature generation pipeline. TCGA somatic mutation data is randomly split into training and validation datasets. Univariate Cox analysis identifies mutated genes associated with survival and only significant genes (FDR ≤ 0.05) are considered further. Bidirectional stepwise model selection for multivariate Cox analysis is used to select the final prognostic somatic mutation gene signature. Risk scores for patients in both training and datasets are computed using the final signature. The 75% percentile risk score of the training dataset is used as the stratification threshold for the KM analysis on both the training and validation datasets
Four different types of somatic mutation profiles were considered: MFR, TMF, BM, and NetNorM profiles. The somatic mutation profile of a single patient is a vector with an element for every gene. The BM profile of a patient consists of a sparse binary vector where an element denotes if a gene is somatically mutated or not. The NetNorM profile was generated from the BM profile by normalizing the number of mutated genes via the removal or addition of somatically mutated genes [8]. While the NetNorM profile remains binary in nature, its process mitigates the sparsity problem of somatic mutation data by incorporating gene-gene network prior knowledge.
Additionally, we propose the usage of MFR and TMF profiles, which to the best of our knowledge, has not be considered previously in the literature to confront the difficulties of working with sparse binary data. TMF profiles incorporate the tumor data on the number of reads supporting the mutation vs. the reference genome. The MFR takes it a step further and considers the mutation frequency ratio of the tumor sample vs. the paired normal tissue sample. Both TMF and MFR profiles use continuous rather than binary values for somatic mutation profile representation.
Individually for each type of somatic mutation profile and tumor type, somatic mutation based prognostic signatures are generated using the pipeline outlined in Fig. 1. Univariate Cox proportional hazards regression is first performed on the training dataset to short list prospective genes with a FDR cutoff of 0.05. The prospective genes are then subjected to bidirectional stepwise multivariate Cox proportional hazards regression model selection to the determine the final prognostic signature (Table 2 and Table 3). We verified that all of the final prognostic signatures do not violate the proportional hazards assumption using the Schoenfeld Residual Test.
Comparison of risk stratification
Kaplan-Meier (KM) survival curves are used to assess and compare the different types of somatic mutation profiles in both the training and validation datasets. Using the final Cox model for risk scoring, the high-risk threshold for stratification in both the training and validation datasets was chosen to be the 75th percentile of the risk scores in the training dataset.
We observed that all somatic mutation profile types achieve significant risk stratification on the training dataset (log rank test p-value ≈ 0) for both LUAD and COAD (Fig. 2, Fig. 3). For both LUAD and COAD, however, only the stratification generated by MFR profiles is statistically significant in the validation datasets (log rank test p-value = 0.0101 for LUAD, 0.0314 for COAD) (Fig. 2a, Fig 3a), while all other profiles, including NetNorM, are not statistically significant (Fig. 2b, c and d, Fig. 3b, c and d). Furthermore, the final prognostic signatures generated by each type of somatic mutation profile only minimally overlap for both LUAD and COAD cases (Fig. 4).

Kaplan-Meier analysis of prognostic somatic mutation gene signatures. KM survival curves for LUAD training and validation datasets using (a) MFR, (b) TMF, (c) BM, and (d) NetNorM somatic mutation profiles

Kaplan-Meier analysis of prognostic somatic mutation gene signatures. KM survival curves for COAD training and validation datasets using (a) MFR, (b) TMF, (c) BM, and (d) NetNorM somatic mutation profiles

Selected somatically mutated genes for signatures. Venn diagrams of selected genes using MFR, TMF, BM, and NetNorM somatic mutation profiles for (a) LUAD and (b) COAD
The results suggest that the MFR profile’s prognostic signature is more robust, while the other types of profiles are subject to harsh overfitting that is typical in contexts with a larger number of covariates than samples. This is consistent with the observation that NetNorM profiles typically do not perform statistically different from binary profiles [8]. De-sparsifying somatic mutation data using gene-gene network prior information does not necessarily lead to improved prognostic and risk stratification performance.
Somatic mutation gene signatures
A PubMed search of the individual genes and a network analysis of the full signatures using Ingenuity Pathway Analysis (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/, accessed: Feb. 14, 2018) was performed to assess the biological relevancy of the final prognostic gene signatures generated by MFR profiles. A network containing 16 of the 20 genes in the LUAD prognostic signature (Table 4) was found (Fig. 5). The network is associated with cell death and survival, and cellular movement. All genes in the prognostic signature are positively associated with risk (denoted in red in Fig. 5). SDHA is the gene with the largest coefficient in the risk model (hazard ratio (HR) = 1.877). SDHA is a tumor suppressor and is implicated in paraganglioma and gastrointestinal stromal tumors [14]. While association of SDHA copy number variation to prognosis was found in lung squamous cell carcinoma [15], we have found no literature exploring the connection of SDHA to lung adenocarcinoma.

Network for somatically mutated genes in LUAD signature. Network containing 16 of the 20 genes in the LUAD prognostic signature using MFR somatic mutation profiles. The network is associated with cell death and survival, and cellular movement. Red nodes represent genes in the final prognostic signature and denote positive association with risk
Four additional genes in the LUAD signature also have known associations with lung cancer. PFKM has mutations associated with survival outcomes in lung squamous cell carcinoma [16]. MADD promotes survival of LUAD cells and is a potential therapeutic target [17]. SERPINI2 is tumor suppressor gene and is associated with squamous cell lung cancer [18]. Finally, it has been found that certain MMP8 mutations are correlated with risk of developing lung cancer [19].
Eight of the remaining genes in the LUAD signature are associated with other cancer types and their connection to LUAD is yet uncharacterized. ABCB6 [20, 21], ZNF768 [22], and the TP53-mediated tumor suppressor gene NDN [23] are all associated with colorectal cancers. MSANTD3 is an oncogene in salivary gland acinic cell carcinoma [24]. FGD3 is implicated in breast cancer [25] and ARHGAP4 in ovarian tumors [26]. It has been observed that increased expression of HSD17B4 is correlated with poor prognosis in prostate cancer [27]. Lastly, correlation of HEATR1 with shorter overall survival has been shown in pancreatic ductal adenocarcinoma [28].
For the COAD prognostic signature (Table 5), we found that 30 of the 32 genes were involved in two different networks. The first network contains 16 of the 32 genes in the COAD prognostic signature (Fig. 6) and is associated with embryonic, organismal, and tissue development. The second network contains 14 of the 32 genes in the COAD prognostic signature (Fig. 7) and is associated with cancer and organismal injury and abnormalities. Unlike the LUAD signature where all genes were positively associated with increased risk, mutations in seven of the genes are associated with reduced risk (USP50, UBTD1, ZNF83, FBX038, C11orf53, IQCH, and CHI3L1) and are denoted in green in Figs. 6 and 7.

First network for somatically mutated genes in COAD signature Network containing 16 of the 32 genes in the COAD prognostic signature using MFR somatic mutation profiles. The network is associated with embryonic, organismal, and tissue development. Red and green nodes represent genes in the final prognostic signature and denote positive and negative association with risk, respectively

Second network for somatically mutated genes in COAD signature Network containing 14 of the 32 genes in the COAD prognostic signature using MFR somatic mutation profiles. The network is associated with cancer and organismal injury and abnormalities. Red and green nodes represent genes in the final prognostic signature and denote positive and negative association with risk, respectively
Ten of the genes in the COAD signature are implicated in colorectal cancers (CRC). MICA has high cell-surface expression in cancers of the digestive system and have been found to be correlated with increased survival [29]. Copy number variation of RERG is correlated with CRC risk [30]. NKD1 is involved in Wnt signaling central to tumor cell growth in CRC and other cancers [31]. Lower expression of UBTD1 correlates with worse prognosis [32]. SERPINB3 has a driving role in more aggressive cellular phenotypes of CRC [33]. DMKN has been previously proposed as a biomarker of early-stage CRC [34]. PDHB diminishes the oncogenic effects of miR-146b-5p on the growth and invasion of CRC [35]. C11orf53 is a potential gene involved in CRC etiology [36]. CHI3L1 promotes macrophage recruitment and angiogenesis in CRC [37]. Lastly, alterations of CDH24 contribute to tumorigenesis, as CDH24 is important to the maintenance of cell adhesion [38].
Another nine genes of the COAD signature have known associations with other types of cancers, but not with CRC yet. DNALI1 [39] and MEOX1 [40] are associated with breast cancer. In particular, MEOX1 is correlated with poor survival of breast cancer patients. MARCH11 has been used as a biomarker in a methylation panel for early cancer detection and prognosis prediction in non-small cell lung cancer [41]. ARHGAP15 is correlated with survival in early-stage pancreatic ductal adenocarcinoma [42]. IGHA1 is associated with gastric tumorigenesis [43]. CER1 is associated with glioma [44]. SDR9C7 promotes lymph node metastasis in esophageal squamous cell carcinoma [45]. PRKG2 is associated with acute mast cell leukemia [46]. Finally, ZNF133 is potential biomarker for osteosarcoma [47].
Discussion
Cancer genomic data is increasingly becoming a hot topic in precision cancer medicine research, including the identification of therapeutic targets, biomarker-based clinical trials, and the study of genomic determinants of therapy response [48]. The signatures found in the present retrospective study are promising and their potential clinical integration should be further investigated with a prospective study.
While the results are promising, there are limitations to this initial work. Demographic and clinical data were not incorporated into the prognostic models. Gene expression data is also available for TCGA LUAD and COAD datasets. Integration of all data types could potentially improve prognostic and risk stratification performance and provide further biological insights. Furthermore, all types of cancer in TCGA should be analyzed for a future pan-cancer study.
The present study was also done at the gene level. There is potential that specific mutations to a gene may have different prognostic effects. However, with the sample size of TCGA data, it is not feasible to observe statistically significant results due to the increased sparsity of somatic mutation data at the specific mutation level. Further data or methods to mitigate the increased sparsity is required for further study.
The present work demonstrated the robustness of prognostic signatures using MFR profiles within TCGA LUAD and COAD VarScan-based somatic mutation data [49] by the partitioning of the data into training and validation datasets. As a result, the experimental and analysis protocols are consistent. The robustness with respect to different somatic mutation calling software within TCGA should be conducted, as calls from MuSE [50], MuTect [51], and SomaticSniper [52] are provided in addition to VarScan. Furthermore, the methods robustness to data generated from different experimental protocols, such as by investigating data generated by different institutions and projects, should be studied in the future.
Conclusions
To improve clinical tools and biological understanding of LUAD and COAD, we demonstrated a methodology to generating robust prognostic somatic mutation-based gene signatures. We demonstrated the robustness of MFR profiles and its potential to be a powerful prognostic tool in cancer, unlike other alternative types of somatic mutation profiles, TMF, BM, and NetNorM, that did not achieve statistically significant risk stratification in validation datasets. The genes selected by the methodology using MFR profiles was shown to be biologically relevant and has potential for use in effective management LUAD and COAD.
Methods
Somatic mutation data and profiles
Controlled TCGA somatic mutation data (VarScan MAF files [49]) were downloaded from NCI’s Genomic Data Commons (https://gdc.cancer.gov/, accessed: Feb. 14, 2018) for LUAD and COAD (Project ID 17109, A Pan-Cancer Analysis of Somatic Mutation Profiles for Tumor Immunogenicity and Prognosis). The data were filtered, keeping only missense mutations. The missense mutations were then aggregated into gene level mutation profiles. For BM profiles, the gene is flagged as mutated if it contains any missense mutation.
The NetNorM normalization method was used as a representative of somatic mutation profiles using gene-gene network information [8]. NetNorM uses networks from Pathway Commons (http://www.pathwaycommons.org), which feature an integrated network data of public pathway and interaction databases. The user-specified parameter for NetNorM is the target number of mutated genes k. This parameter is set to the median number of mutated genes in the training dataset, which is 193 and 151 for LUAD and COAD, respectively. NetNorM ranks genes based on their mutation status and network connectedness. A patient’s somatic mutation profile is normalized by setting only the top k genes as being mutated. Since mutated genes are always ranked higher than non-mutated genes, patients with more than k mutated genes will have lower ranked mutated genes set to unmutated, while patients with less than k mutated genes will obtain artificial proxy mutated genes.
Mutation frequency ratio and tumor frequency profiles
For patient i, the MAF files contain the number of reads supporting the reference allele for mutation j, TRCij and NRCij for tumor and normal samples, respectively. Analogously, denote the number of reads supporting the alternate allele, TACij and NACij for tumor and normal samples, respectively. The tumor and normal sample mutation frequencies, TMFij and NMFij, are computed using Eqs. (1) and (2), respectively. The mutation frequency ratio MFRij is then simply the ratio of the tumor to normal sample mutation frequencies. To generate a patient’s gene level MFR and TMF profiles, the mutations are aggregated by gene using the mean ratio or frequency within that gene.
Signature generation and statistical analysis
TCGA clinical data were downloaded from NCI’s Genomic Data Commons (https://gdc.cancer.gov/, accessed: Feb. 14, 2018) for LUAD and COAD. These data were partitioned randomly into training (n = 328 for LUAD and n = 286 for COAD) and validation (n = 167 for LUAD and n = 141 COAD) datasets. Rarely mutated genes in somatic mutation profiles were omitted when less than 1% of patients in a sample have the mutation. MFR and TMF profiles, which are continuous valued, were log2 transformed. Univariate Cox proportional hazards regression was used to assess association with overall survival using R survival package (R v3.4.0, survival v2.41–3) with a Benjamini-Hochberg FDR cutoff of 0.05. Multivariate Cox proportional hazards regression was performed using bidirectional stepwise model selection with the R MASS package (MASS v7.3–47). Kaplan-Meier analysis was used to assess risk stratification with R survival and GGally packages (GGally v1.3.2). Pathway and network analysis weres performed with Ingenuity Pathway Analysis.
Abbreviations
- BM:
-
Binary mutation
- COAD:
-
Colorectal adenocarcinoma
- HR:
-
Hazard ratio
- KM:
-
Kaplan-Meier
- LUAD:
-
Lung adenocarcinoma
- MFR:
-
Mutation frequency ratio
- TCGA:
-
The Cancer Genome Atlas
- TMF:
-
Tumor mutation frequency
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30.
Sanoff HK, Sargent DJ, Campbell ME, Morton RF, Fuchs CS, Ramanathan RK, et al. Five-year data and prognostic factor analysis of oxaliplatin and irinotecan combinations for advanced colorectal cancer: N9741. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26:5721–7.
Shukla S, Evans JR, Malik R, Feng FY, Dhanasekaran SM, Cao X, et al. Development of a RNA-Seq based prognostic signature in lung adenocarcinoma. J Natl Cancer Inst. 2017;109. https://doi.org/10.1093/jnci/djw200.
Xue W, Li J, Wang F, Han P, Liu Y, Cui B. A long non-coding RNA expression signature to predict survival of patients with colon adenocarcinoma. Oncotarget. 2017;8:101298–308.
Yuan Y, Van Allen EM, Omberg L, Wagle N, Amin-Mansour A, Sokolov A, et al. Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat Biotechnol. 2014;32:644–52.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–58.
Le Morvan M, Zinovyev A, Vert JP. NetNorM: capturing cancer-relevant information in somatic exome mutation data with gene networks for cancer stratification and prognosis. PLoS Comput Biol. 2017;13:e1005573.
Yang Y, Han L, Yuan Y, Li J, Hei N, Liang H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat Commun. 2014;5:3231.
He Z, Zhang J, Yuan X, Liu Z, Liu B, Tuo S, et al. Network based stratification of major cancers by integrating somatic mutation and gene expression data. PLoS One. 2017;12:e0177662.
Kuijjer ML, Paulson JN, Salzman P, Ding W, Quackenbush J. Cancer subtype identification using somatic mutation data. Br J Cancer. 2018;118:1492–501.
Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50.
Network TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330.
Schaefer I-M, Hornick JL, Bovée JVMG. The role of metabolic enzymes in mesenchymal tumors and tumor syndromes: genetics, pathology, and molecular mechanisms. Lab Investig J Tech Methods Pathol. 2018.98:414–26.
Liu L, Huang J, Wang K, Li L, Li Y, Yuan J, et al. Identification of hallmarks of lung adenocarcinoma prognosis using whole genome sequencing. Oncotarget. 2015;6:38016–28.
Lee SY, Jin CC, Choi JE, Hong MJ, Jung DK, Do SK, et al. Genetic polymorphisms in glycolytic pathway are associated with the prognosis of patients with early stage non-small cell lung cancer. Sci Rep. 2016;6:35603.
Bi W, Wei Y, Wu J, Sun G, Guo Y, Zhang Q, et al. MADD promotes the survival of human lung adenocarcinoma cells by inhibiting apoptosis. Oncol Rep. 2013;29:1533–9.
Boelens MC, van den Berg A, Fehrmann RSN, Geerlings M, de Jong WK, te Meerman GJ, et al. Current smoking-specific gene expression signature in normal bronchial epithelium is enhanced in squamous cell lung cancer. J Pathol. 2009;218:182–91.
González-Arriaga P, López-Cima MF, Fernández-Somoano A, Pascual T, Marrón MG, Puente XS, et al. Polymorphism +17 C/G in matrix metalloprotease MMP8 decreases lung cancer risk. BMC Cancer. 2008;8:378.
Boswell-Casteel RC, Fukuda Y, Schuetz JD. ABCB6, an ABC transporter impacting drug response and disease. AAPS J. 2018;20:8.
Hlavata I, Mohelnikova-Duchonova B, Vaclavikova R, Liska V, Pitule P, Novak P, et al. The role of ABC transporters in progression and clinical outcome of colorectal cancer. Mutagenesis. 2012;27:187–96.
O’Reilly J-A, Fitzgerald J, Fitzgerald S, Kenny D, Kay EW, O’Kennedy R, et al. Diagnostic potential of zinc finger protein-specific autoantibodies and associated linear B-cell epitopes in colorectal cancer. PLoS One. 2015;10:e0123469.
Hu Y-H, Chen Q, Lu Y-X, Zhang J-M, Lin C, Zhang F, et al. Hypermethylation of NDN promotes cell proliferation by activating the Wnt signaling pathway in colorectal cancer. Oncotarget. 2017;8:46191–203.
Barasch N, Gong X, Kwei KA, Varma S, Biscocho J, Qu K, et al. Recurrent rearrangements of the Myb/SANT-like DNA-binding domain containing 3 gene (MSANTD3) in salivary gland acinic cell carcinoma. PLoS One. 2017;12:e0171265.
Ou Yang T-H, Cheng W-Y, Zheng T, Maurer MA, Anastassiou D. Breast cancer prognostic biomarker using attractor metagenes and the FGD3-SUSD3 metagene. Cancer Epidemiol Biomark Prev Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol. 2014;23:2850–6.
L’Espérance S, Popa I, Bachvarova M, Plante M, Patten N, Wu L, et al. Gene expression profiling of paired ovarian tumors obtained prior to and following adjuvant chemotherapy: molecular signatures of chemoresistant tumors. Int J Oncol. 2006;29:5–24.
Ko H-K, Berk M, Chung Y-M, Willard B, Bareja R, Rubin M, et al. Loss of an androgen-inactivating and isoform-specific HSD17B4 splice form enables emergence of castration-resistant prostate cancer. Cell Rep. 2018;22:809–19.
Liu T, Fang Y, Zhang H, Deng M, Gao B, Niu N, et al. HEATR1 negatively regulates Akt to help sensitize pancreatic cancer cells to chemotherapy. Cancer Res. 2016;76:572–81.
Zhao Y, Chen N, Yu Y, Zhou L, Niu C, Liu Y, et al. Prognostic value of MICA/B in cancers: a systematic review and meta-analysis. Oncotarget. 2017;8:96384–95.
Thean LF, Low YS, Lo M, Teo Y-Y, Koh W-P, Yuan J-M, et al. Genome-wide association study identified copy number variants associated with sporadic colorectal cancer risk. J Med Genet. 2017.55:181–8.
Waaler J, Machon O, von Kries JP, Wilson SR, Lundenes E, Wedlich D, et al. Novel synthetic antagonists of canonical Wnt signaling inhibit colorectal cancer cell growth. Cancer Res. 2011;71:197–205.
Zhang X-W, Wang X-F, Ni S-J, Qin W, Zhao L-Q, Hua R-X, et al. UBTD1 induces cellular senescence through an UBTD1-Mdm2/p53 positive feedback loop. J Pathol. 2015;235:656–67.
Terrin L, Agostini M, Ruvoletto M, Martini A, Pucciarelli S, Bedin C, et al. SerpinB3 upregulates the Cyclooxygenase-2 / β-catenin positive loop in colorectal cancer. Oncotarget. 2017;8:15732–43.
Tagi T, Matsui T, Kikuchi S, Hoshi S, Ochiai T, Kokuba Y, et al. Dermokine as a novel biomarker for early-stage colorectal cancer. J Gastroenterol. 2010;45:1201–11.
Zhu Y, Wu G, Yan W, Zhan H, Sun P. miR-146b-5p regulates cell growth, invasion, and metabolism by targeting PDHB in colorectal cancer. Am J Cancer Res. 2017;7:1136–50.
Biancolella M, Fortini BK, Tring S, Plummer SJ, Mendoza-Fandino GA, Hartiala J, et al. Identification and characterization of functional risk variants for colorectal cancer mapping to chromosome 11q23.1. Hum Mol Genet. 2014;23:2198–209.
Kawada M, Seno H, Kanda K, Nakanishi Y, Akitake R, Komekado H, et al. Chitinase 3-like 1 promotes macrophage recruitment and angiogenesis in colorectal cancer. Oncogene. 2012;31:3111–23.
An CH, Je EM, Yoo NJ, Lee SH. Frameshift mutations of cadherin genes DCHS2, CDH10 and CDH24 genes in gastric and colorectal cancers with high microsatellite instability. Pathol Oncol Res POR. 2015;21:181–5.
Parris TZ, Danielsson A, Nemes S, Kovács A, Delle U, Fallenius G, et al. Clinical implications of gene dosage and gene expression patterns in diploid breast carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2010;16:3860–74.
Sun L, Burnett J, Gasparyan M, Xu F, Jiang H, Lin C-C, et al. Novel cancer stem cell targets during epithelial to mesenchymal transition in PTEN-deficient trastuzumab-resistant breast cancer. Oncotarget. 2016;7:51408–22.
Ooki A, Maleki Z, J-CJ T, Goparaju C, Brait M, Turaga N, et al. A panel of novel detection and prognostic methylated DNA markers in primary non-small cell lung cancer and serum DNA. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23:7141–52.
Liao X, Huang K, Huang R, Liu X, Han C, Yu L, et al. Genome-scale analysis to identify prognostic markers in patients with early-stage pancreatic ductal adenocarcinoma after pancreaticoduodenectomy. OncoTargets Ther. 2017;10:4493–506.
Rajkumar T, Vijayalakshmi N, Gopal G, Sabitha K, Shirley S, Raja UM, et al. Identification and validation of genes involved in gastric tumorigenesis. Cancer Cell Int. 2010;10:45.
Idbaih A, Carvalho Silva R, Crinière E, Marie Y, Carpentier C, Boisselier B, et al. Genomic changes in progression of low-grade gliomas. J Neuro-Oncol. 2008;90:133–40.
Tang S, Gao L, Bi Q, Xu G, Wang S, Zhao G, et al. SDR9C7 promotes lymph node metastases in patients with esophageal squamous cell carcinoma. PLoS One. 2013;8:e52184.
Wang RC, Ward D, Dunn P, Chang C-C. Acute mast cell leukemia associated with t(4;5)(q21;q33). Hum Pathol. 2017;67:198–204.
Li Y, Liang Q, Wen Y, Chen L, Wang L, Liu Y, et al. Comparative proteomics analysis of human osteosarcomas and benign tumor of bone. Cancer Genet Cytogenet. 2010;198:97–106.
AACR Project GENIE Consortium. AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7:818–31.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
Fan Y, Xi L, Hughes DST, Zhang J, Zhang J, Futreal PA, et al. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 2016;17:178.
Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213.
Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–7.
Acknowledgements
Not applicable.
Funding
MM was supported by INBRE (4P20GM103466) and COBRE (5P30GM114737). YD was supported by NIH grants 1R01CA223490 and 1R21CA164764, Bears Care Foundation and Hawaii Community Foundation to Youping Deng. This work was also supported by the NIH Grant U54MD007584, the NIH Grant 4P30CA071789 and the NIH Grant 2U54MD007601. Publication of this article was sponsored by NIH Grant 5P30GM114737.
Availability of data and materials
The datasets analyzed are available in the TCGA repository [https://portal.gdc.cancer.gov].
About this supplement
This article has been published as part of BMC Medical Genomics Volume 12 Supplement 1, 2019: Selected articles from the International Conference on Intelligent Biology and Medicine (ICIBM) 2018: medical genomics. The full contents of the supplement are available online at https://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-12-supplement-1.
Author information
Authors and Affiliations
Contributions
MM conducted all bioinformatics and statistical analyses and interpreted the data; YD developed the new method and designed whole project. MM and YD wrote the manuscript. YZ, YW. JZ and BJ helped to interpret the data and revise the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Data obtained from the TCGA open-access database was collected from tumors of patients who provided informed consent based on the guidelines from the TCGA Ethics, Law and Policy Group.
Consent for publication
All patients included in the TCGA public domain database consented for publication as detailed in [https://cancergenome.nih.gov/abouttcga/policies/informedconsent].
Competing interests
Author Youping Deng is a Section Editor for BMC Medical Genomics. All other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Menor, M., Zhu, Y., Wang, Y. et al. Development of somatic mutation signatures for risk stratification and prognosis in lung and colorectal adenocarcinomas. BMC Med Genomics 12 (Suppl 1), 24 (2019). https://doi.org/10.1186/s12920-018-0454-7
Published:
DOI: https://doi.org/10.1186/s12920-018-0454-7