
Abstract
Background
microRNAs (miRs) are small non-coding RNAs involved in the fine regulation of several cellular processes by inhibiting their target genes at post-transcriptional level. Osteosarcoma (OS) is a tumor thought to be related to a molecular blockade of the normal process of osteoblast differentiation. The current paper explores temporal transcriptional modifications comparing an osteosarcoma cell line, Saos-2, and clones stably transfected with CD99, a molecule which was found to drive OS cells to terminally differentiate.
Methods
Parental cell line and CD99 transfectants were cultured up to 14 days in differentiating medium. In this setting, OS cells were profiled by gene and miRNA expression arrays. Integration of gene and miRNA profiling was performed by both sequence complementarity and expression correlation. Further enrichment and network analyses were carried out to focus on the modulated pathways and on the interactions between transcriptome and miRNome. To track the temporal transcriptional modification, a PCA analysis with differentiated human MSC was performed.
Results
We identified a strong (about 80 %) gene down-modulation where reversion towards the osteoblast-like phenotype matches significant enrichment in TGFbeta signaling players like AKT1 and SMADs. In parallel, we observed the modulation of several cancer-related microRNAs like miR-34a, miR-26b or miR-378. To decipher their impact on the modified transcriptional program in CD99 cells, we correlated gene and microRNA time-series data miR-34a, in particular, was found to regulate a distinct subnetwork of genes with respect to the rest of the other differentially expressed miRs and it appeared to be the main mediator of several TGFbeta signaling genes at initial and middle phases of differentiation. Integration studies further highlighted the involvement of TGFbeta pathway in the differentiation of OS cells towards osteoblasts and its regulation by microRNAs.
Conclusions
These data underline that the expression of miR-34a and down-modulation of TGFbeta signaling emerge as pivotal events to drive CD99-mediated reversal of malignancy and activation of differentiation in OS cells. Our results describe crucial and specific interacting actors providing and supporting their relevance as potential targets for therapeutic differentiative strategies.
Similar content being viewed by others


Background
MicroRNAs (miRNAs) are small non-coding RNAs that act as gene regulators at post-transcriptional level. At present, it is largely established that they have a central role in both physiological and pathological conditions. In particular, miRNAs have a central role in cancer as key regulators of a multitude of processes [1], like cell differentiation [2], cell proliferation, and apoptosis [3]. Their mechanism of action is exerted through the binding of their 6–7 nt seed sequence to 3’UTR of target mRNAs which thus lead to degradation or transcriptional repression, depending on partial or perfect sequence match. Currently, more than 2.5 thousand mature miRNAs have been discovered in humans (miRbase v.20); if we consider the small matching miRNA-target sequence, the entire transcriptome could be a putative target of miRNAs. Several predicting target algorithms, like TargetScan, miRanda, PicTar and Diana MicroT [4–7] are now a routine start point to target definition. However, determination of miRNA-mRNA interactions still remain a difficult task, as these algorithms introduce a very high number of false positives [8]. Furthermore, microRNAs have also been described as positive regulators at transcriptional level of mRNA expression [9–11], with a strict dependency on the cellular context [12]. New methods together with new computational approaches are continuously being developed. Among high-throughput methods, a strategy focused on expression correlation between genes and miRNAs microarrays has been defined [13, 14], based on the evidence that degradation of a mRNA target is preferred to inhibition of its translation [15, 16]. In specific contexts like time-series experiments, integration of miRNA-mRNA expression may add valuable information on dynamic changes in gene regulation with respect to data focused on a single time point. Analysis of differentiative processes by integration of gene and miRNA time-series data may thus result particularly helpful in identifying the set of regulatory interactions at different time-points, and assignment of different microRNAs to specific differentiative phases or processes.
Sarcomas are rare tumors caused by disruptions of mesenchymal cell differentiation [17–19]. In particular, osteosarcoma (OS) is a bone cancer caused by multiple and complex genetic alterations that ultimately result in a blockage of osteoblast differentiation. Although several pathways and genes related to development, such as Wnt signaling [20, 21], TGFbeta signaling [22], Notch signaling [23], Hedgehog signaling [24], have been found to be frequently dysregulated in this sarcoma, a more comprehensive view of the processes that are aberrantly modified during OS differentiation is still missing. Highly regulated expression of genes accomplishes the process of normal osteoblastogenesis during differentiation and development [25]. Considering that OS cells appear to be somehow ‘frozen’ in a state of incomplete osteogenic differentiation [26–28], a better insight into specific gene regulation during OS differentiation may help remove this block and may have therapeutic value. In this paper we investigated integration of time-series miRNome and transcriptome to provide a better comprehension of the potential role that miRNAs may have in reprogramming genome activity coupled with OS differentiation. Recently we found that re-expression of CD99, a cell surface molecule present in osteoblasts but generally lost in OS [29], can inhibit malignancy [29, 30] and reactivate terminal differentiation [28] of OS cells. We took advantage of this experimental model and compared miRNAs-mRNAs interactions of the parental Saos-2 OS cells and osteoblast differentiation-prone Sa/CD99 cell variants at basal and differentiating conditions. Multiple bioinformatics approaches were used: integration of target prediction and expression correlation methods identified modulated genes and pathways that are directly or indirectly under control of miRNAs reprogramming; network visualization clarified differential processes where modulated miRNAs act at each time point; PCA analysis described temporal transcriptional reversion of OS cells towards the osteoblastic phenotype.
Results
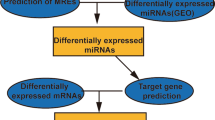
By Affymetrix GeneChip and miRNA Agilent arrays we compared gene and miRNA expression profiles of CD99-transfected clones versus the respective parental cell line; four samples at each time point (two samples for Sa/CD99 and two for Saos-2) were profiled at basal conditions and after 7 and 14 days in osteoblast differentiation medium. A flowchart of the analyses is shown in Fig. 1 and matching numbers of each phase are resumed in Table 1.

Flowchart of bioinformatic analyses. Gene and miRNA profiling of differentially expressed genes/miRs were initially integrated by correlation score of expression data or by prediction target databases. The two approaches were subsequently integrated to identify most significant miRNAs targets (d.e. = differentially expressed; FC = fold change of absolute values; p = p-value; r = Pearson’s product moment correlation coefficient; D0, D7, D14 = respectively day 0, day 7 and day 14)
CD99 over-expression fosters a massive down-regulation in gene expression of osteosarcoma cells and modulates specific pathways
Gene expression analysis identified 250 to 317 probes differentially expressed, depending on the time point (Additional file 1: Table S1). Differentially expressed genes are pointed out in a volcano plot in Additional file 2: Figure S1a. Interestingly, we observed a marked and time-constant down-regulation in gene expression when CD99 is re-expressed and cells are prone to terminal differentiation. From 78 % to 89 % of genes resulted under expressed (|logFC| ≥ 0.585 and p-value ≤ 0.05) with respect to the more aggressive and less differentiated parental cell line. Enrichment analysis showed a significant modulation of 32 pathways at day 0, 54 at day 7, and 17 at day 14 (Additional file 3: Table S2 for the 30 most significant pathways). Among the most significant modulated pathways we observed: “TGF-beta dependent induction of EMT via SMADs” (p-value = 3.78 E-08) and “Regulation of epithelial to mesenchymal transition” (p-value 2.78 E-06) (Fig. 2). TGF-beta dependent induction of EMT via SMADs pathway increased its significance during differentiation, with, over time, an increase of down-regulated genes (i.e., occludin, fibronectin). Genes involved in the apoptotic mechanism were constantly enriched but with wider involvement at initial phases (days 0 and 7), while enrichment in genes regulating the cell cycle (“regulation of G1/S transition” p-value = 1.8 E-08) was found only at day 0, in keeping with the functional role of these pathways in the reversion of malignancy and induction of differentiation. Considering single genes, a total of 64 genes were constantly modulated across all differentiative process and all but two constantly down-modulated. These genes were involved in several cellular processes like protection from apoptosis or survival (AKT1, TOX3, SMAD2, BAG5), chemoresistance, (MGST1), Notch pathway (HEY2, and HES1), histone modifications (SNCA) and transcriptional regulation (SMAD4, PAX3). To define network hubs of CD99-mediated differentiation on the 64 genes constantly modulated, a network analysis by GeneGO was performed: 16 genes out of 64 were found to have a direct modulation, with a changeless cascade signaling involving SMAD2 and SMAD4 genes with AKT1 (Fig. 3). Interestingly, there is a variable sequence of gene patterns that interacts with this core network (Additional file 4: Figure S2): (i) at day 0, THEM4, PPP2R5C and C13orf15 (alias RGC32) (ii) at day 7, GAB2, BCL2, GRB10, JAK1 (iii) at day 14, MAPKAPK2, LYN ICAM1, LIF, FN1, HMGA2, VDR, ID1.

Enrichment analysis of biologically modulated pathways. Analysis first shows a significant enrichment in TGFbeta signaling modulated by SMAD proteins: as the majority of genes are down-regulated, we have inhibition of this pathway in Saos clones, with an effect that increases over time. Other important and significantly modulated are the EMT and Apoptotic pathways. (min (p-value) = most significant p-value among the 3 time points after multiple correction test)

Network analysis of differentially expressed genes at all time points. Among the 64 genes significantly modulated at both basal and differentiative conditions, 16 have direct biological interaction according to literature (positive or activation, in green, negative or inhibition, in red, or unspecified effects, in gray) and participate in the network. All drawn genes are down-regulated
PCA meta-analysis with human mesenchymal stem cells (hMSCs) expression data was performed to see CD99 impact on the program of transcriptional differentiation. Due to array platform type and comparable experimental conditions limitations, we only recovered a single dataset of 3 human osteoblast cell lines [31]; these cells were derived from bone marrow hMSCs obtained from iliac crest and directed to osteoblast differentiation [32]. Expression profile of genes differentially expressed at basal (day 0, Fig. 4a and Additional file 5: Table S3) or in differentiative conditions (day 14, Fig. 4b) shows that OS cells prone to have a terminally differentiated phenotype (Sa/CD99 cells) tend to converge to osteoblasts cells. This confirms that our model may reflect a physiological process, thus providing valuable information on reversion toward an osteoblast-like phenotype of OS transfected cells.

Meta-analysis with human osteoblast cells. Meta-analysis including expression profile of Saos-2, Sa/CD99 cells and human osteoblasts differentiated from bone marrow hMSCs (BM-MSCs) at day 0 (a) and day 14 (b). Transfected Sa/CD99 cells progressively shift from an OS profile (day 0) towards a less aggressive and more osteoblast-like phenotype (day 14)
miRNAs modulation during differentiation emerges as a specific mechanism to define changes in gene profiling
Considering that widespread gene down-modulation was observed at all time points in consequence of the re-expression of CD99, we evaluated involvement of miRNAs as a possible epigenetic mechanism of gene regulation. We thus analyzed miRNAs profile in transfectants versus parental cells and identified 7, 4, and 16 miRs with a significant differential expression at days 0, 7, and 14 respectively (Table 2 for miRNAs modulation and Additional file 2: Figure S1b for corresponding volcano plot). At days 0 and 14, the majority of miRs showed an up-regulation (respectively 6/7 and 14/16), in line with the hypothesis that gene expression down-regulation could at least partially be controlled by miRNAs. On the contrary, miR up-regulation is not evident at day 7, maybe because of the lower number of significant miRs. In basal conditions (day 0), up-regulated miRNAs were: miR-1225-3p, miR-1305, miR-1238, miR-425, miR-191* and miR-34a, while miR-378 was the only down-regulated miR. This profile is therefore associated with a less aggressive phenotype more prone to differentiation. Among these miRs only miR-1305 was maintained significantly modulated over osteoblast-like differentiation. Increased expression of miR-34a was significantly up-regulated at days 0 and 7, in keeping with the oncosuppressive role of this miR that negatively regulates cell proliferation while increasing apoptosis [33]. When Sa/CD99 cells are terminally differentiated (day 14) [28], we observed a modulation of a different set of miRs: miR-342-3p was the most down-regulated, while miR-139-3p, miR-1288 and miR-1914* were the most up-regulated.
Dynamic interactions between mRNA and miRNA profiles defines miR-34a as a leading player of TGFbeta signaling in CD99 cells
As suggested in recent papers, different high-throughput methods may reduce the number of false positives in miRNA target definition [34]. Thus, we first integrated expression data of genes and microRNAs expression by Pearson’s correlation, then we defined putative target regions in the 3’ UTR of target genes by miRNA-mRNA sequence comparison. For expression correlations, both negative and positive r scores were considered. We finally integrated expression correlations and miRNAs target information approaches to identify the most interesting targets of differentially expressed miRs. We only used genes and miRNAs significantly modulated by CD99 transfection instead of calculating correlations or predictions for all or for moderately modulated probe:miRNA couples in arrays [35], since we considered these genes and miRNAs as the most informative for the differentiative program.
Previous expression analysis defined a total of 536 probes and 22 miRs differentially modulated in at least one out of three time points. To determine the putative target genes for each microRNA, we merged the predictions from 4 different prediction target algorithms: Diana MicroT, TargetScan, PicTar and miRanda. We obtained 96, 181, and 461 predicted targets at the 3 time points (Additional file 6: Table S4). There is at least one putative target for each microRNA at days 0 and 7, instead only 12 out of 16 miRs have predicted targets at day 14, because 4 miRs (miR-1181, miR-1288, miR-1914, miR-1469) are missing in the prediction target databases. The analysis showed that mir-34a has the highest number of predicted targets, respectively 44 and 73 genes at days 0 and 7. As expected, several genes are predicted targets of multiple microRNAs. A pathway enrichment analysis by GeneGO on predicted targets was performed to identify the putatively regulated biological pathways. We confirmed in this way the significant enrichment of genes related to TGF-Beta signaling pathway (Additional file 7: Figure S3). Several microRNAs seem to regulate multiple genes of the TGFbeta pathway, like miR-760: SMAD4/SMURF1, miR-378: SMAD2/SMAD4/SOS1, miR-26b: SMAD2/4, miR-520e: AKT1/SMAD4. miR-34a in particular shows a target site on the majority of these (SMAD2, SMAD4, AKT1, SMURF1).
Associations between miRNAs/mRNA were subsequently analyzed by correlative expression analysis. We plotted the frequency distribution of the global correlations across the 3 time points (Additional file 8: Figure S4). Notably, the 3 distributions have 3 distinct shapes and basically deviate from a normal distribution but related frequencies of p-values are in line with previously published studies [36]. On a total number of 6954 correlations, 510 (7.3 %) are significant (|r| ≥ 0.7 and p-value <0.05) (Additional file 9: Table S5), including both the positive and negative (see Table 1 for details of positive and negative percentages at the 3 time points). Since the analysis was performed at probe level, a total of 52 significant correlations of miRNA:gene were predicted more than once. In detail, we identified 159, 136, and 215 significant correlations at days 0, 7, and 14. The number of significant correlations for each microRNA is strongly heterogeneous (range: 4–207, mean: 23, median: 9). miR-34a show the highest number of negatively correlated probes (n = 70). Positive and significant correlations are 25.2 % and 22.1 % at days 0 and 7 respectively, while this percentage almost triplicates at day 14 (67.4 %) where a single miR, miR-26b, holds the majority of positive correlations (86/145) (Additional file 10: Figure S5). Among negative and significant correlations (70/215), we observed a strong correlation (−0.903) between ID1, one of the main down-stream targets of TGFbeta signaling [37], and miR-520e, a microRNA belonging to the miR-373/520 family: its over-expression has been recently described to have a tumor suppressive role in breast cancer, also by down-regulation of the TGFbeta signaling [38]. To further define interactions among miRNAs and related targets, we represented the resulting network of miRNA:gene by Cytoscape. At basal condition, we observed two main distinct subnetworks (Fig. 5) where part of the genes (56, 68 %) show a perfect star topology with miR-34a (network centralization = 1). The rest of genes are principally interconnected across 5 of 6 remaining miRs (network centralization = 0.552). To verify that the two separate networks were only partially related to the parameters we used for correlation, we tested less stringent combinations of Pearson’s score and p-value (data not shown). However, due to the very limited overlap between the two networks (7.5 % at the most) and the consequent increase in the number of false positives, we maintained the initial thresholds to optimize information. The presence of two separated subnetworks suggests a separate mechanism of action of miR-34a respect to the other miRs. miR-34a centrality is more evident at day 7, where almost all correlations concentrate on miR-34a itself. At advanced differentiation stages we observed a more interconnected mesh-like network, although miR-26b polarizes the majority of correlations.

Network representation of significantly correlated miRNA: gene couples. At initial (day 0) and middle (day 7) phases of differentiation miR-34a shows the highest number of correlated probes, instead miR-26b shows the highest number at an advanced stage of differentiation (day 14). Interestingly, miR-34a in particular seems to have a distinct action because of the completely separated network compared to the other miRs
Finally, to define the most interesting couples miR:target, intersection of prediction and correlation approaches were used, identifying 62 couples miR:gene (Table 3). This analysis strongly reduced the number of miRs with a potential to regulate the system, only 9 out of the 22 differentially expressed microRNAs have at least one target significantly predicted and correlated. Five miRs have one single target gene (miR-202: CBFA2T3, miR-500: NEBL, miR-378: KIF26A, miR-892b: BACH2, miR-1305:ID1), with a restricted impact on gene expression modulation. Among the remaining 4 miRs (miR-26b, miR-34a, miR-342-3p, miR-520e), attention still points to miR-34a, which has the highest number (24) of predicted and correlated genes. Most correlations are negative (17/24) and, as expected, involve genes of TGFbeta pathway, such as SMAD4 (r = −0.904), AKT3 (r = −0.909) and GRB10 (r = −0.842). In contrast, the most down-regulated miRNAs, miR-342-3p and miR-26b, showed a prevalence of positive correlations with their targets (respectively 8/9 and 20/22), which indicate a more complex and possible indirect relationship with the regulated genes.
Discussion
Etiology of OS still remains unclear although recent papers have related this tumor to a molecular blockade of the normal process of osteoblast differentiation [18]. OS cells are thought to derive from mesenchymal stem cells already committed towards osteblast differentiation and thus displaying features of osteo-progenitors [26]. Transformation hampers progression of malignant cells towards terminal osteoblastic differentiation while maintaining cell proliferation.
High-throughput screening techniques appear the most refined tool to identify pivotal players in complex biological processes: combination of different approaches to integrate several platforms can help better explore their fine tuning activity. Some authors have recently integrated a considerable number of high-throughput data from OS cell lines, either by miRNA vs mRNA expression profiles [39, 40] or vs CGH arrays [41] or by protein arrays [42], identifying miR-17/92 cluster [39] or TGFbeta pathway [42] as crucial mediators. However, all these studies focalized on the events at basal conditions and their expression during the differentiative process was not explored.
Besides alterations in Rb, p53 [18], and oncogene MET pathways, CD99 transmembrane antigen was also found to regulate differentiation of OS cells [26, 28]. In this paper we used CD99 transfected OS cells to explore the temporal transcriptional changes that couple with malignancy reversal and modulation of differentiation. CD99 is generally lost in OS cells and, when its expression is restored, the molecule switches cells from proliferation status to cell cycle withdrawal, favoring the achievement of a terminally osteoblast-differentiated phenotype, thus resembling the fate of mature osteoblasts [28]. This phenotype is confirmed by PCA meta-analysis, which shows the shift of Saos-2 cell genetic profile towards normal osteoblasts when CD99 is re-expressed. Gene expression analysis indicated that CD99 forced expression induced broad gene expression down-regulation: almost 80 % of genes showed decreased expression in CD99 transfected cells. Enrichment analysis revealed a significant modulation in Epithelial to Mesenchymal Transition (EMT), apoptosis and TGFbeta signaling pathways.
EMT has received considerable attention in the last years as a paradigm to explain invasive and metastatic behavior of cancer cells. Firstly described in carcinomas, the hallmarks of this program are the disruption of cell adhesion structures between adjacent cells, a dramatic remodeling of the cytoskeleton, and the acquisition of a mesenchymal phenotype. Reduced expression of epithelial markers, such as E-cadherin, and a simultaneous increase in mesenchymal marker expression, such as N-cadherin and vimentin characterize EMT, whose master regulators, SNAIL1 and SNAIL2 are direct transcriptional targets of the TGFbeta pathway SMADs mediators [43]. OS is characterized by the expression of EMT-related transcription factors, which are involved in the complex pathogenesis of the tumor [44]. Over-expression of CD99 determines a down-regulation of mesenchymal markers, such as fibronectin, and the transcription factor snail1. In keeping with the less malignant behavior, Sa/CD99 OS cells also exhibited down-modulation in genes belonging to TGFbeta signaling, a pathway that plays fundamental roles in carcinogenesis. TGFbeta, one of the most abundant growth factors stored and released by bone, is known to regulate a broad range of biological processes, including cell proliferation, cell survival, cell differentiation, cell migration, production of extracellular matrix molecules and regulation of cell stemness [45]. The combined actions of these cellular responses mediate the global effects of TGF-beta pathway on cancer, immune responses, angiogenesis, wound healing, development, and bone formation. In cancer, several studies have clearly demonstrated that TGFbeta signaling pathway can either foster or suppress tumor progression [46, 47]: depending on the cellular context and the type of TGF-beta signaling pathway that is initiated (Smad-dependent or Smad-independent pathway), the cell is directed to undergo either proliferation, differentiation or apoptosis. In the bone environment TGFbeta signaling is reported to inhibit osteoblast differentiation [48] while inducing proliferation and migration in OS cells [49, 50]. Together with hypoxia, TGFbeta has also been shown as an important element that prompts OS cells toward cancer stem cell phenotype [51]. In addition, Yang and Franchi et al. also found higher TGFbeta1 expression in the patients with high-grade osteosarcoma and lung metastasis [52, 53], indicating that TGF-beta signaling promoted the chemoresistance, tumorigenicity, and metastatic potential of OS. Decreased expression of genes belonging to TGFbeta signaling in the Sa/CD99 OS, which are reverted in malignancy and prone to differentiation, is thus consistent with the potential value of therapies targeting TGFbeta signaling. Reversing the tumorigenicity of OS cells and placing them back on the road to normal osteoblasts differentiation means not only the induction of progressive loss of proliferative capacities but generation of apoptosis, which is part of the program cell fate toward terminal osteoblast differentiation to osteocytes. According to these processes, network analysis of genes modulated in Sa/CD99 cells identified two core hubs composed of AKT1 and SMAD 2/4 proteins. In particular, SMAD4, one of the leading mediators of TGFbeta signaling [54], resulted to be the main hub with the highest number of connected genes (8/16). This protein has been found to have an oncogenic role in sarcomas [55] and in glioblastoma [56], while SMAD4 silencing induced growth inhibition and apoptosis in rhabdomyosarcoma cells [57]. Observation of the network analysis depicted a changeable entourage of important mediators that act at different stages. In particular, silencing of ID1, which interacts directly with SMAD4 at day 14, is essential for activation of terminal differentiation [37], while SMAD2, a partner of SMAD4 in TGFbeta signaling, enhances SMAD4 expression and suppresses osteocalcin [58], a marker of late osteoblast differentiation. SMAD2 is also expressed in the majority (85 %) of OS clinical samples [59], and its repression by microRNA mimics inhibits proliferation and invasion in OS cells [50]. The other component of the hub, AKT1 plays an important role in proliferation, survival, migration and metastatization of many cancers including OS [60]. Its widespread activation has been observed in OS patients with pulmonary metastatic disease [60], and AKT1 inhibition by Akt-siRNA or allosteric specific inhibitors was found to decrease cell migration and/or inhibit proliferation in several OS experimental models [61, 62], supporting the therapeutic attractiveness of AKT-targeted inhibitors. Besides direct activation, multiple studies have also suggested the existence of indirect mechanisms for TGFbeta activation of PI3K/AKT, where TGFbeta may act in concert with other stimuli. On the other hand, PI3K/AKT antagonizes TGFbeta-induced cytostatic responses and causes the shift in TGFbeta/SMAD signaling to its tumor-promoting mode during malignant tumor progression, thus indicating the existence of a signaling interplay between TGFbeta and PI3K/AKT pathways.
Taken together, annotation analysis of Sa/CD99 gene expression with respect to parental cells clearly identifies in down-regulation of TGFbeta/Smad4/Akt signaling a crucial event in the reversion of OS malignancy. Interestingly, miRNAs expression profiling also indicated a modulation of miRNAs that converge on TGFbeta pathway. A general miRNA up-regulation was observed after CD99 transfection. A signature of 22 modulated miRNAs was defined and their expression correlated with significantly modulated genes to detect important associations at each phase of cell differentiation. Network analysis identified in miR-34a and miR-26b the two main regulators in early (day 0 and day 7) or in late (day 14) phases of differentiation. These two miRs exhibit an opposite mechanism in regulating their respective targets: miR-26 mainly shows a direct correlation, which introduces to an indirect and difficult to unravel mechanism of regulation, whereas miR-34a displays a canonical down-regulation of its targets.
Enrichment analysis on predicted targets of significantly modulated miRNAs confirmed that TGFbeta signaling and apoptosis-related mechanisms could be miRNA-driven. In particular, miR-34a emerges as the main putative modulator of several genes of TGFbeta signaling. In our model, SMAD4 resulted to be the best candidate target at transcriptional level, in line with regulation of SMAD4 by miR-34a that has been recently shown in glioblastoma [56]. miR-34a is a well-known oncosuppressor miRNA found to induce cell-cycle arrest and apoptosis thorough negative regulation of proteins directly involved in the regulation of cell proliferation and/or cell death, such as E2F, cyclin D1, CDK4, CDK6, cyclin E2, and bcl-2 [63, 64]. miR-34, whose expression is generally reduced in most tumors [33] including OS [65], also displays a role in osteogenic differentiation [66]. Our results further support these evidences, indicating miR-34a as a leader player in the reversal of malignancy and reactivation of differentiation of OS cells by TGFbeta signaling down-modulation.
Complexity of the genetic landscape in OS cells together with its rarity make any targeted therapy difficult to be defined. In this context, the multiple approaches here adopted may represent a powerful tool to unravel and better characterize the genetic background associated with malignant phenotype of this tumor, thus offering identification of critical hubs for the design of differentiative therapeutic strategies.
Conclusions
Our intent was to define the transcriptional modifications that characterize reversion of malignancy and induction of terminal osteoblast differentiation in OS. A global gene down-regulation was observed across the 14 days of in vitro differentiation and down-modulation of the TGFbeta signaling pathway, together with involvement of several important mediators like AKT1 and SMADs proteins, were defined as crucial events. The use of multiple analyses supported the interactions between miRNome and transcriptome and helped to define miRNAs impact on gene down-modulation. miR-34a was clearly identified as key regulator at initial and middle phases of OS differentiation toward osteoblast. To our knowledge, this is the first study where deciphering of the miRNAs role in the differentiative block of OS is tracked in time. miR-34a up-regulation followed by TGFbeta/SMAD4 signaling inhibition appeared as two crucial players able to induce malignant reversion and osteogenic differentiation of OS cells. These new insights could drive future efforts to investigate the relevance of miR-34a and TGFbeta signaling as potential targets for innovative therapeutic strategies against OS.
Methods
Cell lines
OS cell line Saos-2 was obtained from American Type Culture Collection (Manassas, VA, Rockville, MD). Stable transfectants expressing CD99 were obtained from Saos-2 cell line (Sa/CD99wt22, Sa/CD99wt36) by using calcium-phosphate transfection method [28] and tested for mycoplasma contamination every three months (last control September 2014) by PCR Mycoplasma detection Set (Takara Bio Inc., Shiga, Japan).
Cells were routinely cultured in Iscove’s modified Dulbecco’s medium (IMDM), supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 % inactivated fetal bovine serum (FBS) and maintained at 37 °C in a humidified 5 % CO2 atmosphere. Transfectants were selected in IMDM containing 10 % FBS and 500 μg/ml neomycin.
Osteoblast differentiation
Four days after seeding cells were exposed to specific osteogenic medium, IMDM supplemented with 2 % FBS, 5 mM β-glycerophosphate and 50 μg/mL ascorbic acid (Sigma-Aldrich, St. Louis, MO) and maintained in differentiative conditions up to 14 days. Medium was renewed every 4 days. Cultures were harvested at various time points to collect RNA. Total RNA was extracted by the TRIzol extraction kit (Life Technologies, Grand Island, NY). Quality and quantity of RNA samples were assessed with NanoDrop analysis (NanoDrop Technologies). Expression of CD99 was verified by real-time PCR at basal and differentiative conditions in all profiled samples (Additional file 11: Figure S6).
Microarray hybridization
Gene expression of Saos-2 and Sa/CD99 cells was profiled by Affymetrix (Santa Clara, CA, USA) GeneChip Human Genome U133A plus 2 according to manufacturer’s instructions and scanned by Affymetrix Scanner 3000 7G to obtain raw data. miRNAs profile was analyzed by Agilent (Santa Clara, CA, USA) Human miRNA microarray platform (v.3) according to manufacturer’s instructions, scanned with Agilent Scanner G2505 and analyzed by Feature Extraction (v10.5) software to obtain raw data. All raw data were inspected for visual and technical artifacts by Bioconductor (v 2.9) packages (simpleaffy [67], affyPLM and AgiMicroRNA [68] for miRNA data) on R (v. 2.15) and were considered of good quality. (see Additional file 12: Figure S7 for density plots of both gene and miRNA expression data, and Additional file 13: Table S6 for gene expression quality metrics). Two samples for Saos-2 and two for Sa/CD99 cells were profiled for each time point, for a total of 12 samples for either gene or miRNA expression profiling.
mRNA and miRNA expression profiling
All microarray analyses were performed by Bioconductor packages on R. Gene expression data were quantified and normalized by rma [69] algorithm and log2 transformed. Probes with low expression (signal intensity of the probe ≤ 100 of absolute values in at least 75 % of samples) and low IQR, Inter-Quartile Range (IQR of log2 signals ≤ median of IQR across all samples) were filtered out. Differentially expressed genes between Sa/CD99 vs Saos-2 were detected using limma [70] package and corrected by FDR according to Benjamini and Hochberg multiple test correction. Genes were considered significant when logFC ≥ 0.585 (logFC, log fold change of absolute normalized values), corresponding to a fold change of 1.5, and p-value ≤ 0.05 after multiple test correction. Only probes matching well annotated genes according to Affymetrix HG_U133 Plus 2.0 array annotation and recovered by hgu133plus2.db Bioconductor package were preserved. MicroRNA expression data were quantified, log2 transformed and normalized in R using AgiMicroRna package [68], that uses an adaptation of rma method for microRNA data. Low or not expressed miRs were filtered out and remaining probes were tested for differential expression using limma modified t-test: miR was considered significant when logFC ≥ 0.485, corresponding to a fold change of 1.4, and p-value ≤ 0.01 in transfected vs parental cell lines. Microarray data are available at GEO database with SuperSeries accession number GSE61930.
Correlations between miRNAs and genes profiling
Correlation was calculated between expression of miRNAs vs gene probes differentially expressed in at least 1 on 3 time points. Analysis was performed on normalized log2 expression data and considered significant when: |r| ≥ 0.7 and p ≤ 0.05, where r is the score according to Pearson’s correlation and p is the asymptotic p-value. Calculation was performed by R scripts using library Hmisc. For gene expression data, correlation was preserved at probe level, thus multiple significant correlations per gene may exist. Both positive and negative correlations were considered with putative biological relevance [10]. No multiple test correction was performed; to control type I error without reducing statistical power we opted for the use of an high Pearson’s r score.
miRNA target prediction
Genes were considered putative target of microRNA when reported in at least 1 of the following databases: Targetscan (v. 6.0), miRanda (v. 3.0), Diana MicroT (v. 3.0), PicTar (v. 4-way). All predictions were downloaded from their respective web sites except for PicTar from UCSC genome browser (v. NCBI35/hg17). Only differentially expressed genes and miRs were taken into account for microRNA target prediction analysis. No score threshold was used for target prediction.
Enrichment and network analyses
Enrichment analysis of pathways was performed using MetaCore in GeneGO (Thomson Reuters, New York, NY, USA) program. Biological pathways were defined according to “GeneGO Pathway Maps” manually curated database. Pathways were tested for significance by modified Fisher’s Exact Test and were corrected by FDR multiple test correction according to the Benjamini and Hochberg method, considered significant when the corrected p-value of enrichment was ≤ 0.1. Network analysis of biologically-related terms was performed with the direct interaction method in GeneGO, were an edge connecting two genes indicates their direct biological relation according to MetaCore database, which includes manually-curated database of human gene and protein interactions, built according to published literature. Network analysis was performed on genes differentially expressed at all time points. Background for statistical analysis by GeneGO is composed of the array gene list. Graphical representation of miRNA-gene from correlation and prediction analyses was performed using Cytoscape (v.3.0) [71].
Meta-analysis
Expression data of human osteoblast derived from bone marrow hMSCs were recovered in GEO database with ID GSE9451 [31]. Expression data from osteosarcoma and mesenchymal cell lines were rma normalized together and log2 transformed before analysis. No further batch correction was performed. Meta-analysis was performed using made4 package [72] on R. Expression data of differentially expressed genes in Sa/CD99 vs Saos-2 at day 0 or day 14 was used for PCA analysis for day 0 or day 14 respectively. The “pca” method for “ord” (ordination method) function and default settings as from made4 introduction file were used.
Abbreviations
- miRNAs or miRs:
-
microRNAs
- OS:
-
Osteosarcoma
- hMSCs:
-
Human mesenchymal stem cells
- EMT:
-
Epithelial-mesenchymal transition
References
Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–69.
Ambros V. The functions of animal microRNAs. Nature. 2004;431(7006):350–5.
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102(39):13944–9.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36(Database issue):D154–158.
Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105.
Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37(5):495–500.
Maragkakis M, Alexiou P, Papadopoulos GL, Reczko M, Dalamagas T, Giannopoulos G, et al. Accurate microRNA target prediction correlates with protein repression levels. BMC Bioinformatics. 2009;10:295.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20.
Majid S, Dar AA, Saini S, Yamamura S, Hirata H, Tanaka Y, et al. MicroRNA-205-directed transcriptional activation of tumor suppressor genes in prostate cancer. Cancer. 2010;116(24):5637–49.
Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A. 2008;105(5):1608–13.
Yu J, Ryan DG, Getsios S, Oliveira-Fernandes M, Fatima A, Lavker RM. MicroRNA-184 antagonizes microRNA-205 to maintain SHIP2 levels in epithelia. Proc Natl Acad Sci U S A. 2008;105(49):19300–5.
Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318(5858):1931–4.
Muniategui A, Nogales-Cadenas R, Vazquez M, Aranguren XL, Agirre X, Luttun A, et al. Quantification of miRNA-mRNA interactions. PLoS One. 2012;7(2):e30766.
Yoon D, Lee EK, Park T. Robust imputation method for missing values in microarray data. BMC Bioinformatics. 2007;8 Suppl 2:S6.
Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, et al. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122(4):553–63.
Wu L, Belasco JG. Let me count the ways: mechanisms of gene regulation by miRNAs and siRNAs. Mol Cell. 2008;29(1):1–7.
Matushansky I, Hernando E, Socci ND, Mills JE, Matos TA, Edgar MA, et al. Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. J Clin Invest. 2007;117(11):3248–57.
Tang N, Song WX, Luo J, Haydon RC, He TC. Osteosarcoma development and stem cell differentiation. Clin Orthop Relat Res. 2008;466(9):2114–30.
Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell. 2007;11(5):421–9.
Cai Y, Mohseny AB, Karperien M, Hogendoorn PC, Zhou G, Cleton-Jansen AM. Inactive Wnt/beta-catenin pathway in conventional high-grade osteosarcoma. J Pathol. 2010;220(1):24–33.
Haydon RC, Deyrup A, Ishikawa A, Heck R, Jiang W, Zhou L, et al. Cytoplasmic and/or nuclear accumulation of the beta-catenin protein is a frequent event in human osteosarcoma. Int J Cancer. 2002;102(4):338–42.
Kloen P, Gebhardt MC, Perez-Atayde A, Rosenberg AE, Springfield DS, Gold LI, et al. Expression of transforming growth factor-beta (TGF-beta) isoforms in osteosarcomas: TGF-beta3 is related to disease progression. Cancer. 1997;80(12):2230–9.
Engin F, Bertin T, Ma O, Jiang MM, Wang L, Sutton RE, et al. Notch signaling contributes to the pathogenesis of human osteosarcomas. Hum Mol Genet. 2009;18(8):1464–70.
Chan LH, Wang W, Yeung W, Deng Y, Yuan P, Mak KK. Hedgehog signaling induces osteosarcoma development through Yap1 and H19 overexpression. Oncogene. 2014;33:4857.
Wei J, Shi Y, Zheng L, Zhou B, Inose H, Wang J, et al. miR-34s inhibit osteoblast proliferation and differentiation in the mouse by targeting SATB2. J Cell Biol. 2012;197(4):509–21.
Dani N, Olivero M, Mareschi K, van Duist MM, Miretti S, Cuvertino S, et al. The MET oncogene transforms human primary bone-derived cells into osteosarcomas by targeting committed osteo-progenitors. J Bone Miner Res. 2012;27(6):1322–34.
Haydon RC, Luu HH, He TC. Osteosarcoma and osteoblastic differentiation: a new perspective on oncogenesis. Clin Orthop Relat Res. 2007;454:237–46.
Sciandra M, Marino MT, Manara MC, Guerzoni C, Grano M, Oranger A, et al. CD99 drives terminal differentiation of osteosarcoma cells by acting as a spatial regulator of ERK 1/2. J Bone Miner Res. 2014;29(5):1295–309.
Manara MC, Bernard G, Lollini PL, Nanni P, Zuntini M, Landuzzi L, et al. CD99 acts as an oncosuppressor in osteosarcoma. Mol Biol Cell. 2006;17(4):1910–21.
Scotlandi K, Zuntini M, Manara MC, Sciandra M, Rocchi A, Benini S, et al. CD99 isoforms dictate opposite functions in tumour malignancy and metastases by activating or repressing c-Src kinase activity. Oncogene. 2007;26(46):6604–18.
Kubo H, Shimizu M, Taya Y, Kawamoto T, Michida M, Kaneko E, et al. Identification of mesenchymal stem cell (MSC)-transcription factors by microarray and knockdown analyses, and signature molecule-marked MSC in bone marrow by immunohistochemistry. Genes Cells. 2009;14(3):407–24.
Matsubara T, Suardita K, Ishii M, Sugiyama M, Igarashi A, Oda R, et al. Alveolar bone marrow as a cell source for regenerative medicine: differences between alveolar and iliac bone marrow stromal cells. J Bone Miner Res. 2005;20(3):399–409.
Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17(2):193–9.
Muniategui A, Pey J, Planes FJ, Rubio A. Joint analysis of miRNA and mRNA expression data. Brief Bioinform. 2013;14(3):263–78.
Fulci V, Colombo T, Chiaretti S, Messina M, Citarella F, Tavolaro S, et al. Characterization of B- and T-lineage acute lymphoblastic leukemia by integrated analysis of MicroRNA and mRNA expression profiles. Genes, Chromosomes Cancer. 2009;48(12):1069–82.
Nunez-Iglesias J, Liu CC, Morgan TE, Finch CE, Zhou XJ. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PLoS One. 2010;5(2):e8898.
Peng Y, Kang Q, Luo Q, Jiang W, Si W, Liu BA, et al. Inhibitor of DNA binding/differentiation helix-loop-helix proteins mediate bone morphogenetic protein-induced osteoblast differentiation of mesenchymal stem cells. J Biol Chem. 2004;279(31):32941–9.
Keklikoglou I, Koerner C, Schmidt C, Zhang JD, Heckmann D, Shavinskaya A, et al. MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting NF-kappaB and TGF-beta signaling pathways. Oncogene. 2012;31(37):4150–63.
Baumhoer D, Zillmer S, Unger K, Rosemann M, Atkinson MJ, Irmler M, et al. MicroRNA profiling with correlation to gene expression revealed the oncogenic miR-17-92 cluster to be up-regulated in osteosarcoma. Cancer Genet. 2012;205(5):212–9.
Namlos HM, Meza-Zepeda LA, Baroy T, Ostensen IH, Kresse SH, Kuijjer ML, et al. Modulation of the osteosarcoma expression phenotype by microRNAs. PLoS One. 2012;7(10):e48086.
Maire G, Martin JW, Yoshimoto M, Chilton-MacNeill S, Zielenska M, Squire JA. Analysis of miRNA-gene expression-genomic profiles reveals complex mechanisms of microRNA deregulation in osteosarcoma. Cancer Genet. 2011;204(3):138–46.
Flores RJ, Li Y, Yu A, Shen J, Rao PH, Lau SS, et al. A systems biology approach reveals common metastatic pathways in osteosarcoma. BMC Syst Biol. 2012;6:50.
Naber HP, Drabsch Y, Snaar-Jagalska BE, ten Dijke P, van Laar T. Snail and Slug, key regulators of TGF-beta-induced EMT, are sufficient for the induction of single-cell invasion. Biochem Biophys Res Commun. 2013;435(1):58–63.
Yang G, Yuan J, Li K. EMT transcription factors: implication in osteosarcoma. Med Oncol. 2013;30(4):697.
Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, et al. TGF-beta: duality of function between tumor prevention and carcinogenesis. J Natl Cancer Inst. 2014;106(2):djt369.
Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10(6):415–24.
Massague J. TGFbeta in cancer. Cell. 2008;134(2):215–30.
Alliston T, Choy L, Ducy P, Karsenty G, Derynck R. TGF-beta-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. Embo J. 2001;20(9):2254–72.
Matsuyama S, Iwadate M, Kondo M, Saitoh M, Hanyu A, Shimizu K, et al. SB-431542 and Gleevec inhibit transforming growth factor-beta-induced proliferation of human osteosarcoma cells. Cancer Res. 2003;63(22):7791–8.
Niu G, Li B, Sun L, An C. MicroRNA-153 inhibits osteosarcoma cells proliferation and invasion by targeting TGF-beta2. PLoS One. 2015;10(3):e0119225.
Zhang H, Wu H, Zheng J, Yu P, Xu L, Jiang P, et al. Transforming growth factor beta1 signal is crucial for dedifferentiation of cancer cells to cancer stem cells in osteosarcoma. Stem Cells. 2013;31(3):433–46.
Franchi A, Arganini L, Baroni G, Calzolari A, Capanna R, Campanacci D, et al. Expression of transforming growth factor beta isoforms in osteosarcoma variants: association of TGF beta 1 with high-grade osteosarcomas. J Pathol. 1998;185(3):284–9.
Yang RS, Wu CT, Lin KH, Hong RL, Liu TK, Lin KS. Relation between histological intensity of transforming growth factor-beta isoforms in human osteosarcoma and the rate of lung metastasis. Tohoku J Exp Med. 1998;184(2):133–42.
Yang G, Yang X. Smad4-mediated TGF-beta signaling in tumorigenesis. Int J Biol Sci. 2010;6(1):1–8.
Wang H, Yang GH, Bu H, Zhou Q, Guo LX, Wang SL, et al. Systematic analysis of the TGF-beta/Smad signalling pathway in the rhabdomyosarcoma cell line RD. Int J Exp Pathol. 2003;84(3):153–63.
Genovese G, Ergun A, Shukla SA, Campos B, Hanna J, Ghosh P, et al. microRNA regulatory network inference identifies miR-34a as a novel regulator of TGF-beta signaling in glioblastoma. Cancer Discov. 2012;2(8):736–49.
Ye L, Zhang H, Zhang L, Yang G, Ke Q, Guo H, et al. Effects of RNAi-mediated Smad4 silencing on growth and apoptosis of human rhabdomyosarcoma cells. Int J Oncol. 2006;29(5):1149–57.
Li J, Tsuji K, Komori T, Miyazono K, Wrana JL, Ito Y, et al. Smad2 overexpression enhances Smad4 gene expression and suppresses CBFA1 gene expression in osteoblastic osteosarcoma ROS17/2.8 cells and primary rat calvaria cells. J Biol Chem. 1998;273(47):31009–15.
Mohseny AB, Cai Y, Kuijjer M, Xiao W, van den Akker B, de Andrea CE, et al. The activities of Smad and Gli mediated signalling pathways in high-grade conventional osteosarcoma. Eur J Cancer. 2012;48(18):3429–38.
Zhu LB, Jiang J, Zhu XP, Wang TF, Chen XY, Luo QF, et al. Knockdown of Aurora-B inhibits osteosarcoma cell invasion and migration via modulating PI3K/Akt/NF-kappaB signaling pathway. Int J Clin Exp Pathol. 2014;7(7):3984–91.
Kuijjer ML, van den Akker BE, Hilhorst R, Mommersteeg M, Buddingh EP, Serra M, et al. Kinome and mRNA expression profiling of high-grade osteosarcoma cell lines implies Akt signaling as possible target for therapy. BMC Med Genomics. 2014;7:4.
Zhang A, He S, Sun X, Ding L, Bao X, Wang N. Wnt5a promotes migration of human osteosarcoma cells by triggering a phosphatidylinositol-3 kinase/Akt signals. Cancer Cell Int. 2014;14(1):15.
Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12(5):414–8.
Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6(13):1586–93.
Yan K, Gao J, Yang T, Ma Q, Qiu X, Fan Q, et al. MicroRNA-34a inhibits the proliferation and metastasis of osteosarcoma cells both in vitro and in vivo. PLoS One. 2012;7(3):e33778.
Sun F, Wan M, Xu X, Gao B, Zhou Y, Sun J, et al. Crosstalk between miR-34a and Notch Signaling Promotes Differentiation in Apical Papilla Stem Cells (SCAPs). J Dent Res. 2014;93(6):589–95.
Wilson CL, Miller CJ. Simpleaffy: a BioConductor package for Affymetrix Quality Control and data analysis. Bioinformatics. 2005;21(18):3683–5.
Lopez-Romero P. Pre-processing and differential expression analysis of Agilent microRNA arrays using the AgiMicroRna Bioconductor library. BMC Genomics. 2011;12:64.
Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–93.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Culhane AC, Thioulouse J, Perriere G, Higgins DG. MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics. 2005;21(11):2789–90.
Acknowledgements
The authors would like to thank A. Balladelli and C. Ghinelli for their help in editing the paper.
Funding
The work was funded by the Italian Association for Cancer Research (IG 14049 to KS) and by grants from Istituto Ortopedico Rizzoli (5xmille contributions to the Rizzoli Institute).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AG: participated in the design of the study, data analysis and drafted the manuscript. MS: carried out cell line preparation, microarray hybridization, data interpretation. MT: carried out cell line transfections and data interpretation. PP: participated in study design. KS: conceived the study, participated in its design and coordination, drafted the manuscript. AG, MS, MT, PP, KS: revised the manuscript content. All authors approved the final manuscript.
Additional files
Additional file 1: Table S1.
Genes differentially expressed in Sa/CD99 vs. Saos-2 cells in at least one time point.
Additional file 2: Figure S1.
Volcano plot of gene (a) and miRNA (b) expression data: for both experiments, we marked out genes and miRNA differentially expressed in Sa/CD99 vs Saos-2 at day 0 (red), day 7 (green) and day 14 (blu).
Additional file 3: Table S2.
Thirty most significant pathways after enrichment analysis on differentially expressed genes.
Additional file 4: Figure S2.
Network analysis of differentially expressed genes at each single time point. Genes from the core network (Fig. 3) are circled in red.
Additional file 5: Table S3.
Euclidean distances of the eigenvector from PCA analysis for Sa/CD99 transfectants, Saos-2 parental and Osteoblast from BM-MSC cells. Distance is calculated on the mean of eigenvectors for each cell type.
Additional file 6: Table S4.
Couples of significant gene-miR from prediction approach.
Additional file 7: Figure S3.
Enrichment analysis of biologically modulated pathways on modulated predicted targets.
Additional file 8: Figure S4.
Frequency histogram of Pearson’s correlation scores (on left) and of related p-values (on right) at the three time points for differentially expressed genes and miRNAs. Dashed lines indicate the threshold (|r| ≥ 0.7 and p-value ≤ 0.05) used to consider significant a correlation.
Additional file 9: Table S5.
Couples of significant gene-miR from correlation approach.
Additional file 10: Figure S5.
Percentage of negative and positive correlations at each time point and at all time points.
Additional file 11: Figure S6.
Expression levels of CD99 were verified by real-time PCR in parental Saos-2 and both clones for all three days analyzed by expression profiling.
Additional file 12: Figure S7.
Density plots of gene and miRNA expression data for all arrays, used for quality control.
Additional file 13: Table S6.
Quality control parameters of gene expression arrays are shown. All values respect suggestion of Affymetrix quality control (for details see simple affy package vignettes).
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Grilli, A., Sciandra, M., Terracciano, M. et al. Integrated approaches to miRNAs target definition: time-series analysis in an osteosarcoma differentiative model. BMC Med Genomics 8, 34 (2015). https://doi.org/10.1186/s12920-015-0106-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-015-0106-0