
Abstract
Background
The study of DNA methylation quantitative trait loci (meQTLs) helps dissect regulatory mechanisms underlying genetic associations of human diseases. In this study, we conducted the first genome-wide examination of genetic drivers of methylation variation in response to a triglyceride-lowering treatment with fenofibrate (response-meQTL) by using an efficient analytic approach.
Methods
Subjects (n = 429) from the GAW20 real data set with genotype and both pre- (visit 2) and post- (visit 4) fenofibrate treatment methylation measurements were included. Following the quality control steps of removing certain cytosine-phosphate-guanine (CpG) probes, the post−/premethylation changes (post/pre) were log transformed and the association was performed on 208,449 CpG sites. An additive linear mixed-effects model was used to test the association between each CpG probe and single nucleotide polymorphisms (SNPs) around ±1 Mb region, with age, sex, smoke, batch effect, and principal components included as covariates. Bonferroni correction was applied to define the significance threshold (p < 5.6 × 10− 10, given a total of 89,217,303 tests). Finally, we integrated our response-meQTL (re-meQTL) findings with the published genome-wide association study (GWAS) catalog of human diseases/traits.
Results
We identified 1087 SNPs as cis re-meQTLs associated with 610 CpG probes/sites located in 351 unique gene loci. Among these 1087 cis re-meQTL SNPs, 229 were unique and 6 were co-localized at 8 unique disease/trait loci reported in the GWAS catalog (enrichment p = 1.51 × 10− 23). Specifically, a lipid SNP, rs10903129, located in intron regions of gene TMEM57, was a re-meQTL (p = 3.12 × 10− 36) associated with the CpG probe cg09222892, which is in the upstream region of the gene RHCE, indicating a new target gene for rs10903129. In addition, we found that SNP rs12710728 has a suggestive association with cg17097782 (p = 1.77 × 10− 4), and that this SNP is in high linkage disequilibrium (LD) (R2 > 0.8) with rs7443270, which was previously reported to be associated with fenofibrate response (p = 5.00 × 10− 6).
Conclusions
By using a novel analytic approach, we efficiently identified thousands of cis re-meQTLs that provide a unique resource for further characterizing functional roles and gene targets of the SNPs that are most responsive to fenofibrate treatment. Our efficient analytic approach can be extended to large response quantitative trait locus studies with large sample sizes and multiple time points data.
Similar content being viewed by others


Background
Genome-wide association studies (GWASs) have discovered hundreds of genetic loci associated with blood lipid levels [1]. Fenofibrate is a widely used medicine to treat high levels of cholesterol and triglycerides. Because treatment response is heterogeneous and heritable, several recent studies have identified genetic determinants of the observed heterogeneity in fenofibrate effects on lipid levels [2] and systemic inflammation involved in lipid metabolism [3, 4]. However, the functional mechanism underlying these findings is largely unknown.
Epigenetic changes such as DNA methylation alteration contribute to phenotypic differences within and between individuals. The study of genetic effects on methylation quantitative trait loci (meQTLs) has emerged to help dissect regulatory mechanisms underlying these GWAS associations [5, 6]. However, the genetic components that determine the nature and strength of meQTL treatment response (re-meQTL) has not yet been studied. Using the GAW20 genotype and methylation data obtained before and after fenofibrate treatment of the same subjects, we conducted a re-meQTL analysis to identify common single nucleotide polymorphisms (SNPs) associated with methylation changes. This study is the first genome-wide examination of genetic drivers of methylation variation in treatment response, providing a unique resource for the community.
In addition to the re-meQTL analysis, we developed an efficient analytic approach that significantly reduced the computational burden. The computation of genome-wide meQTL analysis is known to be intensive, especially for re-meQT study which requires more than one time-point data. In this work, we analyzed the pre- and post- treatment methylation data in one model. We believe our approach is more efficient and improves on the recently reported analysis of response-expression QTLs (re-eQTLs) in which different time-point data were first analyzed separately, then the re-eQTLs were defined based on the effect size difference between post stimulus and basal levels [7].
Methods
Sample demography
The original samples are from the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study (details at https://dsgwebwp.wustl.edu/labs/province-lab/goldn/), which aimed to characterize the genetic impact on response to interventions that affects triglycerides (TGs) [8]. The GOLDN study was done through a collaboration of 6 university-based medical centers and all participants were whites of European ancestry. Our study only used GAW20 real data, which contains participants in response to 4 visits in 3 weeks for an open-label clinical trial of fenofibrate 160 mg. Data from visits 1 and 2 reflected the basal levels for participants before the fenofibrate treatment, whereas visits 3 and 4 were collected after fenofibrate treatment. For our re-meQTL analysis, we included 429 subjects (male = 221, female = 208) from 140 families with genotype and both pre- (visit 2) and posttreatment (visit 4) methylation data available.
Sample quality control
The methylation of the whole blood was measured by two different probes on the methylation chip, they were adjusted using a beta-mixture quantile normalization (BMIQ) to fit the beta values of Type II design probes of Illumina 450 K into a statistical distribution characteristic of Type I probes [9]. To avoid bias, cytosine-phosphate-guanine (CpG) sites with SNP markers underneath that could be compromised by genotype [10], cross-reactive probes possibly co-hybridizing to alternate genomic sequences [11], and polymorphic probes were removed according to the procedures outlined in Price et al. [10] and Chen et al. [11]. Only subjects with genotype and both post- and pre-treatment methylation data that have complete age, sex, center, and smoking information were included in the analysis (n = 429). The SNP genotypes were first filtered to include only cis SNPs to each CpG site, and then filtered to only include common variants with a minor allele frequency (MAF) > 0.05. The principal components (PCs) of the genotypes were computed using the GENESIS R package to fit PCs on a maximal unrelated subset and then project all samples to those PCs.
Association analysis of identifying cis re-meQTLs
Cis re-meQTL association analysis was done with 208,449 CpG sites, where SNPs within ±1 Mb around the CpG probe were tested in an additive linear mixed-effects model. Log transformed post−/pretreatment methylation values were regressed on SNP genotypes (coded as 0, 1, 2) after adjusting for age, sex, batch effect, smoking status, and first 10 PCs using the lmekin function from the coxme R package with pedigree structure as random effects. For a given CpG probe, the model can be explained as below for a SNP within 1 Mb around the CpG:
in which, Yi is log10 (post/pre) values of cluster/family i; γi represent the estimates of random effects for pedigree structure that is based on known pedigree relationships; β represent the estimates of fixed effects as the following:
The Bonferroni correction was applied to define statistical significance, resulting in p < 5.6 × 10− 10, given a total of 89,217,303 tests. A total of 208,449 CpG sites were tested, so 89,217,303 is the summation of all the SNPs that are within ±1 Mb around each CpG site. The average number of SNPs per CpG site is 428 (89,217,303/208,449). Regional plots of all significant SNPs associated with CpGs were generated using LocusZoom.
Enrichment analysis
Identified cis re-meQTL SNPs (p < 5.6 × 10− 10) from the above association tests were compared with disease−/trait-associated SNPs in the National Human Genome Research Institute–European Bioinformatics Institute (NHGRI-EBI) GWAS Catalog (downloaded on 01/09/2017) [12] by rsID. A binomial test was conducted to test the enrichment of our unique cis re-meQTL SNPs in the GWAS catalog. Regional plots were generated for cis re-meQTL SNPs that were also found to be associated with lipid traits in the GWAS catalog using our re-meQTL results and published lipid GWAS results [1] for comparison using LocusZoom.
Results
CpG-SNP association
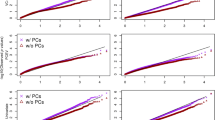
Quantile–quantile (Q-Q) plots (Fig. 1) for SNPs with a MAF > 5% did not show evidence of substantive overdispersion/genomic inflation in our CpG-SNP association test (λ = 1.097), especially considering that SNPs within a ± 1 Mb of each CpG site were analyzed so that some of the correlated SNPs within that region were analyzed more than once. Because sample relatedness was controlled as a random effect in the linear mixed-effects model, a model that has proven to successfully control the genome-wide error rate [12], we optimized for controlling of the relatedness. Population structure also was controlled by adding PCs to the model.

Q-Q plot of cis re-mQTLs, Expected p value of a null distribution was plotted on a negative log scale (black line) and the p values of the re-mQTLs was plotted on a negative log scale (black dots).The 95% confidence interval of the observed p value is shown in gray. The genomic inflation lambda is 1.097
At a p value threshold of 5.6 × 10− 10, 1087 SNPs located in 351 unique gene regions were identified as cis re-meQTLs associated with 610 CpG sites. Among these 1087 cis re-meQTL SNPs, 229 are unique. Figure 2 highlights in red the cis re-meQTL SNPs with 6 loci found in the GWAS catalog. In addition, the most significant cis re-meQTL, rs3733749, is associated with cg00514575 (p = 8.06 × 10− 73) and is upstream of MGAT1, a gene related to TG/lipid accumulation [13], suggesting that a novel genetic variant might regulate a nearby lipid gene, MGAT1, through DNA methylation.

The Manhattan plot of cis re-mQTLs (p < 5.6 × 10− 10) identified in this study with GWAS SNPs reported in the National Human Genome Research Institute (NHGRI) Catalog (highlighted in red)
GWAS catalog look-up and enrichment analysis
Among 1087 cis re-meQTLs, 6 unique SNPs were co-localized at 8 unique disease/trait loci reported in the NHGRI GWAS catalog (Table 1).As reported, a lipid SNP, rs10903129, was associated with total cholesterol (p = 7.0 × 10− 11), and low-density lipoprotein (LDL) cholesterol (p = 2.0 × 10− 19) [1]. As Table 1 shows, SNP rs10903129, located in an intron of gene TMEM57, is a re-meQTL (p = 3.20 × 10− 36) associated with cg09222892, which is upstream of the gene RHCE, indicating a new function target gene for this reported lipid SNP rs10903129. Figure 3a shows the top re-meQTLrs10903129 results in this region, whereas Fig. 3b displays published LDL GWAS results of the same SNP in this region [1, 14]. Among these 1087 cis re-meQTL SNPs, 229 are unique and 6 are also found in the GWAS catalog (enrichment p = 1.51 × 10− 23), indicating that the re-meQTLs identified in our study are highly relevant to human diseases/traits.

Regional plot of SNP rs10903129. a Associated with cg09222892 probe (chr1: 25861512–25,861,513). b Associated with total cholesterol GWAS results in which the SNP is mapped to TMEM57
Co-localization of re-meQTL with GWAS results of fenofibrate effects
Among the GWAS results of fenofibrate effects reported by the GOLDN study, we are interested in whether the cis re-meQTLs from our study could serve as epigenetic evidence of how genetic loci associated with fenofibrate treatment can be explained by the variation of methylation changes, and whether there is methylation change variation relevant to the genetic loci associated with fenofibrate effects on phenotype variation such as the changes of lipid or inflammatory biomarkers. As shown in Fig. 4, we found a SNP (rs12710728) that has a suggestive association with a CpG site (cg17097782, p = 1.77 × 10− 4), which is in high linkage disequilibrium (LD) (R2 > 0.8) with the GWAS-reported SNP rs7443270 for TG-lowering effects of fenofibrate (p = 5.00 × 10− 6) [3].

Regional plot of SNP rs12710728–associated cg17097782 probe (annotation: OSR1)
Discussion
Epigenetic modifications such as DNA methylation can account for phenotypic differences within and between individuals. Specifically, meQTL studies can help dissect regulatory mechanisms underlying GWAS loci. In this short study, we identified thousands of cis re-meQTLs that link to the methylation differences in fenofibrate treatment responses, which could provide genetic and epigenetic determinants for TG level differences between pre- and post-treatment in individuals. As part of our results, we have helped to further validate disease-relevant genes such as MGAT1 [13] and RHCE [15]. In addition, we established a link between meQTL and reported fenofibrate GWAS SNPs via LD. As a result of the coverage of the 450 K Illumina chip, none of the reported fenofibrate GWAS SNPs were found in our genotype data after quality control. However, we did identify a suggestive significant meQTL, rs12710728, which is in high LD with a reported fenofibrate treatment SNP (rs7443270). Our findings provide a unique drug-response–specific meQTL resource for further characterizing the potential functional roles of GWAS SNPs that show response differences to fenofibrate treatment. Finally, as the first response-specific meQTL study of its kind, it will also serve as an important resource for future studies.
Because of the large number of SNPs and CpG sites involved, the computation for meQTL analysis can be quite intensive, especially when looking at both pre- and posttreatment data, or even both cis and trans SNPs. We believe our novel analytic approach to the computation improves on the method used in the Quach et al. study [7], which focused on response-specific expression quantitative trait locus (eQTL). We used the log difference of the pre- and posttreatment states, which can be more efficient than looking at the basal and posttreatment states separately, as done in the Quach et al. study [7]. In the future, we can further validate our approach by optimizing our pipeline and conducting analysis on pre and post states separately, using an interaction model, as well as analyzing both cis and trans re-meQTLs.
Conclusions
In this study, we aim to investigate cis-methylation loci that are responsible for lipid lowering drug fenofibrate treatment. Thousands of cis re-meQTLs that link to the methylation differences were identified, which provide genetic and epigenetic determinants for TG level differences between pre- and post- treatment among individuals. As the first response-specific meQTL study of this kind, these results will serve as an important resource for future studies of the potential functional roles of GWAS SNPs that show response differences to fenofibrate treatment.
References
Teslovich T, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;446(7307):707–13.
Irvin MR, Rotroff DM, Aslibekyan S, Zhi D, Hidalgo B, Motsinger-Reif A, Marvel S, Srinivasasainagendra V, Claas SA, Buse JB, et al. A genome-wide study of lipid response to fenofibrate in Caucasians: a combined analysis of the GOLDN and ACCORD studies. Pharmacogenet Genomics. 2016;26(7):324–33.
Aslibekyan S, Kabagambe EK, Irvin MR, Straka RJ, Borecki IB, Tiwari HK, Tsai MY, Hopkins PN, Shen J, Lai CQ, et al. A genome-wide association study of inflammatory biomarker changes in response to fenofibrate treatment in the genetics of lipid lowering drug and diet network. Pharmacogenet Genomics. 2012;22(3):191–7.
Aslibekyan S, An P, Frazier-Wood A, Kabagambe E, Irvin M, Straka R, Tiwari HK, Tsai MY, Hopkins PN, Borecki IB, et al. Preliminary evidence of genetic determinants of adiponectin response to fenofibrate in the genetics of lipid lowering drugs and diet network. Nutr Metab Cardiovasc Dis. 2013;23(10):987–94.
Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6(5):e1000952.
Chen L, Ge B, Casale FP, Vasquez L, Kwan T, Garrido-Martín D, Watt S, Yan Y, Kundu K, Ecker S, et al. Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell. 2016;167(5):1398–414.
Quach H, Rotival M, Pothlichet J, Loh YE, Dannemann M, Zidane N, Laval G, Patin E, Harmant C, Lopez M, et al. Genetic adaptation and Neandertal admixture shaped the immune system of human populations. Cell. 2016;167(3):643–56.
Irvin MR, Zhi D, Aslibekyan S, Claas SA, Absher DM, Ordovas JM, Tiwari HK, Watkins S, Arnett DK. Genomics of post-prandial lipidomic phenotypes in the genetics of lipid lowering drugs and diet network (GOLDN) study. PLoS One. 2014;9(6):e99509.
Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2012;29(2):189–96.
Price ME, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6(1):4.
Chen Y-A, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–9.
Eu-Ahsunthornwattana J, Miller EN, Fakiola M; Wellcome Trust Case Control consortium 2, Jeronimo SM, Blackwell JM, Cordell HJ: Comparison of methods to account for relatedness in genome-wide association studies with family-based data. PLoS Genet 2014, 10(7): e1004445.
Lee YJ, Ko EH, Kim JE, Kim E, Lee H, Choi H, et al. Nuclear receptor PPARγ -regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc Natl Acad Sci U S A. 2012;109(34):13656–61.
Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, Penninx BW, Janssens AC, Wilson JF, Spector T, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009;41(1):47–55.
Sidore C, Busonero F, Maschio A, Porcu E, Naitza S, Zoledziewska M, Mulas A, Pistis G, Steri M, Danjou F, et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet. 2015;47(11):1272–81.
Acknowledgements
The authors are grateful to the participants of the GOLDN study for the time and specimens they contributed, and we thank the GAW20 for providing the data. The genetic and DNA methylation data used in this study were collected by the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study. Details regarding study recruitment, design, ethic approval and consent to participate have been reported previously (see https://www.gaworkshop.org/data-sets).
Funding
Publication of this article was supported by NIH R01 GM031575. This work was supported by the United States National Institutes of Health, National Institute on Aging (NIH-NIA) through the following grants: U01-AG032984, UF1-AG046198, and R01-AG048927.
Availability of data and materials
The data that support the findings of this study are available from the Genetic Analysis Workshop (GAW), but restrictions apply to the availability of these data, which were used under license for the current study. Qualified researchers may request these data directly from GAW.
About this supplement
This article has been published as part of BMC Proceedings Volume 12 Supplement 9, 2018: Genetic Analysis Workshop 20: envisioning the future of statistical genetics by exploring methods for epigenetic and pharmacogenomic data. The full contents of the supplement are available online at https://bmcproc.biomedcentral.com/articles/supplements/volume-12-supplement-9.
Author information
Authors and Affiliations
Contributions
JW performed the analyses, wrote the manuscript and interpreted the results. DP, JC and CZ assisted with the analysis. SL, VF, and AP planned and coordinated access to all of the genetic and methylation data from the GAW20. LF and XZ supervised the study. XZ designed the study, interpreted the results, and wrote the manuscript. All the authors contributed to the review of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cox, J.W., Patel, D., Chung, J. et al. An efficient analytic approach in genome-wide identification of methylation quantitative trait loci response to fenofibrate treatment. BMC Proc 12 (Suppl 9), 44 (2018). https://doi.org/10.1186/s12919-018-0152-7
Published:
DOI: https://doi.org/10.1186/s12919-018-0152-7