
Abstract
Background
Chickens are major sources of human nutrition worldwide, but the chicken intestinal microbiota can be a source of bacterial infection. The microbiota has potential to regulate the colonization of pathogens by competitive exclusion, production of antimicrobial compounds, and stimulation of the mucosal immune system. But information on the microbiota in commercial broiler chickens is limited because of the difficulty of conducting studies at commercial farms. To obtain fundamental information that can be used to control pathogens in chickens, we determined the 6-week dynamics of microbiota in chicken cecal droppings from commercial broiler farms.
Results
Cecal droppings from four chickens were collected once a week from 1 to 6 weeks of age at three commercial broiler farms. A total of 168 samples were collected from 7 flocks and subjected to 16S rRNA amplicon sequencing. Despite the farms have distinctly different climate conditions, the microbiota in the same growth stages were similar among farms. Moreover, as the chickens grew and the feed types were switched, the richness and diversity of the microbiota gradually increased and convergence of the composition of the microbiota was apparent. Notably, minor bacterial taxa (i.e. OTUs with relative abundance < 0.05%) within the microbiota were changed by the chicken age, switching of feed types, and presence of Campylobacter. In particular, the effects of switching of feed types on the microbiota were larger than the effects of age and Campylobacter.
Conclusions
Irrespective of the locations of the farms, the microbiota of chicken cecum, especially minor bacteria, was successively changed more affected by feed types than by ages. Switching of feed types inducing the alteration of the microbiota may be associated with the colonization of pathogens in the chicken gut. These results will also help with extrapolation of studies in experimental animals to those in the commercial farms.
Similar content being viewed by others


Background
Chickens are common domesticated animal worldwide, and chicken has become the leading meat consumed due to the short lifecycle and high feed conversion ratio [1]. On the other hand, chickens can also be sources of foodborne bacterial pathogens which can disseminate to humans or act as a pool for antimicrobial resistance and transmission [2,3,4]. Since the colonization of major foodborne pathogens, such as Campylobacter and Salmonella enterica serovars Enteritidis and Typhimurium, is often asymptomatic in broilers [5, 6], chickens with foodborne pathogens cannot be distinguished from the others. Therefore, it is necessary to prevent the colonization of foodborne pathogens in the gut to raise the pathogen-free chickens and supply safe chicken.
In recent years, an understanding of the mechanisms through which gut microbiota influence colonization of pathogens has become important for control of diseases for chickens. The chicken gut microbiota plays a key role in preventing invasion of pathogens by competitive exclusion, production of antimicrobial compounds, and stimulation of the mucosal immune system [7, 8]. In the gastrointestinal tract of chickens, the number and variety of bacteria are highest in the cecum, which contains up to 1011 bacteria/g [9, 10], and several studies have examined protection of chickens from pathogens by modification of the cecal microbiota [11,12,13]. However, the colonization of foodborne pathogens is ongoing problems in commercial farms. A major limitation is that most studies have used experimentally reared chickens, rather than commercial chickens, and the experimental models cannot fully mimic the actual conditions of commercial farms. In contrast, it is difficult to control the experimental conditions at farms, as complex environmental factors, such as biosecurity level, house type, and climate, may affect the composition of the chicken intestinal microbiota [14]. Thus, there have been few studies on the intestinal microbiota at commercial farms, with only two reports from one farm [15, 16], and the common factors affecting the cecal microbiota, excluding age, remain unclear.
In this study, we aimed to reveal the factor affecting cecal microbiota of commercial chickens and examined the microbiota of cecal droppings collected at 3 commercial broiler farms focusing on the differences between farms, ages, and feed types. Our findings will contribute to understand the fundamental and comprehensive feature of the cecal microbiota of commercial chickens and design experiments that mimic the conditions at commercial farms.
Results
Richness and diversity of the microbiota
On the basis of 97% sequence identity, a total of 3451 operational taxonomic units (OTUs) were obtained from 168 cecal dropping samples. The OTUs were assigned to 240 bacterial taxa; however, 26.7% (64 taxa) of the OTUs could not be assigned at the genus level. Rarefaction curves reached the saturation phase, and Good’s coverage was > 99% for all samples, which suggested that the reads obtained from the samples represented a sufficient number of sequences for analysis of the microbiota. The number of OTUs in cecal dropping samples significantly increased with the chicken age (Fig. 1a), and was also increased by switching of feed types (Fig. 1a). The Chao1 index differed significantly between all combinations of ages and feed types, except between samples collected in weeks 3 and 4, and weeks 5 and 6 (Fig. 1b). In contrast, the number of OTUs and Chao1 index showed no significant differences between farms (Fig. 1a and b). There were significant increases in the Simpson and Shannon indexes with the chicken age and with switching of feed types (Fig. 1c and d). The Shannon index also differed significantly between samples from farms A and C (Fig. 1d).

Differences in microbial richness and diversity among ages, feed types, and farms. Box plots show the number of OTUs (a), Chao1 index (b), Simpson index (c), and Shannon index (d). *P < 0.05; **P < 0.01; ***P < 0.001
Dominant bacterial taxa in the microbiota
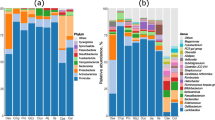
The composition of the microbiota was examined for 6 age categories (1–6 weeks), 3 feed types (starter, grower, and finisher feeds), and 3 farms (farms A, B, and C), giving 12 categories in total. The major bacterial taxa and their proportions in cecal dropping samples were similar regardless of age, feed type, and farm, except that the level of Lachnospiraceae_unclassified significantly decreased in samples from weeks 1 to 2 and from use of starter to grower feeds (Steel-Dwass test, P < 0.001, Fig. 2). The bacterial taxa with > 5% average relative abundance in one or more of the 12 categories were Lachnospiraceae_unclassified (11.8 to 30.7%), Lactobacillus (11.8 to 21.9%), Ruminococcaceae_unclassified (9.7 to 16.3%), Bacteroides (3.1 to 9.5%), Faecalibacterium (1.3 to 9.5%), Clostridiales_unclassified (4.2 to 6.0%), and Streptococcus (0.9 to 5.7%) (Fig. 2). Lachnospiraceae_unclassified, Lactobacillus, and Ruminococcaceae_unclassified were dominant, and these 3 bacterial taxa accounted for almost 40% of the microbiota (Fig. 2).

Genus-level distribution of the microbiota. Bacterial taxa with > 5% relative abundance in one or more of 12 categories (6 for age, 3 for feed type, and 3 for farms) are shown in different colors
Successive changes of the microbiota
Samples were divided into ages (weeks 1–2, 3–4, and 5–6), feed types (starter, grower, and finisher feed), and farms (farms A, B, and C), and non-metric multidimensional scaling (NMDS) was performed for these subgroups (Fig. 3). Using unweighted UniFrac distances, samples at weeks 5–6 were clustered more closely than those at weeks 1–2 and 3–4, resulting in a smaller 95% confidence ellipse for the samples at weeks 5–6 (Fig. 3a). Similarly, samples collected during use of finisher feed were clustered more closely than those collected during use of starter and grower feeds (Fig. 3a). Ellipses for samples at weeks 5–6 and with use of finisher feed almost overlapped with those for samples at weeks 3–4 and with use of grower feed, respectively. In contrast, ellipses for samples collected during use of finisher and starter feeds were separated from each other, despite the overlap of the ellipses for samples from weeks 1–2 and 5–6 (Fig. 3a).

NMDS plots of the microbiota. NMDS of the microbiota based on unweighted (a) and weighted (b) UniFrac distance matrices of OTUs. Circles, squares, and triangles show each sample at farms a, b and c, respectively. The samples are indicated by different colors: red (weeks 1–2, starter feed, and farm a), green (weeks 3–4, grower feed, and farm b), and blue (weeks 5–6, finisher feed, and farm c). A 95% confidence ellipse is shown for each subgroup
Using weighted UniFrac distances, ellipses for samples at weeks 5–6 and with use of finisher feed were inside those for samples at weeks 3–4 and with use of grower feed, respectively (Fig. 3b). Ellipses for samples at weeks 1–2 and with use of starter feed partially overlapped with those for the other 2 categories (Fig. 3b). Ellipses for samples from each farm overlapped with each other in NMDS plots using unweighted and weighted UniFrac distances (Fig. 3a and b).
The compositional variation of the microbiota was measured by beta-dispersion based on unweighted and weighted UniFrac distances. Using unweighted UniFrac distances, the beta-dispersion analysis showed significant differences between samples at weeks 5–6 and those at other ages (Table 1), and between samples with use of finisher feed and other feed types. The compositional variation of the microbiota also differed significantly between samples from farms A and C. In the permutational multivariate analysis of variance (PERMANOVA), the R2 values for the percentage of variation explained by age, feed types and farms were 17.6, 19.7, and 6.47%, respectively (Table 1). With weighted UniFrac distances, the beta dispersion had no significant difference between samples from different ages, but did differ significantly between samples collected during use of grower and finisher feeds, and between samples from farm A and the other farms (Table 1). The R2 values for age, feed types and farms were 8.2, 8.8, and 5.9%, respectively (Table 1).
Abundance of bacteria among ages and feed types
OTUs with significantly different abundance among the subgroups of age and feed types are depicted in Fig. 4. Forty-eight and 190 OTUs were exclusively found in the age and feed type subgroups, respectively (Fig. 4a and Additional files 1 and 2: Tables S1 and S2), and the 48 OTUs involved 3 particular genera (Fig. 4b and Additional file 1: Table S1). In contrast, 10 genera, for example Enterococcus, had significantly different abundance among the subgroups of feed types, but did not vary among the subgroups of age (Fig. 4b and Additional file 2: Table S2). Among these genera, Anaeroplasma and Enterococcus (except for OTU 90) had decreased levels, and 6 genera increased with switching of feed types (Additional file 2: Table S2). There were 31 genera, including Campylobacter, that had significantly different abundances in both subgroups (Fig. 4b), but the relative abundance of the genus Salmonella did not differ in either subgroup. The relative abundance of these 31 genera were shown in Additional file 3: Table S3.

Venn diagrams representing the numbers of significantly different OTUs and bacterial genera. The numbers of significantly different OTUs (a) and bacterial genera (b) among ages or feed types (ANOVA, Bonferroni, P < 0.05). The numbers of minor OTUs (relative abundance in 168 samples < 0.05%) are shown in parentheses
The number of minor OTUs (i.e. OTUs with relative abundance < 0.05%) was 3153 of 3451 OTUs (Additional file 4: Fig. S1), and the total average abundance of the minor OTUs accounted for 18.8% of that of the total OTUs. Among the OTUs with significantly different abundance among ages or feed types, 41 of 48 and 155 of 190, respectively, were minor OTUs (Fig. 4a and Additional files 1 and 2: Tables S1 and S2).
Salmonella and campylobacter in the microbiota
The genus Salmonella was detected in 17 of 168 cecal dropping samples (Additional file 5: Table S4), which was too few to perform statistical analysis. The genus Campylobacter was detected in 21 of 168 samples, and 15 of them were detected at 6 weeks of age (Additional file 6: Table S5). There was no sample which was both positive for Salmonella and Campylobacter. In cecal dropping samples at 6 weeks of age, the relative abundance of 81 OTUs differed significantly by Welch t-test between Campylobacter-positive and -negative samples (Additional file 7: Table S6). Among the 81 OTUs, 64 (79.0%) were minor OTUs (relative abundance < 0.05%) (Additional file 7: Table S6).
Discussion
In this study, we determined the 6-week dynamics of the composition of the microbiota in chicken cecal droppings. At commercial farms, chickens cannot be slaughtered for sampling because they are to be shipped as products. Therefore, cecal droppings were considered to be the best alternative to monitor the cecal microbiota because its diversity, richness and bacterial composition are reported to be similar to the microbiota of cecal contents [17]. To collect samples without disturbing the microbiota and to avoid stress on the chickens, we made the farmers collect cecal droppings. The composition of microbiota was similar with the previous study [17], and Lachnospiraceae_unclassified, Lactobacillus, and Ruminococcaceae_unclassified were dominant in the microbiota (Fig. 2), indicating that no obvious contamination was occurred at the sampling.
The richness and diversity of the microbiota increased with the chicken age (Fig. 1), as reported previously [18,19,20,21]. The richness and diversity were also increased by switching of feed types (Fig. 1), which reflects the effects of age because feed types were switched with growth of the chickens. In contrast, differences in microbial richness and Simpson’s diversity were not observed among farms (Fig. 1). Moreover, the R2 value of PERMANOVA for the farms was lower than those for ages and feed types (Table 1), indicating that the different farms had less effect on microbial variation. On the other hand, the differences of geographic location among the farms were explained by the significant difference in beta dispersion (Table 1). Farm A, which is located in the northern part of Japan, is about 1300 km from farms B and C, which are located in the southern part of Japan. Northern and southern Japan have distinct climates with different ranges of temperature, different amounts of total rain and snow fall, and different levels of humidity. The broiler house types also differed between farm A (windowless type for preparing for cold weather) and the other farms (open-sided type for preparing for hot weather). Previous studies have suggested that chickens of the same age have differences in their composition of cecal microbiota due to geographic location [22, 23], climate [18], and season [24], whereas significant differences caused by feed additives were not found [25]. Our findings indicate only small effects of location and climate of farms on the microbiota, with these effects being less important than those of age or feed types.
The composition of the microbiota converged with increased age in the NMDS plot based on unweighted UniFrac distances (Fig. 3a), which is consistent with previous studies [13, 26]. Moreover, the composition of the microbiota converged with switching of feed types (Fig. 3a). These observations suggest that the microbiota may become stable and show less variation between individual chickens as a consequence of acquisition and replacement of new bacteria through growth. In contrast, using weighted UniFrac distances, a difference in composition of the microbiota was not clear in all samples (Fig. 3b). In these distances, low abundance taxa have a low impact on the total distance metrics [27]. Therefore, weighted UniFrac distances are mainly affected by OTUs comprising the major bacterial taxa whose abundance was similar regardless of age and feed type (Fig. 2). This finding suggests that minor bacterial taxa mainly shifted within the microbiota with increased age and switching of feed types. Consistent with this idea, the microbiota was mostly minor OTUs (Additional file 4: Fig. S1), and the abundance of these OTUs differed significantly among feed types as well as ages (Fig. 4a). Therefore, it is plausible that the composition of the microbiota changes due to acquisition and replacement of minor bacterial taxa throughout the growth of chickens.
This study also showed that chicken age and switching of feed types may have contributed differently to the changes in the composition of the microbiota. Despite some differences in feeding schedules among the farms, switching of feed types was related to increased age. This relationship makes comparisons complex, and thus, the effects of switching of feed types on the microbiota have yet to be completely clear. NMDS plots based on unweighted UniFrac distances suggested that changes in this composition have coincided with switching of feed types, rather than age (Fig. 3a). Furthermore, a Venn diagram showed many significantly different OTUs among feed types (Fig. 4a). Thus, the effects of switching of feed types on the microbiota seemed to be larger than those of age.
The abundance of minor OTUs was changed by the presence of Campylobacter, as shown by a Welch t-test (Additional file 7: Table S6). There was a significantly lower abundance of Lactobacillus and a higher abundance of Streptococcus in Campylobacter-positive samples than in negative samples (Additional file 7: Table S6), as also reported previously [28, 29]. However, most minor OTUs were not characterized, and the correlation between the minor OTUs and colonization of Campylobacter was not clear. Previous evidence suggested that minor bacteria had a beneficial influence on host animals and breeding success [30, 31]; thus, these bacteria should not be ignored due to their influence on host health and their beneficial role in chickens.
The relative abundance of Campylobacter was significantly increased by age and by switching of feed types (Fig. 4b). Several studies have shown an association between age and colonization of Campylobacter [32,33,34]. However, little is known about the effects of switching of feed types, with only one report [20]. Interestingly, the amount of Enterococcus was decreased by switching of feed types (Additional file 2: Table S2). Enterococcus faecium and Enterococcus faecalis cause enterococcal infections in poultry and humans, and acquisition of resistance to antibiotics by these bacteria is a serious problem [35, 36]. In commercial farms, Enterococcus cecorum causes diseases of chickens that lead to economic losses for farmers [37]. Early establishment of a mature gastrointestinal microbiota has been associated with prevention of colonization by some pathogens [38, 39]. Thus, a stable microbiota with high diversity and low variation during use of finisher feed may have decreased the Enterococcus level (Figs. 1 and 3a). Taken together, these results suggest that feed types have a major effect on changes of abundance of specific bacteria, including pathogens, within 50 days of ages. Switching of feed types that induces the alteration of the microbiota which could affect colonization of pathogens in the chicken gut, although other various factors, such as immunity of the chickens, gut health, broiler strains, climatic conditions, and the surrounding environment could also affect the colonization. As the microbiota in a certain growth stage was similar among farms, our results are not limited to this study and may represent the general features of the microbiota in chicken cecal droppings.
Conclusions
We obtained fundamental information on the microbiota in chicken cecal dropping from commercial farms. The composition of the microbiota seemed to change due to acquisition and replacement of minor bacterial taxa with increasing chicken age, switching of feed types, and presence of Campylobacter. The composition of minor bacteria may have affected colonization of pathogens in the gut, therefore, understanding the role of minor bacteria is considered important. Especially, switching of feed types may have a major impact on changes of the composition of minor bacteria in the microbiota. Therefore, replacement of feed types throughout the growth of chickens may be an important factor for mimicking commercial broiler chickens under experimental conditions. Furthermore, it may be better to start administration of probiotics during use of starter feed before the chicken cecal microbiota becomes diverse and stable. Further studies are needed to identify the effects of switching of feed types on the chicken cecal microbiota for utilizing the feeds to prevent colonization by pathogens.
Methods
Animals and sample collection
From October 2016 to January 2018, cecal dropping samples from 4 clinically healthy broiler chickens (Chunky breed) per flock were collected by farmers once a week from 1 to 6 weeks of age. Four cecal droppings in each sampling was the maximum number that the farmers could collect. The cecal droppings were collected randomly from 2 flocks at farms A and B, and from 3 flocks at farm C (Additional file 8: Fig. S2), and a total of 168 samples were obtained. Farm A is located in Tohoku, in the north of Japan’s main island, and farms B and C are located in Kyushu, the southern island of Japan. The area of chicken houses and the approximate number of chickens per flock were as follows: farm A, 463 m2 and 10,000 chickens; farm B, 660 m2 and 9000 chickens; farm C, 1040 m2 and 17,000 chickens. The type of broiler house was windowless in farm A, and open-sided in farms B and C. Each owner gave consent for collection of samples. At the 3 farms, all chickens were reared for 50 days until shipment, except that parts of 2 flocks at farm A were shipped to another client at 33 days of age (Additional file 8: Fig. S2). After all chickens were shipped, the houses were cleaned and disinfected, and the litter between the flocks within the same house was changed at the 3 farms.
Poultry feeds were categorized into 3 types: starter, grower, and finisher feeds. These were fed to chickens depending on growth, and the ages of switching of feeds varied among the farms (Additional file 8: Fig. S2). According to information from the farms, the amount of protein in chicken feeds decreased in the order of starter, grower, and finisher feeds, whereas the amount of lipids in the feeds increased in the same order. The detailed information on nutrition contained in the feeds was confidential.
The cecal droppings were collected by the same farmers at each farm, based on our instructions for sampling. For collection of cecal droppings, the farmers did not touch the animals and simply picked up the cecal droppings. The Institutional Animal Care and Use Committee of the Graduate School of Agriculture and Life Sciences, the University of Tokyo, confirmed that the study did not require approval. Each cecal dropping was immediately collected using a sterilized 150-mm polypropylene spatula (As One, Osaka, Japan) and immersed in 500 μl of RNAlater Stabilization Solution (Thermo Fisher Scientific, Waltham, MA) in a conical tube to prevent bacterial growth and degradation of DNA. Cecal dropping samples were transported to our laboratory under refrigeration within 2 days and stored at − 20 °C until use.
Extraction of DNA from cecal dropping samples
A total of 168 samples were used for DNA extraction. Frozen samples were thawed and centrifuged at 13,000×g for 5 min at 4 °C. The pellet in each tube was washed twice with sterile 0.85% saline [40] and total DNA was extracted using a PowerFecal® DNA Isolation kit (Qiagen, Hilden, Germany). To increase the extraction efficiency, 400 μl of 0.5-mm diameter zirconia beads (Toray, Tokyo, Japan) and two 5-mm diameter zirconia beads (Toray) were used instead of the beads in the kit [40], with a μT-12 bead crusher (Taitec, Saitama, Japan). Total DNA was eluted in 100 μl of the elution buffer in the kit and stored at − 20 °C until use. The DNA concentration was measured with a Quantus™ Fluorometer with a QuantiFluor® dsDNA System (Promega, Madison, WI).
16S rRNA gene amplicon sequencing
The V3-V4 regions of 16S rRNA genes in the extracted DNA were amplified with S-D-Bact-0341-b-S-17 (5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG-3′), and S-D-Bact-0785-a-A-21 (5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATC C-3′) primers [41], include the Illumina overhang adapter sequence (Illumina, San Diego, CA). Prior to PCR amplification, the amount of total bacterial genome DNA contained in the extracted DNA was estimated by quantitative real-time PCR [40]. The 25 μl reaction volume in the PCR contained 12.5 μl of 2× KAPA HiFi HotStart ReadyMix DNA polymerase (Kapa Biosciences, Woburn, MA), 0.2 μM of forward and reverse primers, and 12.5 ng of DNA template (amount of total bacterial DNA). The PCR conditions were as follows: 95 °C for 3 min; 25 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s; and a final extension step at 72 °C for 5 min. The PCR amplification and the quality of the PCR products were checked using an Agilent 2100 Bioanalyzer (Agilent Technologies Japan, Tokyo, Japan). The products were purified with Agencourt AMpure XP beads (Beckman Coulter Inc., Brea, CA). Dual indexes and sequencing adapters were attached by subsequent PCR using a Nextera XT Index Kit (Illumina). The indexed products were further purified using AMpure XP beads (Beckman Coulter Inc.) and checked with the Bioanalyzer (Agilent Technologies Japan). The prepared libraries were quantified by qPCR with a Library Quantification Kit for Illumina (Kapa Biosciences). Libraries of equimolar DNA molecules were pooled and diluted in hybridization buffer. Sequencing was performed using the 2 × 300 bp paired-end method on the MiSeq platform with a MiSeq v3 Reagent Kit (Illumina) and a 50% phiX spike.
Sequence data processing and taxonomy assignment
Fastq reads were processed using the analysis pipeline Illinois Mayo Taxon Organization from RNA Dataset Operations ver. 2.0.3.2 [42] with default parameters, except for the following parameters in Trimmomatic: LEADING:20, TRAILING:20, and MINLEN:180. Trimmomatic was used to trim low-quality sequences. For ensuring use of high-quality reads when assigning OTU representation, singletons and chimeric reads were discarded in the pipeline. Mothur [43] was used for OTU clustering at 100% sequence identity, and the OTUs were sorted by cluster size and processed in USEARCH using the UPARSE algorithm to detect OTU representatives using a de novo OTU picking strategy. OTUs were picked and assigned using the Ribosomal Database Project (RDP) naive Bayesian classifier [44] at 97% similarity against the RDP database (v.11; Ribosomal Database Project, Michigan State University, East Lansing, MI) [45]. The number of sequences was normalized to 25,000 for each sample with the core_qiime_analyses.py script from Quantitative Insights Into Microbial Ecology (QIIME) ver. 1.9.1 [46] and used in the following analyses; this means that the detection limit was relative abundance of 0.004% in each sample. In addition, a sample containing at least one sequence classified as Salmonella or Campylobacter was considered positive for these bacteria.
Statistical analysis
Indices of Chao1, Simpson, Shannon, and Good’s coverage of samples were calculated and rarefaction curves were constructed using QIIME. Significant differences (P < 0.05) of the alpha-diversity indices between samples were examined by a nonparametric multiple comparison test (Steel-Dwass) using the pSDCFlig function in the NSM3 package in R [47].
From the downstream analysis, the sequencing data from cecal dropping samples were divided into subgroups by age of chickens (weeks 1–2, 3–4 and 5–6), feed types (starter, grower, and finisher feed) or farms (farms A, B, and C). The unweighted and weighted UniFrac distance [48] for each subgroup was calculated from the normalized composition of microbiota in cecal dropping samples using QIIME. NMDS based on the obtained distance matrix was conducted using the vegan package in R, and visualized with 95% confidence ellipses. The ellipses were drawn using ggplot2 stat_ellipse (type = “norm”) function, and represent the 95% confidence interval of the multivariate t-distribution.
Beta-dispersions measuring the compositional variation of the microbiota among subgroups were analyzed using the vegan package in R. The average distance of each sample to the centroid of the subgroup in multivariate space was calculated based on unweighted and weighted UniFrac distances. A Tukey honestly significant difference test was then conducted for pairwise comparisons of mean dispersions among subgroups. Differences in bacterial communities among the subgroups were evaluated with PERMANOVA with 9999 random permutations using the vegan package.
To identify significant differences in OTU abundances among ages and feed types (Bonferroni P < 0.05), an analysis of variance (ANOVA) was conducted using QIIME, and Venn diagrams were generated to compare the OTUs between ages and feed types. OTUs with an average relative abundance in 168 samples < 0.05% were defined as minor OTUs, and the frequency distribution was created for the number of OTUs.
A Welch t-test was performed to test the significance of differences between samples with and without Campylobacter (P < 0.05).
Availability of data and materials
Datasets generated by 16S rRNA gene amplicon sequencing in this study have been deposited in the DNA Data Bank of Japan under accession number DRA007956.
Abbreviations
- ANOVA:
-
analysis of variance
- NMDS:
-
non-metric multidimensional scaling
- OTU:
-
operational taxonomic unit
- PERMANOVA:
-
permutational multivariate analysis of variance
- QIIME:
-
Quantitative Insights Into Microbial Ecology
- RDP:
-
Ribosomal Database Project
References
Food and agriculture organization of the united nations. http://www.fao.org/livestock-systems/production-systems/chicken/en. Accessed 14 Jan 2020.
Zhou W, Wang Y, Lin J. Functional cloning and characterization of antibiotic resistance: genes from the chicken gut microbiome. Appl Environ Microbiol. 2012;78:3028–32.
Hegde NV, Kariyawasam S, DebRoy C. Comparison of antimicrobial resistant genes in chicken gut microbiome grown on organic and conventional diet. Vet Anim Sci. 2016;1:9–14.
Shang Y, Kumar S, Oakley B, Kim WK. Chicken gut microbiota: importance and detection technology. Front Vet Sci. 2018;5:254.
Doyle MP, Erickson MC. Reducing the carriage of foodborne pathogens in livestock and poultry. Poult Sci. 2006;85:960–73.
Kogut MH, Arsenault RJ. Immunometabolic phenotype alterations associated with the induction of disease tolerance and persistent asymptomatic infection of Salmonella in the chicken intestine. Front Immunol. 2017;8:372.
Oakley BB, Lillehoj HS, Kogut MH, Kim WK, Maurer JJ, Pedroso A, Lee MD, Collett SR, Johnson TJ, Cox NA. The chicken gastrointestinal microbiome. FEMS Microbiol Lett. 2014;360:100–12.
Clavijo V, Flórez MJV. The gastrointestinal microbiome and its association with the control of pathogens in broiler chicken production: a review. Poult Sci. 2018;97:1006–21.
Bjerrum L, Engberg RM, Leser TD, Jensen BB, Finster K, Pedersen K. Microbial community composition of the ileum and cecum of broiler chickens as revealed by molecular and culture-based techniques. Poult Sci. 2006;85:1151–64.
Yeoman CJ, Chia N, Jeraldo P, Sipos M, Goldenfeld ND, White BA. The microbiome of the chicken gastrointestinal tract. Anim Health Res Rev. 2012;13:89–99.
Nishiyama K, Seto Y, Yoshioka K, Kakuda T, Takai S, Yamamoto Y, Mukai T. Lactobacillus gasseri SBT2055 reduces infection by and colonization of Campylobacter jejuni. PLoS One. 2014;9:e108827.
Wang L, Li L, Lv Y, Chen Q, Feng J, Zhao X. Lactobacillus plantarum restores intestinal permeability disrupted by Salmonella infection in newly-hatched chicks. Sci Rep. 2018;8:2229.
Thomas M, Wongkuna S, Ghimire S, Kumar R, Antony L, Doerner KC, Singery A, Nelson E, Woyengo T, Chankhamhaengdecha S, Janvilisri T, Scaria J. Gut Microbial Dynamics during Conventionalization of Germfree Chicken. mSphere. 2019;4:1–12.
Kers JG, Velkers FC, Fischer EAJ, Hermes GDA, Stegeman JA, Smidt H. Host and environmental factors affecting the intestinal microbiota in chickens. Front Microbiol. 2018;9:235.
Park SH, Lee SI, Kim SA, Christensen K, Ricke SC. Comparison of antibiotic supplementation versus a yeast-based prebiotic on the cecal microbiome of commercial broilers. PLoS One. 2017;12:e0182805.
Johnson TJ, Youmans BP, Noll S, Cardona C, Evans NP, Karnezos TP, Ngunjiri JM, Abundo MC, Lee C-W. A consistent and predictable commercial broiler chicken bacterial microbiota in antibiotic-free production displays strong correlations with performance. Appl Environ Microbiol. 2018;84:e00362–18.
Pauwels J, Taminiau B, Janssens GPJ, De Beenhouwer M, Delhalle L, Daube G, Coopman F. Cecal drop reflects the chickens’ cecal microbiome, fecal drop does not. J Microbiol Methods. 2015;117:164–70.
Awad WA, Mann E, Dzieciol M, Hess C, Schmitz-Esser S, Wagner M, Hess M. Age-related differences in the luminal and mucosa-associated gut microbiome of broiler chickens and shifts associated with Campylobacter jejuni infection. Front Cell Infect Microbiol. 2016;6:154.
Danzeisen JL, Kim HB, Isaacson RE, Tu ZJ, Johnson TJ. Modulations of the chicken cecal microbiome and metagenome in response to anticoccidial and growth promoter treatment. PLoS One. 2011;6:e27949.
Ijaz UZ, Sivaloganathan L, McKenna A, Richmond A, Kelly C, Linton M, Stratakos AC, Lavery U, Elmi A, Wren BW, Dorrell N, Corcionivoschi N, Gundogdu O. Comprehensive longitudinal microbiome analysis of the chicken cecum reveals a ahift from competitive to environmental drivers and a window of opportunity for Campylobacter. Front Microbiol. 2018;9:2452.
Ocejo M, Oporto B, Hurtado A. 16S rRNA amplicon sequencing characterization of caecal microbiome composition of broilers and free-range slow-growing chickens throughout their productive lifespan. Sci Rep. 2019;9:2506.
Zhou X, Jiang X, Yang C, Ma B, Lei C, Xu C, Zhang A, Yang X, Xiong Q, Zhang P, Men S, Xiang R, Wang H. Cecal microbiota of Tibetan chickens from five geographic regions were determined by 16S rRNA sequencing. Microbiologyopen. 2016;5:753–62.
Pandit RJ, Hinsu AT, Patel NV, Koringa PG, Jakhesara SJ, Thakkar JR, Shah TM, Limon G, Psifidi A, Guitian J, Hume DA, Tomley FM, Rank DN, Raman M, Tirumurugaan KG, Blake DP, Joshi CG. Microbial diversity and community composition of caecal microbiota in commercial and indigenous Indian chickens determined using 16s rDNA amplicon sequencing. Microbiome. 2018;6:1–13.
Oakley BB, Vasconcelos EJR, Diniz PPVP, Calloway KN, Richardson E, Meinersmann RJ, Cox NA, Berrang ME. The cecal microbiome of commercial broiler chickens varies significantly by season. Poult Sci. 2018;97:3635–44.
Oakley BB, Buhr RJ, Ritz CW, Kiepper BH, Berrang ME, Seal BS, Cox NA. Successional changes in the chicken cecal microbiome during 42 days of growth are independent of organic acid feed additives. BMC Vet Res. 2014;10:1–8.
Lu J, Idris U, Harmon B, Hofacre C, Maurer JJ, Lee MD. Diversity and succession of the intestinal bacterial community of the maturing broiler chicken. Appl Environ Microbiol. 2003;69:6816–24.
Wong RG, Wu JR, Gloor GB. Expanding the UniFrac toolbox. PLoS One. 2016;11:e0161196.
Dicksved J, Ellström P, Engstrand L, Rautelin H. Susceptibility to Campylobacter infection is associated with the species composition of the human fecal microbiota. MBio. 2014;5:1–7.
Kaakoush NO, Sodhi N, Chenu JW, Cox JM, Riordan SM, Mitchell HM. The interplay between Campylobacter and Helicobacter species and other gastrointestinal microbiota of commercial broiler chickens. Gut Pathog. 2014;6:18.
Troussellier M, Escalas A, Bouvier T, Mouillot D. Sustaining rare marine microorganisms: macroorganisms as repositories and dispersal agents of microbial diversity. Front Microbiol. 2017;8:1–17.
Antwis RE, Edwards KL, Unwin B, Walker SL, Shultz S. Rare gut microbiota associated with breeding success, hormone metabolites and ovarian cycle phase in the critically endangered eastern black rhino. Microbiome. 2019;7:1–12.
Gregory E, Barnhart H, Dreesen DW, Stern NJ, Corn JL. Epidemiological study of Campylobacter spp. in broilers: source, time of colonization, and prevalence. Avian Dis. 1997;41:890–8.
Newell DG, Fearnley C. Sources of Campylobacter colonization in broiler chickens. Appl Environ Microbiol. 2003;69:4343–51.
Han Z, Pielsticker C, Gerzova L, Rychlik I, Rautenschlein S. The influence of age on Campylobacter jejuni infection in chicken. Dev Comp Immunol. 2016;62:58–71.
Giraffa G, Carminati D, Neviani E. Enterococci isolated from dairy products: a review of risks and potential technological use. J Food Prot. 1997;60:732–8.
Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10:266–78.
Jung A, Chen LR, Suyemoto MM, Barnes HJ, Borst LB. A review of Enterococcus cecorum infection in poultry. Avian Dis. 2018;62:261–71.
Nurmi E, Rantala M. New aspects of Salmonella infection in broiler production. Nature. 1973;241:210–1.
Crhanova M, Hradecka H, Faldynova M, Matulova M, Havlickova H, Sisak F, Rychlik I. Immune response of chicken gut to aatural colonization by gut microflora and to Salmonella enterica serovar Enteritidis infection. Infect Immun. 2011;79:2755–63.
Arai S, Kim H, Watanabe T, Tohya M, Suzuki E, Ishida-Kuroki K, Maruyama F, Murase K, Nakagawa I, Sekizaki T. Assessment of pig saliva as a Streptococcus suis reservoir and potential source of infection on farms by use of a novel quantitative polymerase chain reaction assay. Am J Vet Res. 2018;79:941–8.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1.
Jeraldo P, Kalari K, Chen X, Bhavsar J, Mangalam A, White B, Nelson H, Kocher J-P, Chia N. IM-TORNADO: a tool for comparison of 16S reads from paired-end libraries. PLoS One. 2014;9:e114804.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–41.
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:141–5.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, Mcdonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314.
Lozupone CA, Knight R. UniFrac : a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–35.
Acknowledgements
We acknowledge the assistance of Teruaki Oshima (Japanese Chicken Association, Tokyo, Japan) and the poultry farmers involved in this study. We are grateful to Masashi Okamura (Kitasato University School of Veterinary Medicine, Aomori, Japan) for valuable comments; to Sakura Arai (National Institute of Health Sciences, Kanagawa, Japan) for helpful advice; and to Kazuyuki Ishihara (Tokyo Dental College, Tokyo, Japan) for providing the MiSeq platform. Supercomputing resources were provided by the Human Genome Center at the Institute of Medical Science (The University of Tokyo; http://sc.hgc.jp/shirokane.html).
Funding
This study was supported by the Japan Society for the Promotion of Science (KAKENHI grant 16 K08015) and a Livestock Promotional Subsidy from the Japan Racing Association. They had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
NT performed the experiments. NT, TW, and KI-K analyzed the data. NT wrote the manuscript with support from TS, TW, and KI-K. TS conceived of the presented idea and supervised the findings of this work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
For collection of cecal droppings, each owner gave consent for collection of samples, and the samples were collected by farmers. The farmers did not touch the animals and simply picked up the cecal droppings. The Institutional Animal Care and Use Committee of the Graduate School of Agriculture and Life Sciences, the University of Tokyo, confirmed that the study did not require approval.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
OTUs which relative abundances were significantly different among the subgroups of age but not among the subgroups of feed type.
Additional file 2.
OTUs which relative abundances were significantly different among the subgroups of feed type but not among the subgroups of age.
Additional file 3.
Bacterial genera which relative abundances were significantly different both among the subgroups of age and feed type.
Additional file 4: Fig. S1.
Frequency distribution of the relative abundance of OTUs. Minor OTUs (relative abundance in 168 samples< 0.05%) are shown in orange. Major OTUs (relative abundance in 168 samples≥0.05%) are shown in blue.
Additional file 5
Prevalence of Salmonella in broiler chickens.
Additional file 6
Prevalence of Campylobacter in broiler chickens.
Additional file 7
OTUs which relative abundances were significantly different between Campylobacter-positive and -negative samples.
Additional file 8: Fig. S2.
Time points of sampling and feeding schedules. Triangles (▼) and squares (■) show time points of sampling and thinning, respectively. Feed types are indicated by different colors: red (starter feed), green (grower feed), and blue (finisher feed), and are separated by circles (●).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Takeshita, N., Watanabe, T., Ishida-Kuroki, K. et al. Transition of microbiota in chicken cecal droppings from commercial broiler farms. BMC Vet Res 17, 10 (2021). https://doi.org/10.1186/s12917-020-02688-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-020-02688-7