
Abstract
Background
Buprenorphine is one of the most used analgesics for postoperative pain in rabbits. The recommended dose in rabbits (0.01–0.05 mg/kg) is the same for intravenous (IV), intramuscular (IM), and subcutaneous (SC) administration, despite lack of pharmacokinetic data. Five male and five female New Zealand White rabbits (mean ± SD body weight 3.1 ± 0.3 kg) were administered 0.05 mg/kg buprenorphine by the IV, IM and SC routes and 0.1 mg/kg by the SC route, in a cross-over design with two-week wash-out periods between treatments. Blood was collected before, and up to 8 h post buprenorphine injection, for determination of serum levels by UPHLC-MS/MS.
Results
The area under the time concentration curve (AUC0-t) was lower after SC (398 ± 155 ng/mL/min) than IM (696 ± 168 ng/mL/min, p < 0.001) and IV (789 ± 189 ng/mL/min, p < 0.001) administration. The maximum serum concentration was lower after SC (2.2 ± 1.4 ng/mL) than after IM (11 ± 3.2 ng/mL) administration (p < 0.001). The bioavailability was lower after SC (50 ± 19%) than after IM (95 ± 21%) administration (p = 0.006). The elimination half-life was longer after SC (260 ± 120 min) than after IM (148 ± 26 min, p = 0.002) as well as IV (139 ± 33 min) injection (p < 0.001). An increase in the SC dose from 0.05 to 0.1 mg/kg resulted in an increase in the area under the time concentration curve of 50% in female (p = 0.022) and 165% in male rabbits (p < 0.001). The bioavailability did not change in the females (36 ± 14%, p = 0.6), whereas it increased in the males (71 ± 23%, p = 0.008).
Conclusions
The lower bioavailability of 0.05 mg/kg buprenorphine after SC administration could explain the lack of efficacy seen in clinical pain studies in rabbits, using this route. For immediate pain relief, IV or IM administration is therefore be recommended, whereas SC administration may be useful to sustain analgesic serum levels, once efficient pain relief has been achieved. The current data do not support an increase in dose to compensate for the lower SC bioavailability.
Similar content being viewed by others
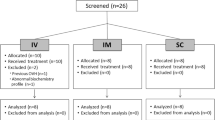


Background
The rabbit is a common pet animal and a common animal model for preclinical studies. In 2016, 350,000 rabbits were used for experimental purposes in the EU [1]. Conditions requiring potent analgesic treatment in rabbits include gastrointestinal disorders and orthopaedic post-operative pain [2, 3]. Buprenorphine is a potent and long acting opioid drug, and the most used analgesic in laboratory rabbits [4]. It is an agonist at the μ-opioid receptor, with approximately 30–40 times the potency, and twice the affinity of morphine [5]. The recommendation for buprenorphine doses in rabbits are the same regardless of administration route; 0.01–0.05 mg/kg IV, SC and IM [2, 6, 7]. There are however few pharmacokinetic studies in rabbits [8,9,10,11] and none on the bioavailability after SC or IM administration. Likewise, there are few published studies on the effect of buprenorphine on clinical pain in rabbits. These however indicate that SC administration of recommended doses lack efficacy [11,12,13], whereas IM and IV administration are reported to have effect [14, 15]. Further, plasma concentrations vary markedly after SC administration of buprenorphine in female New Zealand White (NZW) rabbits [11]. From studies in cats it is known that the bioavailability of recommended doses of SC administered buprenorphine is low [16], and that postoperative pain scores are higher after SC than IM or IV administration of the same dose [17]. The SC dose may result in an inadequate concentration gradient for the buprenorphine to leave the SC tissues, resulting in a smaller than predicted exposure [18].
Pharmacokinetic (PK) data is important for choosing doses and routes of drugs. The aim of the present study was to establish the bioavailability of SC and IM administered buprenorphine in rabbits, and if necessary, revise the current recommendations of administration.
The study was designed to test the hypotheses that a recommended dose of buprenorphine leads to: 1) a lower total exposure over time, as measured by the area under the concentration curve (AUC); 2) lower maximum serum levels (Cmax) after SC than after IM administration and that; 3) a higher dose can compensate for the lower AUC and Cmax of SC administration.
Results
All rabbits were clinically healthy and easy to handle. Hematological data were within normal ranges.
Pharmacokinetics
Individual data on buprenorphine serum concentration over time are shown in Fig. 1a-d. The data from rabbit #6 after 0.05 mg/kg IM and rabbit # 3 after 0.05 mg/kg SC were significantly different from the others and therefore excluded from the data analysis. Buprenorphine mean resident time (MRT) was 180 min ± 54 min, volume of distribution at steady state (Vdss) was 10.2 ± 3.2 L/kg and clearance (CL) 38 ± 11 mL/kg/min.

a-d: Buprenorphine serum concentrations over time in 10 rabbits after administration of 0.05 mg/kg buprenorphine a) by the IV route, b) by the IM route and c) by the SC route and ) after administration of 0.1 mg/kg by the SC route
Comparison of administration routes (0.05 mg/kg)
See Table 1 and Fig. 2. The administration route had an overall effect on the AUC0-t (p < 0.001), the AUC0-∞ (p = 0.003), Cmax (p < 0.001), bioavailability (F, p = 0.006) and elimination half-life (t½, p < 0.001), but not on tmax (p = 0.379). The AUC0-t and AUC0-∞ were higher in females than males (p = 0.0495 and p = 0.02, respectively): 20% after SC and IM, and 40% higher after IV administration. There was an overall effect of treatment day on Cmax (p = 0.013) and of sex on t½ (p = 0.041). There was no consistent effect of treatment day across the responses.

Semilogarithmic graph of buprenorphine serum concentrations in 10 NZW rabbits after administration of 0.05 mg/kg by the IV, IM and SC routes (mean ± SD)
The AUC0-t and AUC0-∞ were lower after SC than IM (p < 0.001 and p = 0.03, respectively) and IV (p < 0.001 and p = 0.001, respectively) administration (Fig. 3) and Cmax was lower after SC than IM administration (p < 0.001), as was F (p = 0.006 Fig. 4). The t½ was longer in females (p = 0.041) and longer in SC than IM injection (p = 0.002) or IV injection (p < 0.001).

Semilogarithmic graph of buprenorphine serum concentrations in 10 NZW rabbits after administration of 0.05 and 0.1 mg/kg by the subcutaneous (SC) route (mean ± SD)

Boxplot of buprenorphine area under the time concentration curves (AUC0-t and AUC0-∞) in 10 NZW rabbits after administration of 0.05 mg/kg by the IV, IM and SC routes. Data displayed as minimum, first quartile, median, mean (X), third quartile, and maximum. (***p < 0.001, two-way ANOVA with route and sex as independent factors and Animal and treatment day as blocking factors This was followed by planned comparisons on the predicted means to compare the IV and IM routes back to SC)
Comparison of SC doses (0.05 and 0.1 mg/kg)
See Table 2 and Fig. 5. AUC0-t and AUC0-∞ were higher after administration of 0.1 than 0.05 mg/kg SC in females (p = 0.022 and p < 0.001, respectively) and males (p < 0.001). Cmax was higher after administration of 0.1 than 0.05 mg/kg SC in males (p < 0.001), but not in females (p = 0.150), as was F (p = 0.008 and p = 0.601, respectively). T½ did not differ between 0.1 and 0.05 mg/kg SC in males (p = 0.075) or females (p = 0.222). Tmax was longer after administration of 0.05 than 0.1 mg/kg SC in males (p = 0.017) but not females (p = 0.871).

Boxplot of buprenorphine bioavailability (F) in 10 NZW rabbits after administration of 0.05 mg/kg by the IM and SC routes. Data displayed as minimum, first quartile, median, mean (X), third quartile, and maximum. (**p < 0.01, two-way ANOVA with route and sex as independent factors and animal and treatment day as blocking factors This was followed by planned comparisons on the predicted means to compare the IV and IM routes back to SC)
There was no evidence of an effect of bodyweight on any of the pharmacokinetic parameters, or a consistent effect of treatment day.
Discussion
Analgesia protocols in rabbits are limited compared with those for cats and dogs, and poor management of pain may be one reason for the high mortality found postoperatively in pet rabbits [19]. There is also a lack of pharmacokinetic data for several analgesic drugs used in rabbits. The current study was aimed at establishing the bioavailability of SC and IM administered buprenorphine in rabbits, because of evidence of poor clinical efficacy after SC administration [11,12,13]. The results show that the Cmax and the bioavailability of a 0.05 mg/kg buprenorphine dose was markedly lower after SC than IM administration. The high bioavailability after IM injection is in contrast with other lipophilic drugs in aqueous solutions, which are poorly and erratically absorbed after both SC and IM administration [20]. It is also in contrast with findings in cats, which show an IM bioavailability of 46% of a recommended dose [16]. If drug absorption is slow or irregular, the peak concentrations will be lower and can occur later, as shown in the current study. Tmax (39 ± 60 min) was long and varied widely between rabbits after SC administration of 0.05 mg/kg. After IM injection, tmax was much shorter and less variable (11 ± 5 min). The amount of subcutaneous fat will influence the absorption rate because fatty tissue is less vascularized [20].
The difference in absorption rate may also affect the t½, as the drug elimination rate is limited by the drug absorption rate. In the present study the t½ was significantly longer after SC than IV and IM injection (p < 0.001). Further, the elimination phases of the IV and SC administration routes are not parallel (Fig. 2). This can be an effect of a delay in SC absorption, which in turn prolongs the elimination phase, a so-called flip-flop phenomenon. The prolonged t½ can extend the duration of action of a drug, but the terminal exponential phase is usually reached only when plasma drug levels are sub-therapeutic. For this reason, the half-life corresponding to the terminal exponential phase cannot always be used in selecting an appropriate dosing interval. The longest elimination half-life in the current study was 717 min in a female rabbit given the high SC dose. In these cases, the absorption/distribution will play a significant role in the terminal phase of the pharmacokinetic profile and thus explains the over 20% difference in AUC0-t compared to AUC0-∞ for the females after SC 0.05 and 0.1 mg/kg. For a more precise calculation of AUC0-∞, blood samples at later time points would have been necessary after SC administration.
From previous studies in rabbits, an IV dose six times higher than in the present study (0.3 mg/kg) gave rise to a plasma concentration of > 6 ng/mL for 4 h, with a t½ of 233 min [10]. The IV t½ in the current study was shorter (160 min). Linhardt et al. [21] administered a three times higher dose (0.15 mg/kg) IV, resulting in an initial concentration of 90 ng/mL at t = 2 min. This is comparable to the 2 min sample in the present study, which was approximately one-third (32 ng/mL). Park et al. [5] administered 0.1 mg/kg SC in 3 male NZW rabbits of unknown age, resulting in Cmax of 17.5 ng/mL, tmax at 15 min and an AUC0-t of 1740 ng/mL/min. In the present study Cmax was 7 ng/mL, tmax 12 min and the AUC0-t 633 ng/mL/min.
With the increase of the SC dose to 0.1 mg/kg, the Cmax and bioavailability increased in the male, but not the female rabbits. The increase in the males could be explained by a higher concentration gradient for the buprenorphine to leave the SC tissues, resulting in a higher-than-predicted Cmax and AUC0-t [18]. Why this was not the case in the females, is not clear. A possible explanation could be that females have a larger amount of less vascularized subcutaneous fat, which reduced the absorption rate.
Studies in cats show that the bioavailability of SC administered buprenorphine in recommended doses is very low, individual variability high, and clinical efficacy poor [16, 17, 22]. Cats receiving buprenorphine SC after ovariohysterectomy have higher pain scores than those receiving IM or IV administration, which has led to the recommendation to use IM or IV administration in the acute setting [17]. Recently however, an aqueous high concentration formulation of buprenorphine has been licensed for SC administration in cats in the USA (1.8 mg/mL, Simbadol, Zoetis, Pasippany, NJ). The SC dose is 10 times higher than previously recommended in cats [18, 23], and provides 24 h of antinociception in thermal threshold tests [24]. So far, there are no published data on the clinical efficacy of this formulation.
In line with the published data in cats, the results of the current study may explain why Weaver et al. [12] and Goldschlager et al. [25] did not find an effect on pain related parameters post ovariohysterectomy after SC administration of 0.02–0.03 mg/kg buprenorphine, whereas Cooper et al. [15] did after IM administration of 0.03 mg/kg. Parameters studied were feed consumption, body weight gain, fecal corticosteroids, locomotion and rearing. Likewise, no effect on facial pain scores (eye closure and ear position) were detected after SC administration of 0.05 mg/kg buprenorphine in a study of postoperative orthopedic pain [12]. There may be other explanations why an effect could not be detected, such as a low sensitivity of the assessment method.
Initial high serum levels of buprenorphine will more likely result in effective analgesia. This can be achieved by IV and IM injections. The onset of analgesia is slow even after IV administration (15–30 min), as shown in rabbits in nociceptive tests [26] and peak effect occurs even later (60–120 min). Buprenorphine rapidly crosses the blood brain barrier, but is slow in binding to the μ-opioid receptor [27]. The benefits of buprenorphine are its high potency and affinity to the μ-opioid receptor [5]. Buprenorphine’s potency and slow dissociation from opioid receptors result in low therapeutic doses in animals and humans with concomitantly low plasma concentrations. Buprenorphine has a biphasic dissociation; a rapid 1st-phase dissociation of approximately 50% of μ-receptor bound drug followed by a slow 2nd-phase dissociation. The fact that analgesia of buprenorphine exceeds its pharmacokinetic half-life is believed to be partly due to this 2nd slow dissociation [28].
Even if there is no direct correlation between plasma levels of buprenorphine and degree of analgesia (hysteresis effect), and variability in analgesic effect between individual animals, it is vital to know the bioavailability of different administration routes and doses. SC injections have been preferred because they seem to cause the animal less discomfort and are easier to perform than IM injections. In the case of buprenorphine, this welfare concern may lead to undertreated pain.
A weakness of the current study was that the randomization procedure resulted in an uneven distribution of treatments between treatment days. This was potentially the reason why an effect of treatment day was seen on the Cmax (p = 0.02). On the first treatment day, randomization led to half of the rabbits receiving buprenorphine by the IM route, which had a big influence on the mean Cmax (8 ng/mL). On the fourth treatment day, half received 0.05 mg/kg buprenorphine by the SC route, which resulted in a much lower mean Cmax that day (3 ng/mL). A block design would most likely have prevented this effect of treament day. There is however no reason to believe that the day had a true impact on the Cmax. The overall effects of route and SC dose were of such magnitude that the results were deemed reliable. The reason why the two excluded cases showed such high serum concentrations could not be determined. Not only were the concentrations more than 3 SD higher than the mean, but the time-concentration curves did not follow a normal pattern. Further, these rabbits did not show an unusual concentration time curve on any of the other treatment days. The only possible explanation imagined is a contamination of the samples during preparation.
Conclusions
IV and IM administration of 0.05 mg/kg of buprenorphine cannot be replaced with SC administration, due to a markedly lower peak serum concentration and bioavailability with this route. Increasing the SC dose to compensate for the lower bioavailability leads to unpredictable serum concentrations and can therefore not be recommended. Initial treatment by the IV or IM routes may be followed by SC administration to uphold serum concentrations.
Methods
Aim
The aim of the study was to determine the bioavailability of buprenorphine after subcutaneous (SC) and intramuscular (IM) injection in New Zealand White (NZW) rabbits.
Study design
In this prospective cross-over study, five female and five male NZW rabbits each received four treatments with buprenorphine (Vetergesic vet, 0.3 mg/mL, Orion Pharma, Danderyd, Sweden), with 2-week wash-out periods between treatments. The order in which the animal received the treatments was randomized (Excel, Microsoft cooperation, Redmond, WA, USA). The treatments were 0.05 mg/kg buprenorphine by the IV, IM and SC routes, and 0.1 mg/kg by the SC route (see supplement).
Animals
The rabbits originated from an SPF breeding colony (Lidköpings kaninfarm, Lidköping, Sweden) free from Clostridium piriforme, Encephalitozoon cuniculi, rabbit hemorrhagic disesase, rotavirus, Pasteurella spp. and Bordetella bronciseptica. They were aged 4–5 months, with a mean ± SD body weight of 3.1 ± 0.3 kg. The females were housed 2 and 3 together and the males singly, in pens of 3 m2. Aspen wood chips (Tapvei, Paekna, Estonia) and autoclaved straw (local farm, Uppsala, Sweden) were used for bedding, and the pens were cleaned weekly. The pens were furnished with combined shelters and resting shelves. The room temperature was 19 ± 2 °C, the light:dark cycle 8 h:16 h. The rabbits were fed a restricted amount of a pelleted diet (K1 special, Lantmännen, Stockholm) and had free access to autoclaved hay (local farm) and tap water. The rabbits were acclimatized for two weeks, during which members of the research team took turns in spending 15 min per day per pen to accustom the rabbits to the presence of the members and to handling. Body weight was recorded daily during a week before study begin and at each treatment. Before each treatment, the rabbits underwent clinical examination (visual inspection, auscultation of heart and lungs). Before study begin, blood was collected for hematology.
Drug administration and sampling
For placements of catheters (Venflon IV PVK, 22G, BD AB, Helsingborg, Sweden), the ears were treated with a local anesthetic cream (EMLA®, AstraZeneca, Södertälje, Sweden). After approximately 30 min, a catheter was placed in the central artery of one ear and on the day that buprenorphine was administered IV, one additional in the lateral ear vein of the contralateral ear. A 2 mL blood sample was collected from the arterial catheter before and at each sampling timepoint after administration of buprenorphine. Timepoints for sampling were 2 min (after IV administration only), 5, 10, 15, 30, 60, 90, 180, 360 and 480 min after injection. The time points were based on previous studies. Prior to blood collection, 0.2 mL of blood was discarded and after collection the catheter was flushed slowly with 2 mL of NaCl (Infusion solution, 9 mg/mL, B. Braun, Danderyd, Sweden) and 0.1 mL of heparinized saline (100 IU /mL, Heparin LEO 5000 IU/mL, LEO Pharma, Malmö, Sweden) deposited in the catheter. A maximum volume 7 mL blood/ kg was collected at each treatment. Depending on the weight of the rabbit, and the treatment, a random sample timepoint was sometimes excluded in order not to exceed the recommended maximum blood volume [15% of blood volume/14 d [29]]. Blood was collected in 2 mL serum tubes and left to coagulate at room temperature for at least 60 min. After centrifugation at 3000 g for 15 min, serum was separated and stored at minus 80 °C until analyzed. At the end of each treatment, 4 mg/kg carprofen (Norocarp vet, 50 mg / mL, N-Vet AB, Uppsala, Sweden) was administered SC for 24 h of post procedural analgesia. After the study end, the rabbis were re-used in a non-recovery anaesthesia study euthanized by injection of pentobarbital (Allfatal) IV.
Buprenorphine analysis
Buprenorphine was quantified in rabbit plasma using ultra-performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) using an Acquity UPLC coupled to a TQS micro tandem mass spectrometer (both Waters Corp, Milford, MA, USA) using an electrospray interface operating in the positive mode. To the plasma samples (250 μL), 50 μL of water, 50 μL of internal standard solution (buprenorphine-d4 40 ng/mL) and 250 μL of sodium carbonate buffer (0.05 M; pH 9.9) were added. Liquid-liquid extraction was performed to 3.0 mL of ter-butyl-methylether for 10 mins. The tubes were centrifuged and frozen at − 70 °C, after which the organic phases were poured into new tubes for evaporation to dryness under nitrogen at 50 °C. The residues were reconstituted in 100 μL of acetonitrile/water/formic acid (9/1/0.01, v/v). The extracts were transferred to vials for UHPLC-MS/MS injection. The column used was an Acquity UPLC BEH C18 (100 × 2.1 mm length x inner diameter, 1.7 μm particle size) from Waters Corp. The mobile phase consisted of (A) 0.1% formic acid in water and (B) 0.1% formic acid in acetonitrile. Gradient elution was performed: initially 15% B for 0.50 min, linear increase to 90% B for 1.0 min, constant at 90% B for 0.50 min, back to 15% B in 0.10 min and constant at 15% B for 0.40 min. The total run time was 2.5 min. The flowrate was 0.40 mL/min and the injection volume was 3.0 μL.
The quantification was performed in the Selective Reaction Monitoring mode (SRM) with the transitions 468 > 396 for buprenorphine [M + H] + (collision energy 36 eV), and 472 > 400 for buprenorphine-d4 [M + H] + (collision energy 38 eV). For quantification, calibrator samples were prepared by spiking standard solutions of the analyte to blank plasma which were analyzed according to the method. The calibration curve was constructed by linear curve fitting using the chromatographic peak area ratio (analyte/internal standard) as a function of analyte concentration with a weighting factor of 1/x. The measurement interval was 0.01 ng/mL – 100 ng/mL. The precision and accuracy of the method were within the acceptance criteria [30].
Pharmacokinetic analysis
For each animal, route and dose the concentration of buprenorphine were plotted vs. time. Different models and weighting factors were assessed by visual inspection of the curve fits and the residuals’ scatter plots, together with the accuracy of fit measures incorporated in the software (e.g. the Akaike criteria). A non-compartmental model was used to compare all routes. The maximal concentration of buprenorphine in serum (Cmax), tmax (time to reach Cmax), t½ (half-life) and AUC (area under the plasma concentration curve) were calculated with a non-compartmental model using the PK Solver add-in for Microsoft Office Excel. For IV administration also a 3-compartment model was used to calculate the maximal concentration at time 0/at the time for administration (C0).
The bioavailability (F%) for the SC and IM administration routes were calculated from the AUC0-t by using the equation:
Statistical analysis
InVivoStat (Version 4.0) was used for the sample size assessment [31]. The number of rabbits used was selected based on a power calculation, with a significance level set at 5%, and a power of at least 80% to detect at least a 25% change from the area under the time-concentration curve (AUC0-t) after IV administration. Data from two animals were removed from the statistical analyses (#3 0.05 mg/kg SC and #6 0.05 mg/kg IM) because the concentration curves did not follow a normal pattern and the concentrations were in part so high than it is unlikely a normal biological variation. All data are included in the supplement data file.
Data were analyzed in two stages. In order to test the overall effect of the administration routes, the data from the 0.05 mg/kg groups were compared by two-way ANOVA with Route and sex as independent factors and animal and treatment day (of administration) as blocking factors (SAS/STAT software, Version 9.4 of the SAS System for Windows 10). This was followed by planned comparisons of the predicted means to compare the IV and IM routes back to SC. In the second stage the additional SC 0.1 mg/kg group was included to allow a comparison between the SC 0.05 mg/kg and SC 0.1 mg/kg groups.
Data are presented as mean ± SD. The level of statistical significance was set to p < 0.05.
Availability of data and materials
The raw data is provided in a supplementary file.
Change history
16 April 2021
A Correction to this paper has been published: https://doi.org/10.1186/s12917-021-02858-1
Abbreviations
- SC:
-
Subcutaneous
- IM:
-
Intramuscular
- IV:
-
Intravenous
- AUC0-t :
-
Area under the curve from t0 to t480 min (last time point sampled)
- AUC0-∞ :
-
Area under the curve from t0 to infinity
- tmax :
-
Time of maximum concentration
- Cmax :
-
Maximum concentration
- C0 :
-
Concentration at time point 0
- t½ :
-
Elimination half life
- F:
-
Bioavailability
- NZW:
-
New Zealand White
References
Commission E. REPORT FROM THE COMMISSION TO THE EUROPEAN PARLIAMENT AND THE COUNCIL 2019 report on the statistics on the use of animals for scientific purposes in the Member States of the European Union in 2015–2017. 2020 5.2.2020.
Varga M. Chapter 4 - Anaesthesia and analgesia. In: Varga M, editor. Textbook of Rabbit Medicine (Second Edition): Butterworth-Heinemann; 2014. p. 178–202.
Varga M. Chapter 8 - Digestive Disorders. In: Varga M, editor. Textbook of Rabbit Medicine (Second Edition): Butterworth-Heinemann; 2014. p. 303–49.
Coulter CA, Flecknell PA, Leach MC, Richardson CA. Reported analgesic administration to rabbits undergoing experimental surgical procedures. BMC Vet Res. 2011;7:1–6.
Park I, Kim D, Song J, In CH, Jeong SW, Lee SH, et al. Buprederm, a new transdermal delivery system of buprenorphine: pharmacokinetic, efficacy and skin irritancy studies. Pharm Res. 2008;25(5):1052–62.
A M. BSAVA small animal formulary 9th ed: British Snall Animal Veterinary Association; 2015.
Fiorello CVDS. Rabbits. In: JW C, editor. Exotic animal formulary. St Louis: Elsevier; 2013. p. 518–59.
Chitty J. Formulary. In: Harcourt-Brown F CJ, editor. Manual of Rabbit Surgery, Denstistry and Imaging. Gloucestershire, UK: BSAVA; 2013. p. 430–1.
Liu SY, Liu KS, Kuei CH, Tzeng JI, Ho ST, Wang JJ. Simultaneous determination of buprenorphine and its prodrug, buprenorphine propionate, by high-performance liquid chromatography with fluorescence detection: application to pharmacokinetic studies in rabbits. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;818(2):233–9.
Ho ST, Wang JJ, Ho W, Hu OY. Determination of buprenorphine by high-performance liquid chromatography with fluorescence detection: application to human and rabbit pharmacokinetic studies. J Chromatogr. 1991;570(2):339–50.
Freijs E. Comparison of plasma levels and analgesic effect between oral transmucosal and subcutaneous administration of buprenorphine in rabbits. [master]. Uppsala: Swedish University of Agricultural Sciences; 2016.
Weaver LA, Blaze CA, Linder DE, Andrutis KA, Karas AZ. A model for clinical evaluation of perioperative analgesia in rabbits (Oryctolagus cuniculus). J Am Assoc Lab Anim Sci. 2010;49(6):845–51.
Hedenqvist P, Trbakovic A, Thor A, Ley C, Ekman S, Jensen-Waern M. Carprofen neither reduces postoperative facial expression scores in rabbits treated with buprenorphine nor alters long term bone formation after maxillary sinus grafting. Res Vet Sci. 2016;107:123–31.
Borkowski R, Karas AZ. Sedation and anesthesia of pet rabbits. Clinical Techniques in Small Animal Practice. 1999;14(1):44–9.
Cooper CS, Metcalf-Pate KA, Barat CE, Cook JA, Scorpio DG. Comparison of side effects between buprenorphine and meloxicam used postoperatively in Dutch belted rabbits (Oryctolagus cuniculus). J Am Assoc Lab Anim Sci. 2009;48(3):279–85.
Steagall PV, Pelligand L, Giordano T, Auberger C, Sear JW, Luna SP, et al. Pharmacokinetic and pharmacodynamic modelling of intravenous, intramuscular and subcutaneous buprenorphine in conscious cats. Vet Anaesth Analg. 2013;40(1):83–95.
Giordano T, Steagall PV, Ferreira TH, Minto BW, de Sa Lorena SE, Brondani J, et al. Postoperative analgesic effects of intravenous, intramuscular, subcutaneous or oral transmucosal buprenorphine administered to cats undergoing ovariohysterectomy. Vet Anaesth Analg. 2010;37(4):357–66.
Taylor PM, Luangdilok CH, Sear JW. Pharmacokinetic and pharmacodynamic evaluation of high doses of buprenorphine delivered via high-concentration formulations in cats. J Feline Med Surg. 2016;18(4):290–302.
Benato L, Rooney NJ, Murrell JC. Pain and analgesia in pet rabbits within the veterinary environment: a review. Vet Anaesth Analg. 2019;46(2):151–62.
Zuidema J, Kadir F, Titulaer HAC, Oussoren C. Release and absorption rates of intramuscularly and subcutaneously injected pharmaceuticals (ii). Int J Pharm. 1994;105(3):189–207.
Lindhardt K, Bagger M, Andreasen KH, Bechgaard E. Intranasal bioavailability of buprenorphine in rabbit correlated to sheep and man. Int J Pharm. 2001;217(1–2):121–6.
Steagall PV, Monteiro-Steagall BP, Taylor PM. A review of the studies using buprenorphine in cats. J Vet Intern Med. 2014;28(3):762–70.
Simon BT, Steagall PV. The present and future of opioid analgesics in small animal practice. J Vet Pharmacol Ther. 2017;40(4):315–26.
Doodnaught GM, Monteiro BP, Benito J, Edge D, Beaudry F, Pelligand L, et al. Pharmacokinetic and pharmacodynamic modelling after subcutaneous, intravenous and buccal administration of a high-concentration formulation of buprenorphine in conscious cats. PLoS One. 2017;12(4):e0176443.
Goldschlager GB, Gillespie VL, Palme R, Baxter MG. Effects of multimodal analgesia with LowDose buprenorphine and meloxicam on fecal glucocorticoid metabolites after surgery in New Zealand white rabbits (Oryctolagus cuniculus). J Am Assoc Lab Anim Sci. 2013;52(5):571–6.
Flecknell PAaL, J.H. Assessment of the analgesic action of opioid agonist-antagonists in the rabbit. J Ass vet Anaesth. 1990;17:24–9.
Boas RA, Villiger JW. Clinical actions of fentanyl and buprenorphine. The significance of receptor binding. Br J Anaesth. 1985;57(2):192–6.
Yassen A, Olofsen E, Romberg R, Sarton E, Danhof M, Dahan A. Mechanism-based pharmacokinetic-pharmacodynamic modeling of the antinociceptive effect of buprenorphine in healthy volunteers. Anesthesiology. 2006;104(6):1232–42.
Diehl KH, Hull R, Morton D, Pfister R, Rabemampianina Y, Smith D, et al. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxicol. 2001;21(1):15–23.
((CHMP) CfMPfHU. Guideline on bioanalytical method validation. London: European Medicines Agent; 2011.
Bate ST CR. InVivoStat [software]. 2020 [updated 13/02/2020. 4.0 [Available from: http://invivostat.co.uk/download/.
Acknowledgements
Technical assistance was provided by Mari Wallbring, Carola Jansson, Ulrika Mattsson and Mathilda Lindholm.
Funding
The study was supported by the American Association for Laboratory Animal Science (AALAS) in the Grants for Laboratory Animal Science (GLAS). The funding body played no part in the design of the study and collection, analysis, or interpretation of data and in writing the manuscript. Open Access funding provided by Swedish University.
Author information
Authors and Affiliations
Contributions
PH and LO designed the study and wrote the grant application. PH, LO, RA, EF and EM performed the experiments and contributed to writing the manuscript, LO performed the pharmacokinetic calculations, MH and UB were in charge of performing the UHPLC-MS/MS and contributed to writing the manuscript, SB performed the statistical analyses and contributed to writing the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animal studies were ethically reviewed and carried out in accordance with European Directive 2010/63/EEC and Swedish legislation on animal experiments (SJVFS 2019:9). Ethics permit was granted from the regional Uppsala animal ethics committee (Uppsala djurförsöksetiska nämnd), permit # 5.2.18–8707/14.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Askar, R., Fredriksson, E., Manell, E. et al. Bioavailability of subcutaneous and intramuscular administrated buprenorphine in New Zealand White rabbits. BMC Vet Res 16, 436 (2020). https://doi.org/10.1186/s12917-020-02618-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-020-02618-7