
Abstract
Background
Norovirus (NoV) is recognized as a highly contagious enteric pathogen of mammals, and bovine norovirus (BNoV) is associated with calf diarrhoea and has caused great economic losses in the cattle industry.
Results
Here, we describe a case of emerging calf diarrhoea on a cattle farm in Henan Province, Central China. BNoV was the only enteric pathogen detected in outbreaks according to tests for enteric viruses, bacteria and parasites. The complete genome of the newly identified strain CH-HNSC-2018 was successfully sequenced and found to be 7342 nucleotides in length. Sequence and phylogenetic analyses revealed that CH-HNSC-2018 belongs to GIII.2 BNoV. Further analysis of the major capsid protein demonstrated that it is separated by specific genetic distances from previous BNoV strains identified in China and has 4 new amino acid (aa) mutations, 134A, 327 T, 380 L and 423A, in the VP1 protein and 11 aa substitutions in the hypervariable P2 subdomain, suggesting that the BNoV strains circulating in China are diverse.
Conclusions
This is the first detection of GIII.2 BNoV in the VP1 region in China. This report should form a basis for further molecular studies on NoV and bovine enteric viruses in China.
Similar content being viewed by others


Background
Norovirus (NoV) is an emerged non-enveloped virus with a positive-stranded RNA genome that widely infects humans and other animals and causes severe gastrointestinal disorders and even fatal bleeding [1]. NoV belongs to the family Caliciviridae and genus Norovirus, and its genome is approximately 7.5–7.7 kb, which encodes for 3 open reading frames (ORF1–3). ORF1 encodes a large polyprotein that is cleaved into 6 nonstructural proteins (N-terminal-NTPase-3A-like-VPg-3C-like-polymerase); ORF2 encodes VP1, the major capsid protein; and ORF3 encodes VP2, a minor structural protein. The pathogenesis of calicivirus remains unclear, and little is known about NoV. One important reason for this lack of knowledge is that NoV has rarely been successfully propagated in cell culture; the sole exception is the culture of a murine isolate.
NoVs are classified into seven genogroups (GI-GVII). Bovine norovirus (BNoV), which belongs to GIII (NoVs-GIII), is divided into two distinct genotypes based on amino acid (aa) diversity of the complete VP1 sequence, genotypes 1 (Bo/Jena/1980/DE) and 2 (Bo/Newbury2/1976/UK), which were originally discovered in Germany [2] and England [3], respectively. Recently, a dual nomenclature system was established to characterize the genomic properties of BNoV strains based on the sequences of RNA-dependent RNA polymerase RdRp (end of ORF1) and major capsid protein (beginning of ORF2). Based on this novel nomenclature system, BNoV GIII is reclassified into GIII.P1_GIII.1 (which contains polymerase and VP1 genes from the Jena strain), GIII.P2_GIII.2 (which contains polymerase and VP1 genes from the Newbury2 strain), GIII.P1_GIII.2 (which contains a polymerase gene from the Jena strain and a VP1 from the Newbury2 (recombinant) strain, e.g., the Bo/Thirsk10/00/UK strain), and GIII.P2_GIII.1 (which contains a polymerase gene from the Newbury2 strain and a VP1 gene from the Jena (recombinant) strain, e.g., the B-1SVD/03/US strain) [4]. Moreover, some studies have employed ORF3 to characterize the phylogenetic topology of BNoV [5]. Until now, the available BNoV strain sequences have been limited [6] and have not provided enough data to establish intensive phylogenetic relationships or rates of evolution of BNoVs. To date, BNoV has been reported worldwide, including in China [7], Iran [8], Egypt [9], Argentina [10], Italy [11], the USA [12], and New Zealand [13], leading to substantial loss to the worldwide bovine industry. Furthermore, serum antibodies against BNoV (GIII) have been detected in humans [14], and sequences of human NoVs have been identified in bovine stool specimens [15], suggesting possible interspecies transmission [16].
Although BNoV plays a role in the aetiology of calf enteritis [17], data on emerging enteric viruses in cattle in China are scarce. Until now, only one report, from 2018, has reported BNoV from calves aged 3 to 4 months, which had a low BNoV GIII.1 infection rate (3/28) in diarrhoea faecal samples from Hebei and Sichuan Provinces, China 7. Here, we detected a newly emerging BNoV strain from a cow farm in Central China, sequenced its whole genome and analysed its phylogeny and mutations. The newly identified strain was classified as genotype 2 and was distantly related to the previously identified Chinese BNoV strains. This study systematically describes the genetic and phylogenetic characteristics of the newly identified BNoV strain and highlights that continuous surveillance is needed to help develop suitable vaccines and reasonable control measures.
Results
Detection of the BNoV
To determine the causative pathogen of the outbreak on the studied cattle farm in Henan Province, Central China, RT-PCR was used to detect the common potential pathogens causing calf diarrhoea. The diarrhoeal faecal samples were positive for BNoV (2/8, 25%), whereas no other viruses were detected, including BRV (A, B, C), BCoV, BVDV, BKV, BAstV, BNebV, or BToV. The samples were further cultured in MDBK cells to isolate the causative agent; no cytopathic effect was observed, and no BNoV nucleic acids were detected in the fifth blind passage. The newly detected viral strain was named CH-HNSC-2018. The results demonstrate that BNoV is an emerging pathogen of calf diarrhoea in this outbreak.
Sequencing the BNoV viral genome
To our knowledge, although the virus was first detected in China in 2018, this study is the first to report the complete genome of GIII.2 and its phylogenetic characteristics in China. CH-HNSC-2018 genomic RNA consists of 7342 nucleotides (nt), with a 19-nt poly (A) tail. The BNoV genome contains three sequential ORFs starting from the 22nd nt: ORF1, 5055 (22–5076) nt; ORF2, 1569 (5063–6631) nt; and ORF3, 651 (6621–7271) nt, which encode a nonstructural polyprotein and the structural VP1 and VP2 proteins, respectively. ORF1, ORF2 and ORF3 overlapped each other by 14 and 10 nt, respectively. The 5′ untranslated region of our strain has as many as 21 nt, similar to Newbury2 (21 nt) and Jena (21 nt) strains. The G + C content of our strain genome was 57.18%, similar to that of the Newbury2 (57%) [18] and Jena (56%) [19] strains. The complete sequence was deposited in the GenBank database, and the accession number is MN122335.
Consistent with other BNoVs, the protease cleavage sites of CH-HNSC-2018 were LQ/GP, LQ/AP, VQ/AP and LE/GG (Additional file 2: Figure S1). Meanwhile, aa motifs typical of caliciviruses were also observed in the CH-HNSC-2018 ORF1 polyprotein in NTPase (490GPPGIGKT497), 3C protease (1134GDCG1137), and RNA-dependent RNA polymerase (RdRp) (1473GLPSG1477 and 1517YGDD1520), as previously described for the Newbury2 and Jena strains.
Homology and phylogenetic analysis of BNoV
In this study, we further analysed the BNoV gene to characterize the enteric pathogen. Nucleotide sequences analysis indicated that CH-HNSC-2018 was closely related to BET-17 (MK159169, CHN) with 87.6% nt identity. Further analysis revealed that the ORF1 gene of BNoV was the most similar region between CH-HNSC-2018 and BET-17, with greater than 95.4% nt and 97.3% aa identity (Table 1). However, the major capsid protein (VP1) was closer to CV186-OH (AF542084, USA), with 90.1% nt identity and 97.3% aa identity. VP2 was closer to SCZ-3 (MK159152, CHN) and SCZ-6 (MK159153, CHN), with 92.6 and 92.5% nt (85.0 and 84.5% aa) identity, respectively, whereas it had 93.9% aa identity to 42FR (MF784576, Egypt).
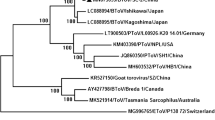
We further generated phylogenetic trees based on the whole genome of our BNoV strain and other previously reported sequences. The results demonstrated that CH-HNSC-2018 was most closely related to GIII.2 BNoV strains (Fig. 1). The BNoV VP1 gene of our strain included an ORF of 1569 nt. These nt sequences encoded predicted proteins containing 523 aa residues. Based on the nt sequence differences of VP1, NoVs could be divided into seven genogroups (GI to GVII), and CH-HNSC-2018 was further confirmed to belong to the GIII 2 group (Fig. 2). This is the first time that a GIII.2 NoV has been detected in the VP1 region in China. Recombination events among BNoV strains were further investigated using RDP4 software, but no correlation was detected. This result showed that no recombination had occurred; however, the available BNoV sequences are limited, so there may not be enough data available to confirm this result. Therefore, further studies are required.

Phylogenetic analyses of the whole genome sequence of CH-HNSC-2018. A phylogenetic tree was generated by MEGA 7.0 software using the neighbour-joining method with 1000 bootstrap replicates

Phylogenetic analyses of VP1 based on the deduced aa sequences. A phylogenetic tree was constructed by MEGA 7.0 software using the neighbour-joining method with 1000 bootstrap replicates. Circle, the strain identified in this study; square, strains detected in China by other groups
To investigate the aetiologic features of the newly identified BNoV strain, we next analysed aa substitution mutations of CH-HNSC-2018. Compared to the other available BNoV strains in GenBank, CH-HNSC-2018 contains several aa substitutions in the VP1 protein, 19 aa substitutions throughout the P domain, and 11 aa substitutions in the P2 domain. Interestingly, 4 new aa mutations, 134A, 327 T, 380 L and 423A, were identified in the VP1 protein of CH-HNSC-2018. (Additional file 1: Table S1). One epitope (E394D) change was found in these substitutions compared to the Newbury2 strain.
Discussion
Similar to other regions throughout the world, diarrhoea is the most important cause of disease symptoms in calves in China and leads to serious economic losses of cattle producers. NoV is one of the leading causes of human and animal gastroenteritis; however, BNoV is not usually included in the diagnostic algorithms of calf diarrhoea. In this study, during pathogen detection of diarrhoeal calves, only BNoV was detected, and other enteric viruses, Coccidium species and Cryptosporidium species were not found. However, the relationship between BNoV infection and diarrhoea in calves requires further epidemiological and experimental studies.
The detection rate of BNoV reported in this study (25.0%) is comparatively higher than that reported in South Korea, 2.8% [20]; Argentina, 3.3% [10]; and Belgium, 7.5% [21]. However, the detection rate of BNoV reported in this study is lower than that reported in other studies: 39.5% in Iran [8], 49.6% in Norway [22] and 72.1% in the USA [12]. Despite these differences, a reliable comparison cannot be made among these frequencies because the samples originated from the same farm in the present study.
The new strain identified in our study was BNoV-GIII 2. Considering the previously reported GIII.1 strains identified in China [7], both genotype GIII.1- and GIII.2-related sequences have been found to circulate in Chinese dairy calves. Due to the limited epidemiological data on BNoV infections in China, the dominant strains cannot be accurately confirmed. More studies on the epidemiology of this emerging pathogen should be carried out in Henan Province and other provinces in China. A BNoV outbreak in China was reported in Hebei and Sichuan Provinces in 2018, and the 3 strains identified were BNoV-GIII.1 based on the partial region of the RdRp polymerase ORF (532 bp) [7]. Nevertheless, the BNoV strain detected in our study was BNoV-GIII 2. Given that strains with different BNoV genotypes co-exist in China, prevention and control are more complex. The results of our study will facilitate further research on the evolution and molecular pathogenesis of BNoV. The importance of BNoV surveillance should be stressed given the cattle industry-intensive area of China.
VP1 is the major structural component of caliciviruses and is involved in receptor recognition, host specificity, strain antigenic diversity, and immunogenicity [23]. X-ray crystallography structures for NoV [24] reveal that the main icosahedral core of the capsid is composed of a conserved S domain and a variable P domain. A sequence comparison and phylogenetic studies were conducted using complete VP1 sequences as phylogenetic markers. It is important to evaluate whether aa changes mainly occur in the P domain or the S domain. The P domain is further divided into a comparatively conserved P1 domain and a highly variable P2 subdomain. The latter has an external localization, and compatible with their functions, both cell receptors are involved in interactions with the host cell membrane [25] and have the most important epitopes [26]. In the present report, we identified four unique aa mutations of VP1 in CH-HNSC-2018 (134A, 327 T, 380 L and 423A) and two (327 T and 380 L) within the P2 subdomain. The hypervariable P2 domain contains putative receptor-binding sites and is responsible for host specificity and strain diversity [23, 24]. Therefore, it was speculated that the large shift in aa in the P2 domain may have major implications for the immunogenic characteristics of this novel genotype as well as significant implications for further vaccine development [27]. These substitutions may have major implications in the immunogenic characteristics of the emerging strain CH-HNSC-2018. Thus, further study of the impact of these aa mutations in the VP1 region on the function of the VP1 protein is required.
Previous studies have shown that the S domain is a conserved region within VP1 and have described the icosahedral scaffold of VP1, and a recent study reported that the S domain of a NoV strain contains strain-specific epitopes that contribute to antigenic diversity [28]. This region in strain CH-HNSC-2018 included a 134A unique aa mutation compared with the other NoV strains. This novel mutation further indicates that the S domain of NoV might possess strain-specific epitopes.
To date, there is no suitable cell culture system for BNoV. The lack of pure viral stocks after culture limits the available information regarding the pathogenesis of BNoV and hampers functional studies of their replication. Although BNoV can infect cattle of different ages, interestingly, in the outbreak investigated in this study, only calves aged less than 1 month were infected. This situation may be linked to the geographical distribution, breeding systems or aa substitutions.
Conclusions
In conclusion, in this study, we detected GIII.2 for the first time in the VP1 region in a diarrhoeal faecal sample in China and sequenced the complete genome of the strain. The results of our study indicated that BNoV was associated with bovine diarrhoea. BNoVs should be considered in the differential diagnosis of calf diarrhoea and are candidates for inclusion in future vaccines in cattle. Further research needs to be conducted to better understand the epidemiology, pathogenic mechanisms, and potential cross-species transmission of this novel BNoV.
Methods
In April 2018, an outbreak of calf diarrhoea occurred on a bovine farm in Henan Province. According to the farm workers’ descriptions, the morbidity of calves less than one month old was 100% (56/56), and the mortality was approximately 12.5% (7/56); the affected calves displayed muddy, watery, or bloody diarrheic stool and were anorexic and lethargic. The outbreak lasted for more than 10 days, despite routine treatment with antibiotics, including sulfonamides, penicillin, cephalosporin, and enrofloxacin, which were less efficient. Meanwhile, no clinical signs were observed in other bovine herd age groups, including young and adult cattle, during the outbreak. To diagnose the enteric pathogens, a total of eight faecal samples were collected from sick calves and used for RNA extraction and virus isolation. The faecal samples were also tested for Coccidium species and Cryptosporidium species using the sucrose floatation method.
Faecal suspensions of each sample were prepared by diluting faeces 1:10 (w/v) in sterile phosphate-buffered saline (pH 7.2). These sample suspensions were mixed for 30 s and centrifuged at 8000 g for 10 min at 4 °C. The supernatants were collected and maintained at − 80 °C until further processing.
Total RNA was extracted from 200 μL of the supernatants using the TaKaRa MiniBEST Viral RNA/DNA Extraction Kit Ver.5.0 (Cat# 9766, TaKaRa Bio Inc.) according to the manufacturer’s instructions, and the RNA was maintained at − 80 °C. Reverse transcription was performed using the PrimeScript II 1st Strand cDNA Synthesis Transcription Kit (Cat# 6210A, TaKaRa Bio Inc.) for first-strand synthesis. All samples were examined for BNoV, BCoV, BRV (A, B, C), BVDV, BKoV, BAstV, BNebV, and BToV, which can cause calf diarrhoea, and were detected by RT-PCR using specific primers (Table 2) as described previously. To confirm the accuracy of the results, all tests were performed three times. The positive samples were then filtered through a 0.22 μm filter and diluted with DMEM containing 1% penicillin and streptomycin. Then, Madin-Darby bovine kidney (MDBK, ATCC) cells were incubated with the dilution to observe the cytopathic effect (CPE) daily. After five blind passages, the whole cell lysates were centrifuged and then analysed for BNoV.
The complete genome of BNoV was amplified from eight overlapping fragments with specific primers and the 3’RACE method (Additional file 1: Table S2). Briefly, eight pairs of primers were designed based on the conserved regions of available BNoV strains in the GenBank database. The PCR products were cloned into the pMD18-T vector and sequenced (Sangon, China). We further designed special primers flanking the genome (3 race-F) and then acquired the 3’UTR region by semi-nested PCR according to the instructions of RACE kits (Cat# 18373–019, Invitrogen).
The sequences were assembled with SeqMan and then aligned using MegAlign software (version 7.0; DNASTAR Inc., WI, USA). Pairwise sequence identity calculations were performed using Clustal W software within DNAStar 7.0 software (available at https://www.dnastar.com/). The phylogenetic tree was analysed using MEGA 7.0 software (available at http://www.megasoftware.net/) with the neighbour-joining method with 1,000 bootstrap replicates. Meanwhile, recombination events among BNoV strains were further investigated using RDP4 software with seven different algorithms, RDP, Bootscan, MaxChi, GeneConv, Chimaera, SiScan, and 3Seq [36].
To further characterize the VP1 sequences, an aa alignment was performed using all 29 complete BNoV VP1 sequences available in the GenBank database. The VP1 protein forms an icosahedral particle with two principal domains, a protruding (P) domain and shell (S) domain. The S domain resides between residues 48 and 218. The more variable, surface-exposed P domain is predicted to be located between residues 219 and 511, with the P1 domain mapped from 219 to 270 and from 397 to 511 and the P2 domain mapped from 271 to 396 of the capsid.
Availability of data and materials
All data generated or analysed during this study are included in the published article and its additional files, and the dataset analysed in the current study is available from the corresponding author upon reasonable request.
Abbreviations
- BAstV:
-
Bovine astrovirus
- BCoV:
-
Bovine coronavirus
- BKV:
-
Bovine kobuvirus
- BNoV:
-
Bovine norovirus
- BRV:
-
Bovine rotavirus
- BToV:
-
Bovine torovirus
- BVDV:
-
Bovine viral diarrhoea virus
- NoV:
-
Norovirus
References
van Der Poel WH, Vinje J, van Der Heide R, Herrera MI, Vivo A, Koopmans MP. Norwalk-like calicivirus genes in farm animals. Emerg Infect Dis. 2000;6(1):36–41.
Gunther H, Otto P. Diarrhea in young calves. 7. "Zackenvirus" (Jena agent 117/80)--a new diarrhea pathogen in calves. Arch Exp Vet. 1987;41(6):934–8.
Woode GN, Bridger JC. Isolation of small viruses resembling astroviruses and caliciviruses from acute enteritis of calves. J Med Microbiol. 1978;11(4):441–52.
Di Felice E, Mauroy A, Pozzo FD, Thiry D, Ceci C, Di Martino B, Marsilio F, Thiry E. Bovine noroviruses: a missing component of calf diarrhoea diagnosis. Vet J. 2016;207:53–62.
Kamel AH, Ali MA, El-Nady HG, de Rougemont A, Pothier P, Belliot G. Predominance and circulation of enteric viruses in the region of greater Cairo, Egypt. J Clin Microbiol. 2009;47(4):1037–45.
Mauroy A, Scipioni A, Mathijs E, Ziant D, Daube G, Thiry E. Complete genome sequence of a novel bovine norovirus: evidence for slow genetic evolution in genogroup III genotype 2 noroviruses. J Virol. 2012;86(22):12449–50.
Guo Z, He Q, Yue H, Zhang B, Tang C. First detection of Nebovirus and norovirus from cattle in China. Arch Virol. 2018;163(2):475–8.
Pourasgari F, Kaplon J, Sanchooli A, Fremy C, Karimi-Naghlani S, Otarod V, Ambert-Balay K, Mojgani N, Pothier P. Molecular prevalence of bovine noroviruses and neboviruses in newborn calves in Iran. Arch Virol. 2018;163(5):1271–7.
Mohamed FF, Mansour SMG, El-Araby IE, Mor SK, Goyal SM. Molecular detection of enteric viruses from diarrheic calves in Egypt. Arch Virol. 2017;162(1):129–37.
Ferragut F, Vega CG, Mauroy A, Conceicao-Neto N, Zeller M, Heylen E, Uriarte EL, Bilbao G, Bok M, Matthijnssens J, et al. Molecular detection of bovine noroviruses in Argentinean dairy calves: circulation of a tentative new genotype. Infect Genet Evol. 2016;40:144–50.
Di Martino B, Di Profio F, Di Felice E, Melegari I, Ceci C, Mauroy A, Thiry E, Martella V, Marsilio F. Genetic heterogeneity of bovine noroviruses in Italy. Arch Virol. 2014;159(10):2717–22.
Smiley JR, Hoet AE, Traven M, Tsunemitsu H, Saif LJ. Reverse transcription-PCR assays for detection of bovine enteric caliciviruses (BEC) and analysis of the genetic relationships among BEC and human caliciviruses. J Clin Microbiol. 2003;41(7):3089–99.
Wolf S, Williamson WM, Hewitt J, Rivera-Aban M, Lin S, Ball A, Scholes P, Greening GE. Sensitive multiplex real-time reverse transcription-PCR assay for the detection of human and animal noroviruses in clinical and environmental samples. Appl Environ Microbiol. 2007;73(17):5464–70.
Widdowson MA, Rockx B, Schepp R, van der Poel WH, Vinje J, van Duynhoven YT, Koopmans MP. Detection of serum antibodies to bovine norovirus in veterinarians and the general population in the Netherlands. J Med Virol. 2005;76(1):119–28.
Menon VK, George S, Shanti AA, Saravanabavan A, Samuel P, Ramani S, Estes MK, Kang G. Exposure to human and bovine noroviruses in a birth cohort in southern India from 2002 to 2006. J Clin Microbiol. 2013;51(7):2391–5.
Mattison K, Shukla A, Cook A, Pollari F, Friendship R, Kelton D, Bidawid S, Farber JM. Human noroviruses in swine and cattle. Emerg Infect Dis. 2007;13(8):1184–8.
Scipioni A, Mauroy A, Vinje J, Thiry E. Animal noroviruses. Vet J. 2008;178(1):32–45.
Oliver SL, Asobayire E, Charpilienne A, Cohen J, Bridger JC. Complete genomic characterization and antigenic relatedness of genogroup III, genotype 2 bovine noroviruses. Arch Virol. 2007;152(2):257–72.
Liu BL, Lambden PR, Gunther H, Otto P, Elschner M, Clarke IN. Molecular characterization of a bovine enteric calicivirus: relationship to the Norwalk-like viruses. J Virol. 1999;73(1):819–25.
Park SI, Jeong C, Park SJ, Kim HH, Jeong YJ, Hyun BH, Chun YH, Kang MI, Cho KO. Molecular detection and characterization of unclassified bovine enteric caliciviruses in South Korea. Vet Microbiol. 2008;130(3–4):371–9.
Mauroy A, Scipioni A, Mathijs E, Saegerman C, Mast J, Bridger JC, Ziant D, Thys C, Thiry E. Epidemiological study of bovine norovirus infection by RT-PCR and a VLP-based antibody ELISA. Vet Microbiol. 2009;137(3–4):243–51.
Jor E, Myrmel M, Jonassen CM. SYBR Green based real-time RT-PCR assay for detection and genotype prediction of bovine noroviruses and assessment of clinical significance in Norway. J Virol Methods. 2010;169(1):1–7.
Chen R, Neill JD, Noel JS, Hutson AM, Glass RI, Estes MK, Prasad BV. Inter- and intragenus structural variations in caliciviruses and their functional implications. J Virol. 2004;78(12):6469–79.
Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. X-ray crystallographic structure of the Norwalk virus capsid. Science. 1999;286(5438):287–90.
Tan M, Hegde RS, Jiang X. The P domain of norovirus capsid protein forms dimer and binds to histo-blood group antigen receptors. J Virol. 2004;78(12):6233–42.
Lindesmith LC, Costantini V, Swanstrom J, Debbink K, Donaldson EF, Vinje J, Baric RS. Emergence of a norovirus GII.4 strain correlates with changes in evolving blockade epitopes. J Virol. 2013;87(5):2803–13.
Tan M, Fang P, Chachiyo T, Xia M, Huang P, Fang Z, Jiang W, Jiang X. Noroviral P particle: structure, function and applications in virus-host interaction. Virology. 2008;382(1):115–23.
Parra GI, Sosnovtsev SV, Abente EJ, Sandoval-Jaime C, Bok K, Dolan MA, Green KY. Mapping and modeling of a strain-specific epitope in the Norwalk virus capsid inner shell. Virology. 2016;492:232–41.
Cho KO, Hasoksuz M, Nielsen PR, Chang KO, Lathrop S, Saif LJ. Cross-protection studies between respiratory and calf diarrhea and winter dysentery coronavirus strains in calves and RT-PCR and nested PCR for their detection. Arch Virol. 2001;146(12):2401–19.
Chang KO, Parwani AV, Saif LJ. The characterization of VP7 (G type) and VP4 (P type) genes of bovine group a rotaviruses from field samples using RT-PCR and RFLP analysis. Arch Virol. 1996;141(9):1727–39.
Barman P, Ghosh S, Das S, Varghese V, Chaudhuri S, Sarkar S, Krishnan T, Bhattacharya SK, Chakrabarti A, Kobayashi N, et al. Sequencing and sequence analysis of VP7 and NSP5 genes reveal emergence of a new genotype of bovine group B rotaviruses in India. J Clin Microbiol. 2004;42(6):2816–8.
Park SJ, Jeong C, Yoon SS, Choy HE, Saif LJ, Park SH, Kim YJ, Jeong JH, Park SI, Kim HH, et al. Detection and characterization of bovine coronaviruses in fecal specimens of adult cattle with diarrhea during the warmer seasons. J Clin Microbiol. 2006;44(9):3178–88.
Givens MD, Heath AM, Carson RL, Brock KV, Edens MS, Wenzel JG, Stringfellow DA. Analytical sensitivity of assays used for detection of bovine viral diarrhea virus in semen samples from the southeastern United States. Vet Microbiol. 2003;96(2):145–55.
Reuter G, Egyed L. Bovine kobuvirus in europe. Emerg Infect Dis. 2009;15(5):822–3.
Tse H, Chan WM, Tsoi HW, Fan RY, Lau CC, Lau SK, Woo PC, Yuen KY. Rediscovery and genomic characterization of bovine astroviruses. J Gen Virol. 2011;92(8):1888–98.
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1(1):vev003.
Acknowledgements
We thank all the cattle farms for submitting the samples to our laboratory.
Funding
This work was funded by the Key Program for Science and Technology Development of Henan (192102110074), the National Key Research and Development Program of China (2018YFD0501700) and the National Beef Cattle Industrial Technology System (CARS-37), as well as the Independent Innovation Fund Project in Henan Academy of Agricultural Science (2018ZC56 and 2017ZC51).
Author information
Authors and Affiliations
Contributions
ZHS, WJW, and ZXX collected the samples and conceived and designed the experiments. ZHS, WJW and YLL performed the experiments. ZHS, XZZ and WJW analysed the data and wrote the paper. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Animal Ethics Committee of Henan Academy of Agricultural Sciences (Approval number SYXK 2014–0007). Experimental protocols for obtaining bovine clinical samples used in this study were carried out in strict accordance with the Animal Ethics Procedures and Guidelines of China. The sample collection work was approved by the cattle farm owner.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Amino acid substitution mutations of VP1 in CH-HNSC-2018 and other BNoV strains worldwide. Table S2. Primers used for complete genome sequencing (DOCX 23 kb)
Additional file 2:
Figure S1. Schematic genomic organization of CH-HNSC-2018. Each polyprotein gene is indicated in a box. The nt position of each gene is shown on each gene-box border. The predicted protease cleavage sites are shown below the gene boxes. The figures on either side of the conserved norovirus aa motifs, shown below the gene boxes, are the locations in CH-HNSC-2018. (TIF 803 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shi, Z., Wang, W., Xu, Z. et al. Genetic and phylogenetic analyses of the first GIII.2 bovine norovirus in China. BMC Vet Res 15, 311 (2019). https://doi.org/10.1186/s12917-019-2060-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-019-2060-0