
Abstract
Background
Bovine viral diarrhea virus (BVDV) causes severe economic losses and is one of the most important viral pathogens of ruminants worldwide. The infection manifests itself in a variety of clinical symptoms. Phylogenetic studies based mainly on 5’UTR of its genome, identified many different subtypes of BVDV. Previous study indicated the predominance of BVDV-1b and BVDV-1d in Poland. The aim of this study was to genotype BVDV isolates currently circulating in Polish dairy herds.
Results
BVDV was detected in 30 herds. Viral subtypes were identified using sequences of the 5’UTR fragment and they were confirmed within a fragment of the Npro region. Seven subtypes of BVDV-1 species have been identified: 1b, 1 g, 1f, 1d, 1r, 1 s and 1e.
Conclusion
The number of subtypes of BVDV in Poland evolves and 2 new subtypes have been identified for the first time. Such studies may have a positive impact on successful eradication of the virus using effective vaccines and diagnostic tests.
Similar content being viewed by others


Background
Bovine viral diarrhea virus (BVDV) belongs to Pestivirus genus in the Flaviviridae family [1]. It consists of four recognized species: bovine viral diarrhea virus type 1 (BVDV-1, Pestivirus A), type 2 (BVDV-2, Pestivirus B), classical swine fever virus (CSFV, Pestivirus C) and border disease virus (BDV, Pestivirus D). A few putative species have been discovered recently which may be classified as members of the Pestivirus genus but they have not been approved as species yet. Among them are: HoBi-like pestiviruses (also called BVDV-3) identified first in batches of contaminated foetal calf serum [2] and then in calves and aborted fetuses [3, 4], giraffe pestivirus associated with the outbreak of mucosal-like disease in Kenyan giraffes [5], Bungowannah virus detected in pig herds in Australia where stillbirth foetuses and neonatal deaths were observed [6] and Pronghorn virus, isolated from a pronghorn antelope in the United States [7]. There are also reports of novel pestiviruses in other animal species like rats and bats [8, 9]. This wide range of pestiviruses infecting different animal species is the proof of genetic plasticity of their genomes, adapting to different hosts.
BVDV is an important pathogen of cattle worldwide with significant economic impact [10]. Infection may lead to a wide array of clinical signs from subclinical to severe acute hemorrhagic syndrome and fatal mucosal disease [11]. BVDV also causes immunosuppression, which increases the severity of clinical picture when other pathogens are involved. BVDV infection of seronegative and pregnant females during the first 40–120 days of pregnancy may lead to the birth of persistently infected (PI) calves. They remain infected for life and shed the virus in high titre, ensuring the persistence of BVDV in the herd if they are not removed immediately after identification.
Viral genome is comprised of a single-stranded positive sense RNA about 12.3 kb in size with one large open reading frame flanked by 5′ and 3′ untranslated regions (5’UTR and 3’UTR respectively) [1]. Pestiviral genome encodes a single polyprotein that is processed into either 11 or 12 proteins: Npro, C, Erns, E1, E2, p7, NS2–3 (NS2, NS3), NS4A, NS4B, NS5A, NS5B. Several regions of BVDV genome have been used to study its genetic diversity [12, 13]. Phylogenetic analysis is mostly based on the comparison of nucleotide sequences from the 5’UTR, Npro or E2 regions of viral genome. Based on genetic studies, 21 subtypes of BVDV-1 (1a - 1u) and 4 subtypes of BVDV-2 (2a – 2d) were identified so far [14, 15]. BVDV-1 is the predominant pestivirus circulating in cattle population in Europe [16]. Similar situation was observed in Poland, where studies encompassing years 2004–2014 revealed the presence of five subtypes of BVDV-1: 1b, 1d, 1f, 1 g [17] and 1e [18] in decreasing frequency. Later, BVDV-2a has been identified but only on one farm [19]. The aim of this study was to genotype BVDV isolates currently circulating in Poland. Such studies are important to understand epidemiology of the virus and they may support the development of successful control and eradication programs, where effective vaccines and reliable diagnostic tests are essential.
Results
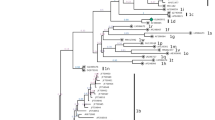
Positive results in RT-PCR test for BVDV were obtained for 63 samples from 30 farms in all 8 provinces tested (overall prevalence of 0.7%). Nucleotide alignment with the reference strains from GenBank using BLAST tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi) showed that all detected strains were characterized as BVDV-1. For phylogenetic tree construction, a 208 nucleotide fragment of the 5′UTR was analyzed and final result with the genetic relatedness of field and reference strains is shown in Fig. 1. One isolate (213-GK/18) was sequenced only in the Npro region (subtype 1f) therefore, sequence analysis in the 5′ untranslated region was based on 62 sequences. Field isolates were separated into seven groups representing seven separate subtypes. Twenty nine isolates were also genotyped within Npro region. The phylogenetic tree of the Npro was constructed based on a 281 nucleotide fragment (Fig. 2) fully confirming classification from 5’UTR even with higher bootstrap values. Analysis revealed that BVDV-1 strains belonged to subtypes 1b detected in 8 herds (n = 17), 1 g in 8 herds (n = 17), 1f in 7 herds (n = 15), 1d in 3 herds (n = 6), 1r in 3 herds (r = 4), 1 s in 2 herds (n = 3) and 1e detected in one herd (n = 1). In order to confirm the allocation of isolates to particular subtypes another tree was constructed using the Bayesian method (Additional file 1 and Additional file 2). Field strains have been assigned to the same subtypes. The list of analyzed isolates is given in Table 1. Animals from the same herd were infected with one subtype only and sequence homology between viral isolates at herd level was very high. The only exception were two farms: one in Wielkopolskie (Farm 10) and another one in Opolskie (Farm 29) province. After initial identification of BVDV-1d (184-KN/17, 185-KN/17, 196-KN/17) in Wielkopolskie farm, another subtype, namely BVDV-1 g (206-KN/17) was identified in the same year. One year later in Opolskie province BVDV-1f was identified (219-KH/18, 220-KH/18, 221-KH/18) followed by identification for the first time in Poland of BVDV-1r (218-KH/18, 222-KH/18) in the same farm. The number of isolates per farm was between 1 and 6, although at more than 80% of farms only 1 or 2 infected individuals were identified (Table 1). The number of subtypes identified anually was 4, 2, 5 and 5 in 2015, 2016, 2017 and 2018, respectively (Fig. 3). The most predominant subtypes of BVDV-1 pear year were: 1f and 1 s (30% each) in 2015, 1b (60%) in 2016, 1b (41%) in 2017 and 1 g (38%) in 2018. The only subtype identified each year was BVDV-1 g while 1 s was identified only in 2015 (like 1e in 2018).

Phylogenetic tree based on 5’UTR fragment of 62 field isolates of BVDV. Black dots indicate field strains

Phylogenetic tree based on Npro fragment of 29 field isolates of BVDV. Black dots indicate field strains

Distribution of BVDV subtypes in Poland between 2015 and 2018 (percentages)
Geographical clustering was observed for subtypes 1d, 1 s and 1e identified in different, single provinces. BVDV-1f was identified in 5 provinces, BVDV-1 g and BVDV-1r in 3 provinces, BVDV-1b in 2 provinces. The highest number of isolates (32) and subtypes (4), was identified in Wielkopolskie with the predominance of BVDV-1 g (41%) and BVDV-1b (37%). Second province with the highest number of positive results was Lubelskie, where 8 isolates of BVDV-1f and 5 of BVDV-1b subtypes were found. Only in two provinces (Podlaskie and Lodzkie), where positive results were obtained, single subtypes were identified. Sequence similarity between various subtypes in 5’UTR ranged from 81 to 93%. The identity percentages within same subtypes 1b, 1 g, 1f, 1d, 1r and 1 s were 91.5–100%, 96.5–100%, 91.4–100%, 92.6–100%, 96.5–98%, 99–100% respectively. Sequence similarity between various subtypes in Npro region ranged from 76.5 to 86.5%. The most diverse sequences within the same subtype in Npro region were identified for BVDV-1b with sequence identity values up to 84.9%. The biggest difference in subtype sequences occurred between BVDV-1b and BVDV-1d, while the tiniest variation was observed between BVDV-1f and BVDV-1 s (Fig. 4).

Matrix of pairwise identity scores generated by alignment of a 371 bp fragment of the Npro gene for 29 Polish isolates and 15 reference strains of BVDV
Sequence identity at the amino acid level in Npro region among isolates tested was 78.8–100% and between various subtypes ranged from 78.8 to 93.4%. The biggest differences were observed between BVDV-1d and BVDV-1r and the smallest one between BVDV-1f and BVDV-1 g and also between BVDV-1f and BVDV-1 s. Nucleotide sequences of the BVDV strains have been submitted to GenBank with the following accession numbers: MK044822-MK044827, MK168328-MK168336, MK381356-MK381402 for 5’UTR and MK381419-MK381447 for Npro region.
Discussion
In this study, we investigated the genetic diversity of BVDV isolates from Polish herds collected between 2015 and 2018. PCR amplified sequences were subjected to sequence-based genotyping in 5′ untranslated region. The Npro phylogenetic analysis confirmed typing results obtained for the 5’UTR. Viral isolates were assigned to seven subtypes in descending order of frequency of appearance: 1b, 1 g, 1f, 1d, 1r, 1 s and 1e. Previous study from years 2004–2011 described the circulation of four subtypes of BVDV-1 in Poland (1b, 1d, 1f, 1 g) with predominance of BVDV-1b and BVDV-1d [17]. In later studies, subtype 1e was also detected [18]. Current phylogenetic studies indicate that the number of BVDV subtypes has increased, however BVDV-1b is still the most often detected subtype. It is the most frequently reported subtype of BVDV worldwide. BVDV-1b is predominant in both Americas, Asia and Europe [16]. A large number of isolates belonging to subtype 1f and some of 1 g have been detected in Austria [20] and Italy [21, 22]. BVDV-1f is the most common subtype in Germany and Slovenia [16, 23]. Several studies indicate that 1f and 1 g subtypes may be unique for Europe. Viruses of BVD-1 g subtype were isolated more frequently now than in the previous study where BVDV-1 g was identified only in two herds [17]. Subtype 1d was predominant in Sweden, in years 2002–2004, when the eradication program was implemented [24]. Strains 183-SY/17, 211-DK/18, 218-KH/18 and 222-KH/18 clustered together with Italian strains belonging to subtype 1r [22]. Three strains (166-KY/15, 167-KY/15, 176-KR/15) form one clade with strains previously identified as 1f (22,146/81) [25] and 1f-like (mousedeer) [26]. Currently, together with the reference strains from Italy [22], they form the 1 s subtype [27]. BVDV-1e represented by strain 217-SM/18 has been identified only in one Polish herd. It had 98% nucleotide similarity to the Italian BVDV-1e strain from Northern Italy [28]. This subtype was found also in Switzerland [29] and France [30]. The results of this study show that the genetic heterogeneity of BVDV viruses infecting cattle in Poland has changed. These differences in subtype distribution in comparison to study from years 2004–2011 could be a result of immune selection due to natural infections and also vaccinations, which became very popular in recent years. In the present work, the evidence for geographical clustering of BVDV subtypes was not clear, unlike Italy, where BVDV-1f was predominant in northern Italy while BVDV-1b was the most frequent subtype in southern part of the country [21, 31].
HoBi-like pestiviruses (BVDV-3) do not seem to circulate in Polish cattle and BVDV-2 was found previously only in one herd [19]. BVDV-2 was first identified in North America and was associated with very high mortalities [32] from where the virus was introduced to the European continent [33]. BVDV-2 was also identified in Europe in several countries like: Italy [14], Germany [34] and Austria [20]. So far, natural infections with BVDV-3 in Europe were identified only in Italy [3]. There are suspicions that the virus has been introduced to the European continent through vaccines or other products which were prepared using contaminated bovine serum. The closest genetically related strains to Polish isolates were identified in Slovenia, United Kingdom and Italy according to blastn analysis. High level of similarity among these viruses may suggest a common ancestor.
Only a few inactivated and recently also modified-live vaccines are commercially available in Poland. In this study BVDV was identified in 6 herds from animals previously vaccinated with killed vaccines. Three herds were infected with BVDV-1b subtype (strains 187-AN/17, 189-JA/17, 190-JA/17 and 194-TC/17), two with BVDV-1d (184-KN/17, 185-KN/17, 188-LS/17) and one herd was infected with BVDV-1 g (215-BK/18). In all these herds protective vaccinations were based on BVDV-1a strain, and they were introduced after PIs removal. Interestingly subtype 1a has never been identified in Poland, which could be the effect of selection force induced by vaccines based on this subtype. Other studies have shown significant differences in antibody levels in serum from calves receiving modified live virus vaccines based on BVDV-1a, with a significantly lower BVDV-1b antibody titres [35]. PI individuals infected with BVDV-1b were identified in one Polish herd vaccinated with a killed vaccine based on BVDV-1a [36]. Although clinical symptoms resembling BVD were not observed in that herd, the protection offered by vaccinal strain did not provide cross protection against BVDV-1b. Vaccination strategy should take into consideration both genetic and antigenic diversity of the virus present in the region where vaccination is implemented and therefore, effective vaccine should include the subtypes of local isolates. For this reason monitoring of newly emerging strains is important for successful control and eradication programs and it requires constant updates. Antigenic differences among individual subtypes of BVDV-1 occur as well [37]. Therefore, more cross-protection studies should be carried out to address the importance of this diversity. It seems reasonable to include a mixture of several viral subtypes present in local herds when designing effective vaccines. Phylogenetic studies with increasing cattle trade can also help to identify potential sources and routes of virus introduction, although such sources were not identified for Polish isolates, probably due to significant diversity of the virus in every country studied.
The genetic diversity is also important for laboratory diagnosis, since it can hamper the ability of diagnostic methods to identify as many viral subtypes as possible. In this study we used specific primers for non-coding 5’UTR and coding Npro region. 5’UTR is highly conserved among the pestiviruses. It contains cis-acting elements required for viral replication and translation [38]. Npro (N-terminal protein) of BVDV encodes for a cysteine protease that cleaves the N-terminus from the core protein. Npro also prevents interferon-α/β induction in infected cells [39]. The validity of 5’UTR classification in this study was confirmed by the parallel analysis of Npro sequences. RT-PCR used in this study [40], which is commonly used for BVDV detection, does not detect or detects with low efficiency strains of HoBi-like viruses due to the presence of a mismatch at the 3′ end of the forward primer which does not allow proper annealing [41]. This disadvantage may lead to false negative results when testing field samples for BVDV-3 and therefore we implemented real-time PCR enabling the detection of all three species of BVDV with high sensitivity. This new method was implemented to study doubtful PCR results although all samples turned negative when tested with real-time PCR.
Conclusion
In summary, the distribution of subtypes in Poland has changed. Two new subtypes 1r and 1 s were detected for the first time. Monitoring of strains circulating in a given country is a useful indicator in the aspect of designing an effective vaccination program or a reliable diagnostic test.
Methods
Sample collection
A total of 9290 serum, tissue homogenate, ear notch and semen samples were collected in years 2015–2018. The animals used in the study came from private farms, where infection with BVDV was suspected based on clinical symptoms or where eradication was under way. The owners of those herds provided local vets with their permissions to collect samples for laboratory testing. Samples were collected in 8 out of 16 provinces of Poland: Kujawsko-Pomorskie, Lubelskie, Łódzkie, Opolskie, Świętokrzyskie, Mazowieckie, Wielkopolskie and Podlaskie. Cattle population in last three provinces comprises 51% of the total population of this ruminant species in Poland. For comparison studies sequences of 81 reference strains of different species and subtypes of BVDV and single strains of BDV and CSFV were retrieved from GenBank (Table 2).
RNA extraction and RT-PCR
Total RNA was extracted using TRI Reagent (Sigma-Aldrich, USA) from 500 μl of serum, tissue homogenates, cell culture medium after overnight soaking of ear notches or from diluted semen following the manufacturer’s instructions and stored at -80 °C until testing. Reverse transcription-polymerase chain reaction (RT-PCR) was carried out using the Transcriptor One-Step RT-PCR Kit (Roche) in a 25 μl reaction mix consisting of PCR buffer 5 μl, water DEPC 15.5 μl, set of primers 1 μl (10 μM), 0.5 μl enzyme mix and 2 μl of template RNA. Reverse transcription was performed at 50 °C for 30 min using reverse primer. cDNA was amplified using primers pair specific for BVDV 5′ untranslated region: 324F (5′-ATGCCCWTAGTAGGACTAGCA-3′) and 326R (5′-TCAACTCCATGT GCCATGTAC-3′) [40]. PCR thermal conditions were the following: initial denaturation at 94 °C for 7 min followed by 35 cycles of denaturation at 94 °C for 10 s, primer annealing at 53 °C for 30 s and elongation at 68 °C for 30 s. The final elongation was extended to 7 min at 68 °C. Primers specific for Npro region: B32-F (TGCTACTAAAAATCTCTGCTGT) and B31-R (CCATCTATrCAyACATArATGTGGT) [23] were used with thermal profile of 94 °C for 15 s, 50 °C for 30s and 68 °C for 1 min for 35 cycles and 10 min in 68 °C for final elongation. Approximate sizes of PCR products were 288 bp and 441 bp for 5’UTR and Npro region respectively.
Sequencing and phylogenetic analysis
The PCR products were sequenced in both directions with the same primers used for amplification using Big Dye Terminator v3.1 Cycle Sequencing Kit with a 3730XL Genetic Analyzer (Applied Biosystems). The DNA fragments were purified using a QIAquick PCR Purification kit (Qiagen), following the analysis in a 16-capillary sequencer ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). The consensus of each genetic region was determined by the alignment of forward and reverse strand sequences using Clustal Omega tool of the European Molecular Biology Laboratory (http://www.ebi.ac.uk). Sequences generated in this study were aligned with the analogous sequences of reference pestivirus strains deposited in the GenBank database (Table 2) using the ClustalW algorithm from Molecular Evolutionary Genetics Analysis software package, version 5.2 (MEGA 5.2). Phylogenetic trees were constructed using neighbor-joining algorithm [42] with a Kimura 2-parameter substitution model [43] with 1000 bootstrap replicates. Phylogenetic trees were also constructed by the Bayes method with the GTR substitution model using the tree-builder tool of the Geneious software [44]. Sequence identity (%) among strains was calculated using the identity matrix in BioEdit v.7.2.5 software [45].
Availability of data and materials
The data sets supporting the results of this article are included within the article.
Abbreviations
- 5’UTR:
-
Untranslated region 5′
- BVDV:
-
Bovine viral diarrhea virus
- Npro :
-
N-terminal protease
- PCR:
-
Polymerase chain reaction
- PI:
-
persistently infected
- RT:
-
Reverse transcription
References
Simmonds P, Becher B, Bukh J, Gould EA, Meyers G, Monath T, Muerhoff S, Pletnev A, Rico-Hesse R, Smith DB, Stapleton JT. ICTV report consortium.: ICTV virus taxonomy profile: Flaviviridae. J Gen Virol. 2017;98:2–3.
Peletto S, Zuccon F, Pitti M, Gobbi E, Marco LD, Caramelli M, Masoero L, Acutis PL. Detection and phylogenetic analysis of an atypical pestivirus, strain IZSPLV_To. Res Vet Sci. 2012;92(1):147–50.
Decaro N, Lucente MS, Mari V, Cirone F, Cordioli P, Camero M, Sciarretta R, Losurdo M, Lorusso E, Buonavoglia C. Atypical pestivirus and severe respiratory disease in calves, Europe. Emerg Infect Dis. 2011;17:1549–52.
Decaro N, Lucente MS, Mari V, Sciarretta R, Pinto P, Buonavoglia D, Martella V, Buonavoglia C. Hobi-like pestivirus in aborted bovine fetuses. J Clin Microbiol. 2012;50(2):509–12.
Plowright W. Other virus diseases in relation to the JP15 programme. In: Joint campaign against rinderpest, proceedings of the 1 st technical review meeting, phase IV. Mogadiscio: Organization of African Unity; 1969. p. 19–23.
Kirkland PD, Frost MJ, Finlaison DS, King KR, Ridpath JF, Gu X. Identification of a novel virus in pigs – Bungowannah virus: a possible new species of pestivirus. Virus Res. 2007;129:26–34.
Vilcek S, Ridpath JF, Van Campen H, Cavender JL, Warg J. Characterization of a novel pestivirus originating from a pronghorn antelope. Virus Res. 2005;108:187–93.
Firth C, Bhat M, Firth MA, Williams SH, Frye MJ, Simmonds P, et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in new York City. MBio. 2014. https://doi.org/10.1128/mBio.01933-14.
Wu Z, Ren X, Yang L, Hu Y, Yang J, He G, et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J Virol. 2012;86(20):10999–1012.
Houe H. Economic impact of BVDV infection in dairies. Biologicals. 2003;31:137–43.
Bachofen C, Braun U, Hilbe M, Ehrensperger F, Stalder H, Peterhans E. Clinical appearance and pathology of cattle persistently infected with bovine viral diarrhea virus of different genetic subgroups. Vet Microbiol. 2010;141(3–4):258–67.
Nagai M, Hayashi M, Sugita S, Sakoda Y, Mori M, Murakami T, Ozawa T, Yamada N, Akashi H. Phylogenetic analysis of bovine viral diarrhea viruses using five different genetic regions. Virus Res. 2004;99:103–13.
Silveira S, Weber MN, Mósena ACS, da Silva MS, Streck AF, Pescador CA, Flores EF, Weiblen R, Driemeier D, Ridpath JF, Canal CW. Genetic diversity of Brazilian bovine Pestiviruses detected between 1995 and 2014. Transbound Emerg Dis. 2015. https://doi.org/10.1111/tbed.12427.
Decaro N, Lucente MS, Lanave G, Gargano P, Larocca V, Losurdo M, Ciambrone L, Marino PA, Casalinuovo F, Elia G, Buonavoglia C. Evidence for circulation of bovine viral diarrhoea virus type 2c in ruminants in southern Italy. Transbound Emerg Dis. 2016. https://doi.org/10.1111/tbed.12592.
Giangaspero M, Harasawa R, Weber L, Bellolo A. Genoepidemiological evaluation of bovine viral diarrhea virus 2 species based on secondary structures in the 5′ untranslated region. J Vet Med Sci. 2008;70(6):571–80.
Yesilbag K, Alpay G, Becher P. Variability and global distribution of subgenotypes of bovine viral diarrhea virus. Viruses. 2017. https://doi.org/10.3390/v9060128.
Kuta A, Polak MP, Larska M, Żmudziński JF. Predominance of bovine viral diarrhea virus 1b and 1d subtypes during eight years of survey in Poland. Vet Microbiol. 2013;166:639–44.
Kuta A, Polak MP, Larska M, Żmudziński JF. Genetic typing of bovine viral diarrhea virus (BVDV) by restriction fragment length polymorphism (RFLP) and identification of a new subtype in Poland. B Vet I Pulawy. 2015;59:19–22.
Polak MP, Kuta A, Rybałtowski W, Rola J, Larska M, Żmudziński JF. First report of bovine viral diarrhoea virus-2 infection in cattle in Poland. Vet J. 2014;202:643–5.
Hornberg A, Fernandez SR, Vogl C, Vilcek S, Matt M, Fink M, Kofer J, Schopf K. Genetic diversity of pestivirus isolates in cattle from Western Austria. Vet Microbiol. 2009;135:205–13.
Cerutti F, Luzzago C, Lauzi S, Ebranati E, Caruso C, Masoero L, Moreno A, Acutis PL, Zehender G, Peletto S. Phylogeography, phylodynamics and transmission chains of bovine viral diarrhea virus subtype 1f in northern Italy. Infect Genet Evol. 2016;45:262–7.
Giammarioli M, Ceglie L, Rossi E, Bazzucchi M, Casciari C, Petrini S, De Mia GM. Increased genetic diversity of BVDV-1: recent findings and implications thereof. Virus Genes. 2015;50:147–51.
Toplak I, Sandvik T, Barlič-Maganja D, Grom J, Paton DJ. Genetic typing of bovine viral diarrhoea virus: most Slovenian isolates are of genotypes 1d and 1f. Vet Microbiol. 2004;99:175–85.
Ståhl K, Kampa J, Baule C, Isaksson M, Moreno-Lopez J, Belak S, Alenius S, Lindberg A. Molecular epidemiology of bovine viral diarrhoea during the final phase of the Swedish BVD-eradication programme. Prev Vet Med. 2005;72:103–8.
Tajima M, Frei HR, Yamato O, Maede Y, Moennig V, Scholz H, Greiser-Wilke I. Prevalence of genotypes 1 and 2 of bovine viral diarrhea virus in Lower Saxony, Germany. Virus Res. 2001;76:31–42.
Grondahl C, Uttenthal A, Houe H, Rasmussen TB, Hoyer MJ, Larsen LE. Characterisation of a pestivirus isolated from persistently infected mousedeer (Tragulus javanicus). Arch Virol. 2003;148:1455–63.
Giangaspero M, Yesilbag K, Apicella C. Who’s who in the bovine viral diarrhea virus type 1 species: genotypes L and R. Virus Res. 2018;256:50–75.
Ebranati E, Lauzi S, Cerutti F, Caruso C, Masoero L, Moreno A, De Mia GM, Peletto S, Zehender G, Luzzago C. Highlighting priority areas for bovine viral diarrhea control in Italy: a phylogeographic approach. Infect Genet Evol. 2018;58:258–68.
Stalder HP, Meier P, Pfaffen G, Wageck-Canal C, Rufenacht J, Schaller P, Bachofen C, Marti S, Vogt HR, Peterhans E. Genetic heterogeneity of pestiviruses of ruminants in Switzerland. Prev Vet Med. 2005;72:37–41.
Jackova A, Novackova M, Pelletier C, Audeval C, Gueneau E, Haffar A, Petit E, Rehby L, Vilcek S. The extended genetic diversity of BVDV-1, typing of BVDV isolates from France. Vet Res Commun. 2008;32:7–11.
Lanave G, Decaro N, Lucente MS, Guercio A, Cavaliere N, Purpari G, Padalino I, Larocca V, Antoci F, Marino PA, Buonavoglia C, Elia G. Circulation of multiple subtypes of bovine viral diarrhoea virus type 1 with no evidence for HoBi-like pestivirus in cattle herds of southern Italy. Infect Genet Evol. 2017;50:1–6.
Pellerin C, van den Hurk J, Lecomte J, Tussen P. Identification of a new group of bovine viral diarrhea virus strains associated with severe outbreaks and high mortalities. Virology. 1994;203:260–8.
Falcone E, Tollis M, Conti G. Bovine viral diarrhea disease associated with a contaminated vaccine. Vaccine. 1999;18:387–8.
Gethmann J, Homeier T, Holsteg M, Schirrmeier H, Saβerath M, Hoffmann B, Beer M, Conraths FJ. BVD-2 outbreak leads to high losses in cattle farms in Western Germany. Heliyon. 2015. https://doi.org/10.1016/j.heliyon.2015.e00019.
Fulton RW, Ridpath JF, Confer AW, Saliki JT, Burge LJ, Payton ME. Bovine viral diarrhoea virus antigenic diversity: impact on disease and vaccination programmes. Biologicals. 2003;31:89–95.
Polak M, Antos A, Rola J, Żmudziński JF. Viral shedders in a herd vaccinated against infection with bovine viral diarrhoea virus (BVDV) without prior testing for the presence of persistently infected animals. J Vet Res. 2016;60:379–84.
Pecora A, Malacari DA, Ridpath JF, Perez Aguirreburualde MS, Combessies G, Odeon AC, Romera SA, Golemba MD, Wigdorovitz A. First finding of genetic and antigenic diversity in 1b-BVDV isolates from Argentina. Res Vet Sci. 2014;96:204–12.
Becher P, Orlich M, Thiel H. Mutations in the 5′ nontranslated region of bovine viral diarrhea virus result in altered growth characteristics. J Virol. 2000;74:7884–94.
Gottipati K, Acholi S, Ruggli N, Choi K. Autocatalytic activity and substrate specificity of the pestivirus N-terminal protease Npro. Virology. 2014. https://doi.org/10.1016/j.virol.2014.01.026.
Vilcek S, Herring AJ, Herring JA, Nettleton PF, Lowings JP, Paton DJ. Pestiviruses isolated from pigs, cattle and sheep can be allocated into at least three genogroups using polymerase chain reaction and restriction endonuclease analysis. Arch Virol. 1994;136:309–23.
Mari V, Losurdo M, Lucente MS, Lorusso E, Elia G, Martella V, Patruno G, Buonavoglia D, Decaro N. Multiplex real-time RT-PCR assay for bovine viral diarrhea virus type 1, type 2 and HoBi-like pestivirus. J Virol Methods. 2016;229:1–7.
Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9.
Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20.
Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A. Geneious v5.5, biomatters. 2010. http://www.geneious.com. Accessed 11 Feb 2013.
Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucl Acid S. 1999;41:95–8.
Acknowledgements
The authors thank Malgorzata Glowacka and Agnieszka Nowakowska for their excellent technical assistance.
Funding
Funded by KNOW (Leading National Research Centre) Scientific Consortium “Healthy Animal - Safe Food”, decision of Ministry of Science and Higher Education No. 05–1/KNOW2/2015. The funding body was solely involved in funding and had no role in the design of the study, the collection, analysis, interpretation of the data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
MP supervised the project. PM conducted and coordinated the study including laboratory and computer analysis and drafted the manuscript. MP drafted and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The material used in this study consisted of field samples collected during clinical examination of animals and these animals were not used for experimental studies. Tissue samples were collected from dead animals by local vets after verbal approvals from the owners for further testing. The approval from ethics committee was not required according to national regulation (“Act on the Protection of Animals Used for Scientific or Educational Purposes” published in the Journal of Laws of 2015, item 266 from 15 January, 2015).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Phylogenetic relationship between field and reference strains inferred by Bayesian analysis in 5’UTR. The figure shows a phylogenetic tree created on the basis of the 5’UTR fragment by the Bayes method with the GTR substitution model. It consists of 62 field isolates and representatives of all known subtypes of the BVDV-1 species, representatives of the BVDV-2, BDV and CSFV species. (PDF 148 kb)
Additional file 2:
Phylogenetic relationship between field and reference strains inferred by Bayesian analysis in Npro region. The figure shows a phylogenetic tree created on the basis of the fragment of the Npro region by the Bayes method with the GTR substitution model. It consists of 29 field isolates and representatives of all known subtypes of the BVDV-1 species, representatives of the BVDV-2, BDV and CSFV species. (PDF 120 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mirosław, P., Polak, M. Increased genetic variation of bovine viral diarrhea virus in dairy cattle in Poland. BMC Vet Res 15, 278 (2019). https://doi.org/10.1186/s12917-019-2029-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-019-2029-z