
Abstract
Background
Fowl adenoviruses (FAdVs) are associated with many diseases, resulting in huge economic losses to the poultry industry worldwide. Since 2015, outbreaks of FAdV infections with high mortality rates have been reported in China. A continued surveillance of FAdVs contributes to understand the epidemiology of the viruses.
Results
We isolated 155 FAdV strains from diseased chickens from poultry in China between 2015 and 2018. PCR analysis determined that 123 samples were FAdV species C, 27 were FAdV species E, and five contained two different FAdV strains. The phylogenetic analysis demonstrates that these sequences of hexon regions were clustered into three distinct serotypes: FAdV-4 (79.4%, 123/155), FAdV-8a (13.5%, 21/155) and FAdV-8b (3.9%, 6/155), of which FAdV-4 was the dominant serotype in China.
Conclusions
The characterization of newly prevalent FAdV strains provides valuable information for the development of an effective control strategy for FAdV infections in chickens.
Similar content being viewed by others
Background
Fowl adenoviruses (FAdVs) are non-enveloped double-stranded DNA viruses with a genome of 43–45 kb in size and a diameter of 70–100 nm, belonging to the genus Aviadenovirus in the family of Adenoviridae [1]. The viruses are clustered into 5 species (FAdV-A to FAdV-E) based on restriction enzyme digest patterns, and they are further classified into 12 serotypes (FAdV-1 to 8a and 8b to 11) by cross-neutralization tests [2]. The hexon protein, as a major surface-exposed capsid structure, harbors group, type and subtype-specific antigenic determinants [3, 4]. Therefore, hexon gene is important for understanding of the genetic relations of FAdV field strains and the epidemiological status of the virus [5, 6].
FAdVs are disseminated both vertically and horizontally, and widely distributed in poultry industry throughout the world [7,8,9]. Most of these viruses are opportunistic pathogens and their infections are subclinical [10]. However, some FAdVs are associated with a wide range of avian diseases, including inclusion body hepatitis (IBH), hepatitis-hydropericardium syndrome (HHS) and gizzard erosion ulceration (GEU), causing substantial economic losses to poultry industry [11,12,13]. Each of the 12 FAdV serotypes has been related to outbreaks of IBH in chickens with 10–30% mortality [5,6,7,8,9,10,11,12,13,14]. HHS is primarily caused by the FAdV-4 serotype and causes a high mortality of 30–80% [15]. In addition, FAdV-1 and FAdV-8 strains are responsible for GEU [16, 17]. In recent years, the outbreaks of IBH and HHS showed an increasing trend in poultry industry [18, 19].
In this study, a total of 280 diagnostic samples from chicken flocks were collected from 15 provinces or cities in China from 2015 to 2018. We identified the FAdV strains using sequencing and phylogenetic analysis of the hexon genes to establish the genetic relationships of FAdV strains. These results would provide us the information on the epidemiology of FAdV infection, and provide an efficacious control strategy for FAdV infections.
Methods
Sample collection and virus isolation
From 2015 to 2018, 280 diagnostic cases were collected from commercial chicken flocks potentially affected by FAdV at ninety-six different chicken farms distributed in 15 provinces or cities in China, including Guangdong, Guangxi, Hunan, Hubei, Jiangxi, Sichuan, Chongqing, Yunnan, Zhejiang, Jiangsu, Henan, Shandong, Hebei, Tianjin and Liaoning. The epidemiological details of the positive isolates are described in Additional file 1: Table S1. Tissue samples (liver, kidney and spleen) were homogenized in phosphate buffered saline (PBS) to obtain a 10% tissue suspension. The suspensions were centrifuged at 5000×g for 10 min after three freeze-thaw cycles. The supernatants were collected, filtered through a 0.22-μm-pore-size syringe filter (Millipore, USA) and inoculated into 7-day-old specific pathogen-free (SPF) chicken embryos which were collected from the SPF Experimental Animal Center (Wens Dahuanong Biotechnology Co., Ltd., Guangdong, China) as previously described [18]. All animals used in this study were unconscious and anaesthesia was maintained with isoflurane in oxygen, and then euthanized by parenteral pentobarbitone at the end of experiment as previously described [20]. Animal experiments were approved by local institutional ethical committee to conduct experiments in chicken embryo and poultry birds as per ethical requirement.
PCR and sequencing
The presence of FAdV in each sample was confirmed by polymerase chain reaction (PCR), according to procedures described previously [21]. Viral DNAs were extracted from the harvested cultures using the MiniBEST Universal Genomic DNA Extraction Kit (TaKaRa, China), in accordance with the manufacturer’s instructions. Different serotypes of FAdVs were tested by a PCR assay, and typing primers were described in Table 1. The published primers 1–2 were used to amplify the complete hexon gene sequences of FAdV-C [19], and the primers 3–4 were used to amplify the complete hexon gene sequences of FAdV-E [22]. Samples of the expected length were sent for sequencing directly by Huada Company (Beijing, China).
Phylogenetic analysis
The nucleotide sequences of the FAdV isolates were assembled and aligned with homologous sequences via the Clustal W method using the Lasergene software (DNAStar, Madison, WI, USA). A phylogenetic tree for the hexon gene was constructed using MEGA 7.0 software by the neighbor-joining method with 1000 bootstrap replicates. 25 reference strains used in this study are summarized in Table 2.
Results
Epidemiological analysis
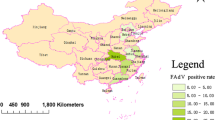
More than 280 suspected clinical cases of IBH or HHS were examined between 2015 and 2018, and these samples were distributed in 15 provinces or cities in China, including Guangdong, Guangxi, Hunan, Hubei, Jiangxi, Sichuan, Chongqing, Yunnan, Zhejiang, Jiangsu, Henan, Shandong, Hebei, Tianjin and Liaoning (Fig. 1). The epidemiological map constructed as a result of current study depicts 280 suspected clinical cases. Among them, 155 samples (55.4%) were positive for FAdV after PCR detection. Temporal analysis showed that the number of positive samples collected from 2015 to 2018 was 10, 73, 38 and 34, respectively, and most of the positive samples were isolated in 2016 (Fig. 2a). Spatial analysis showed that FAdV strains were collected from 15 provinces or cities: 10 provinces were in southern China, and the others were in northern China. In total, 70 strains were isolated in Guangdong Province (Fig. 2b).

Geographic distribution of collected FAdV samples. The samples were collected from 15 provinces or cities in China. The colours correspond to the number of collected samples

Summary of isolated FAdV strains. (a) the distribution of FAdV positive samples from 2015 to 2018, (b) the distribution of FAdV positive samples in different regions, (c) FAdV serotypes and (d) the age of the affected birds and complications
Serotyping of FAdV isolates
Hexon gene was sequenced for all 155 positive samples with Sanger method. In 155 cases, 123 (79.4%, 123/155) of the isolates were related to FAdV-C, and 27 (17.4%, 27/155) were identified as FAdV-E, whereas 5 (3.2%, 5/155) of the cases were proven to include two different FAdV strains (FAdV-C with FAdV-E) (Fig. 2c). Phylogenetic analysis based on the hexon gene showed that these isolates were clustered into three distinct serotypes: FAdV-4 (79.4%, 123/155), FAdV-8a (13.5%, 21/155) and FAdV-8b (3.9%, 6/155), of which FAdV-4 was the dominant serotype in China. Clinical cases of FAdV-4, FAdV-8a and FAdV-8b infections happened in chickens that were 1–73 days old (average 39.1 days), 1–7 days old (average 1.7 days) and 35–45 days old (average 39.8 days) respectively, and mix infections occurred in 1-day-old chicken (Fig. 2d).
Phylogenetic analysis of FAdV isolates
The results of the phylogenetic analysis of FAdV isolated strains and reference strains are shown in Fig. 3. The FAdV strains clustered into two major groups. By computing the pairwise distance of full-length hexon coding sequence, 123 strains from species FAdV-C had 98.7–100% similarities with FAdV-4 reference strains, 21 FAdV-E strains were found to share 99.3–100% similarities with the FAdV-8a reference strains, and six FAdV-E strains possessed 98.3–99.5% identities with FAdV-8b reference strains at the nucleotide level.

Phylogenetic tree based on hexon gene sequences of 155 field strains and other representative FAdV strains. The tree was constructed using MEGA 7.0 software by the neighbor-joining method (1000 replicates for bootstrap).‘ ’ indicate FAdV-C hexon gene sequences and‘
’ indicate FAdV-C hexon gene sequences and‘ ‘indicate FAdV-E hexon gene sequences
‘indicate FAdV-E hexon gene sequences
Discussion
Recently, more and more clinical cases of FAdV infection have been reported worldwide, and multiple FAdV strains have been isolated from dead or sick animals [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23]. In addition, epidemics with mixed serotypes have been observed in different regions, for instance, FAdV-4 and -8 in Asia [10, 18, 23, 24], FAdV-2 and -8b in South Africa [25, 26], FAdV-2, -11, -7 and -8 in Europe and North America [6, 27, 28]. FAdV infections are associated with some avian diseases, such as IBH, HHS and GEU. In China, the prevalence of IBH is sporadic in chickens without resulting in a high mortality rate. However, starting from 2015, outbreaks of HHS have been reported with high mortality rates in several broiler-producing provinces in China due to new FAdV variants. Subsequently, the disease spread throughout the country and lead to tremendous economic losses to the fast-developing poultry industry [18]. FAdV hexon gene plays an important part in the molecular epidemiology and in the genetic variation of field strains. In order to prevent and control FAdV infection, it is necessary for us to further investigate the prevalence of FAdV and the molecular characteristics of the hexon genes of Chinese FAdV field strains from 2015 to 2018. In this study, the genetic epidemiology of 155 FAdV samples collected between 2015 and 2018 in China was investigated.
It was previously reported that IBH and HHS mainly affected 3–5-week-old chickens, although outbreaks have occasionally occurred among layers [29, 30]. The present epidemiological survey showed that some clinical cases of FAdV infections have affected various ages of chickens and it might be caused by both horizontal and vertical transmission, although we were unable to know whether the route of transmission was horizontal or vertical in each case [18]. It also showed that FAdV infections were associated with different FAdV species in China, and at least two species of FAdVs (species C and E) were detected. In our surveillance of FAdV, IBH outbreaks have been related to FAdV-8a and FAdV-8b, and HHS cases have been related to FAdV-4. In addition, 79.4% FAdV strains were genetically related to serotype 4, proving that this serotype was dominant in China between 2015 and 2018. In a previous epidemiological survey, FAdV-11 was the predominant serotype in some regions of China from 2007 to 2014 [31]. Thus, it is necessary for us to further investigate the prevalence of FAdV, and understand the genetic epidemiology of viruses associated with IBH and HHS.
In our survey, five cases were detected with mixed infections of FAdV strains (FAdV-C with FAdV-E), which confirmed that mixed infections with different FAdV serotypes could happen in the same bird as previously reported [18, 27]. The possible reason for this was that commercial chickens often transmitted vertically and horizontally by fecal route. In this case, multiple infections might occur in a chicken flock due to the reactivation of latent infection virus. So far, it is ambiguous whether mixed infections of adenovirus could aggravate the severity of the clinical diseases [18]. In fact, the control and prevention of disease often centered on the primary epidemic viral serotypes and ignored previous mixed infection of FAdV, this may result in outbreaks of other serotypes in China. Thus, mixed infection of FAdV with multiple serotypes will be a big challenge to prevent and control the disease.
Conclusions
Fowl adenoviruses (FAdVs) have a worldwide distribution and are associated with a variety of diseases, causing huge economic losses to the poultry industry. In this study, we isolated 155 FAdV strains from diseased chickens from poultry in China between 2015 and 2018, and investigated the molecular characteristics by performing phylogenetic analyses based on the hexon genes. It showed that these isolates were clustered into three distinct serotypes: FAdV-4 (79.4%, 123/155), FAdV-8a (13.5%, 21/155) and FAdV-8b (3.9%, 6/155), of which FAdV-4 was the dominant serotype in China. Taken together, these results provided us a clear landscape of genetic epidemiology of FAdV circulating in China during 2015–2018 and would help us for the FAdV prevention.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- FAdV-4:
-
Serotype 4 fowl adenovirus
- FAdV-8a:
-
Serotype 8a fowl adenovirus
- FAdV-8b:
-
Serotype 8b fowl adenovirus
- FAdVs:
-
Fowl adenovirus
- GEU:
-
Gizzard erosion ulceration
- HHS:
-
Hepatitis-hydropericardium syndrome
- IBH:
-
Inclusion body hepatitis
- PCR:
-
Polymerase chain reaction
- SPF:
-
Specific pathogen free
References
Harrach B, Benko M. Phylogenetic analysis of adenovirus sequences. Methods Mol Med. 2007;131:299–334.
Hess M. Detection and differentiation of avian adenoviruses: a review. Avian Pathol. 2000;29:195–206.
Toogood CI, Murali R, Burnett RM, Hay RT. The adenovirus type 40 hexon: sequence, predicted structure and relationship to other adenovirus hexons. J Gen Virol. 1989;70(Pt 12):3203–14.
Russell WC. Adenoviruses: update on structure and function. J Gen Virol. 2009;90(Pt 1):1–20.
Mase M, Mitake H, Inoue T, Imada T. Identification of group I-III avian adenovirus by PCR coupled with direct sequencing of the hexon gene. J Vet Med Sci. 2009;71:1239–42.
Niczyporuk JS. Phylogenetic and geographic analysis of fowl adenovirus field strains isolated from poultry in Poland. Arch Virol. 2016;161:33–42.
Schachner A, Matos M, Grafl B, Hess M. Fowl adenovirus-induced diseases and strategies for their control - a review on the current global situation. Avian Pathol. 2018;47:111–26.
Grafl B, Aigner F, Liebhart D, Marek A, Prokofieva I, Bachmeier J, et al. Vertical transmission and clinical signs in broiler breeders and broilers experiencing adenoviral gizzard erosion. Avian Pathol. 2012;41:599–604.
Grgic H, Philippe C, Ojkic D, Nagy E. Study of vertical transmission of fowl adenoviruses. Can J Vet Res. 2006;70:230–3.
Choi KS, Kye SJ, Kim JY, Jeon WJ, Lee EK, Park KY, et al. Epidemiological investigation of outbreaks of fowl adenovirus infection in commercial chickens in Korea. Poult Sci. 2012;91:2502–6.
Mase M, Nakamura K, Minami F. Fowl adenoviruses isolated from chickens with inclusion body hepatitis in Japan, 2009-2010. J Vet Med Sci. 2012;74:1087–9.
Domanska-Blicharz K, Tomczyk G, Smietanka K, Kozaczynski W, Minta Z. Molecular characterization of fowl adenoviruses isolated from chickens with gizzard erosions. Poult Sci. 2011;90:983–9.
Lim TH, Lee HJ, Lee DH, Lee YN, Park JK, Youn HN, et al. Identification and virulence characterization of fowl adenoviruses in Korea. Avian Dis. 2011;55:554–60.
Ojkic D, Martin E, Swinton J, Vaillancourt JP, Boulianne M, Gomis S. Genotyping of Canadian isolates of fowl adenoviruses. Avian Pathol. 2008;37:95–100.
Kim JN, Byun SH, Kim MJ, Kim J, Sung HW, Mo IP. Outbreaks of hydropericardium syndrome and molecular characterization of Korean fowl adenoviral isolates. Avian Dis. 2008;52:526–30.
Okuda Y, Ono M, Shibata I, Sato S, Akashi H. Comparison of the polymerase chain reaction-restriction fragment length polymorphism pattern of the fiber gene and pathogenicity of serotype-1 fowl adenovirus isolates from gizzard erosions and from feces of clinically healthy chickens in Japan. J Vet Diagn Investig. 2006;18:162–7.
Okuda Y, Ono M, Shibata I, Sato S. Pathogenicity of serotype 8 fowl adenovirus isolated from gizzard erosions of slaughtered broiler chickens. J Vet Med Sci. 2004;66:1561–6.
Niu Y, Sun Q, Zhang G, Sun W, Liu X, Xiao Y, et al. Epidemiological investigation of outbreaks of fowl adenovirus infections in commercial chickens in China. Transbound Emerg Dis. 2018;65:e121–6.
Zhang T, Jin Q, Ding P, Wang Y, Chai Y, Li Y, et al. Molecular epidemiology of hydropericardium syndrome outbreak-associated serotype 4 fowl adenovirus isolates in Central China. Virol J. 2016;13:188.
O'Kane PM, Connerton IF, White KL. Pilot study of long-term anaesthesia in broiler chickens. Vet Anaesth Analg. 2016;43:72–5.
Niu YJ, Sun W, Zhang GH, Qu YJ, Wang PF, Sun HL, et al. Hydropericardium syndrome outbreak caused by fowl adenovirus serotype 4 in China in 2015. J Gen Virol. 2016;97:2684–90.
Ruan SF, Zhao J, Ren YC, Feng JL, Zhang GZ. Phylogenetic analyses of fowl adenoviruses (FAdV) isolated in China and pathogenicity of a FAdV-8 isolate. Avian Dis. 2017;61:353–7.
Mittal D, Jindal N, Tiwari AK, Khokhar RS. Characterization of fowl adenoviruses associated with hydropericardium syndrome and inclusion body hepatitis in broiler chickens. Virus disease. 2014;25:114–9.
Park HS, Lim IS, Kim SK, Kim TK, Park CK, Yeo SG. Molecular analysis of the hexon, penton base, and fiber-2 genes of Korean fowl adenovirus serotype 4 isolates from hydropericardium syndrome-affected chickens. Virus Genes. 2017;53:111–6.
Joubert HW, Aitchison H, Maartens LH, Venter EH. Molecular differentiation and pathogenicity of Aviadenoviruses isolated during an outbreak of inclusion body hepatitis in South Africa. J S Afr Vet Assoc. 2014;85:1058.
Maartens LH, Joubert HW, Aitchison H, Venter EH. Inclusion body hepatitis associated with an outbreak of fowl adenovirus type 2 and type 8b in broiler flocks in South Africa. J S Afr Vet Assoc. 2014;85:e1–5.
Kajan GL, Kecskemeti S, Harrach B, Benko M. Molecular typing of fowl adenoviruses, isolated in Hungary recently, reveals high diversity. Vet Microbiol. 2013;167:357–63.
Schachner A, Marek A, Grafl B, Hess M. Detailed molecular analyses of the hexon loop-1 and fibers of fowl aviadenoviruses reveal new insights into the antigenic relationship and confirm that specific genotypes are involved in field outbreaks of inclusion body hepatitis. Vet Microbiol. 2016;186:13–20.
Naeem K, Niazi T, Malik SA, Cheema AH. Immunosuppressive potential and pathogenicity of an avian adenovirus isolate involved in hydropericardium syndrome in broilers. Avian Dis. 1995;39:723–8.
Afzal M, Muneer R, Stein G. Studies on the aetiology of hydropericardium syndrome (Angara disease) in broilers. Vet Rec. 1991;128:591–3.
Changjing L, Haiying L, Dongdong W, Jingjing W, Youming W, Shouchun W, et al. Characterization of fowl adenoviruses isolated between 2007 and 2014 in China. Vet Microbiol. 2016;197:62–7.
Acknowledgements
No applicable.
Funding
Funding information is not applicable.
Author information
Authors and Affiliations
Contributions
LC and LY carried out most of the experiments and drafted the manuscript. QZ participated in the design of the study. PP, YD and LL collected the samples and carried out the sequencing. YZ and CX analyzed the data. YC conceived the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal experiments were performed strictly in accordance with the guidelines of Sun Yat-sen University Institutional Animal Care and Use Committee (Permit number: IACUC DD-15-0905). The research was conducted in the compliance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. And the ownership of the animals used in this study were research animals.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Epidemiological findings of flocks that were positive for FAdV. (DOC 404 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, L., Yin, L., Zhou, Q. et al. Epidemiological investigation of fowl adenovirus infections in poultry in China during 2015–2018. BMC Vet Res 15, 271 (2019). https://doi.org/10.1186/s12917-019-1969-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-019-1969-7