
Abstract
Background
Spinocerebellar ataxia also referred to as hereditary ataxia comprises different forms of progressive neurodegenerative diseases. A complex mode of inheritance was most likely in Parson Russell Terriers (PRT) and in Jack Russell Terriers (JRT). Recently, the missense mutation KCNJ10:c.627C > G was shown to be associated with the spinocerebellar ataxia (SCA) in JRT and related Russell group of terriers, whereas the missense mutation CAPN1:c.344G > A was associated with late onset ataxia (LOA) in PRT.
Results
We performed a genome-wide association study (GWAS) in PRT including 15 cases and 29 controls and found a statistically strong signal in the genomic region on dog chromosome 38 (CFA38) where KCNJ10 is located. We tested the CAPN1:c.344G > A and KCNJ10:c.627C > G (Transcript XM_545752.4) mutations in a sample of 77 PRT and 9 JRT from Germany as well as further 179 controls from 20 different dog breeds. All cases and controls genotyped carried the wild-type for the CAPN1:c.344G > A mutation. Among the PRT, 17/77 (22.1 %) dogs were homozygous for the mutant KCNJ10 allele and 22/77 (28.6 %) dogs were heterozygous. Three cases of PRT had the homozygous KCNJ10 wild-type. In JRT, 1/3 cases did show the mutant KCNJ10 allele homozygous. Thus, we sequenced the KCNJ10 exons with their adjacent regions from 10 PRT and 3 JRT including the animals with imperfect co-segregation of the c.627C > G mutation. We identified a total of 45 genetic variants within KCNJ10. The most likely variant explaining the cases appeared a 1-bp-insertion in a C-stretch within exon 3 (KCNJ10:g.22141027insC). In silico analysis showed that this indel may influence the regulation of gene expression.
Conclusions
In the present study, 16/21 cases of hereditary ataxia perfectly co-segregated with the KCNJ10:c.627C > G mutation. The CAPN1:c.344G > A mutation could not be validated and seems to be a rare variant in the samples screened. Screening KCNJ10 for further mutations did result in a genetic variant explaining 2 JRT cases but further 3 cases with a non-mutant homozygous c.627C > G genotype could not be resolved. Breeders have to be aware that DNA-testing for hereditary ataxia in PRT and JRT does not capture all cases of hereditary ataxia in these dog breeds. At least one further form of hereditary ataxia not yet resolved by a mutation may occur in PRT and JRT.
Similar content being viewed by others

Background
In dogs, spinocerebellar ataxia (hereditary ataxia) is characterized by progressive incoordination of gait, loss of balance, hypermetric and spastic movements. This condition may be associated with changes of brainstem auditory evoked potentials, myokymia, neuromyotonia and muscle fasciculation or seizures [1–7].
Hereditary ataxia is classified according to different aspects like age of onset (neonatal, juvenile and adult) and location of the pathomorphological lesions (spinal cord and/or cerebellar cortex) [8]. In Parson (PRT) and Jack Russell Terriers (JRT) lesions were found in the spinal cord and in the brain [2]. In Fox Terriers, lesions were only present in the spinal cord [9], whereas a more recent study reported histopathological degenerative changes in the brainstem [10]. Histopathological examinations of the central nervous system in PRT and JRT demonstrated a bilateral symmetrical myelopathy, predominantly a neuronal axonopathy in the dorsal and ventral or ventromedial funiculi [1, 2]. Loss of axons and myelin and astrogliosis were primarily seen in the spinocerebellar tracts of the cervical cord but were also present in all parts of the brain [1, 2, 7].
A complex segregation analysis using regressive logistic models was performed to elucidate the mode of inheritance in PRT and JRT [2]. The data included 3 pedigrees with a total of 115 dogs, either JRT or PRT, and 27 affected with the typical clinical signs and 9/27 with a histopathological examination showing a bilateral symmetric myelopathy in the nervous system. A monogenic mode of inheritance could be clearly ruled out. Mixed models with a major gene effect and a polygenic model were most likely for these pedigrees. The analysis could not differentiate how many loci may segregate in these two dog breeds.
In JRT, PRT and related breeds, the missense mutation c.627C > G in the KCNJ10 (potassium inwardly-rectifying channel, subfamily J, member 10) gene (Transcript XM_545752.4) was found to be strongly associated with spinocerebellar ataxia (SCA) [7, 11]. This KCNJ10 variant was identified screening a whole-genome sequence of a single JRT with spinocerebellar ataxia and myokymia under the assumptions of an autosomal recessive inheritance and a mutation altering the protein structure. Data filtering included other 81 canids that had not shown SCA. Affected animals showed ataxia, myokymia and/or seizures beginning from 2–12 months of age. The spinal cord sections revealed a bilateral myelopathy. Brain magnetic resonance imaging showed no abnormalities and hearing loss was not evident [7].
A missense mutation in the CAPN1 gene (c.344G > A) was associated with a late-onset spinocerebellar ataxia phenotype (LOA) in a cohort of PRT [12]. Signs of symmetric spinocerebellar ataxia had been noticed in affected dogs between 6 and 12 months of age. The CAPN1 missense mutation c.344G > A was identified using a genome-wide association study (GWAS) followed by the massively parallel sequencing of affected and control PRT for the LOA-associated chromosomal interval.
The objectives of this study were to perform a genome-wide association study (GWAS) in PRT for hereditary ataxia using the canine Illumina high density bead chip and then to show whether the previously reported SCA- and LOA-associated KCNJ10 and CAPN1 genes in PRT and JRT are in windows of significantly associated genomic regions. In addition, we sequenced KCNJ10 and validated the previously reported KCNJ10 and CAPN1 mutations in PRT and JRT and further control samples of other breeds of dog.
Methods
We collected EDTA-blood samples from 77 PRT and 9 JRT whereof 16 PRT and 3 JRT were clinically affected by SCA (Additional file 1). All 19 cases were presented at the clinic for Small Animals, University of Veterinary Medicine Hannover. Controls did not show any signs of ataxia, myokymia or seizures based on medical records of practitioners and clinical examinations at the clinic for Small Animals, Hannover. In addition, controls had to be >4 years of age. For 17 PRT a pedigree could be constructed. A subsample of the cases for the present study had been previously described elsewhere [2]. In brief, all clinically affected dogs showed symmetric generalized ataxia with hypermetric and spastic movements in all 4 legs at 2–9 months of age. Generalized seizures were observed in some cases. In all affected dogs clinical signs worsened and had to be euthanized at an age <3.5 years. All dogs had their origin in Germany. Out of the 16 clinically SCA-affected PRT cases, 14 were confirmed through a histopathological examination. In all examined 14 PRT, a bilateral symmetrical myelopathy was evident. Predominantly an axonopathy combined with myelin loss, in the dorsal and ventral or ventromedial funiculi was seen. Swelling of axons and dilatation of myelin sheaths with loss of myelin adjacent to a mild astrogliosis were observed primarily in the spinocerebellar tracts of the cervical cord but were also observed in all parts of the brain.
Genomic DNA was isolated using standard methods with RBC (Red Blood Cell) lysis buffer and SE (sodium EDTA) buffer. The DNA concentration of the samples was adjusted to 50 ng/μl using the Nanodrop ND-1000 (Peqlab Biotechnology, Erlangen, Germany) and quality control was performed by gel electrophoresis using 1 % agarose gels (peqGold Universal Agarose, Peqlab Biotechnologie).
We performed a GWAS in 44 PRT using the canine Illumina high density beadchip (Illumina, San Diego, CA, USA) containing 173,662 single nucleotide polymorphisms (SNPs). The data set included 15 cases and 29 controls. Out of these 15 cases, 12 had a histopathological examination confirming the clinical diagnosis. Controls had to be unrelated with the cases at the parent level and >4 years of age. Among controls, 11 were male and 18 female. Cases included 7 males and 8 females. After quality control (genotyping rate per SNP and animal >0.90), filtering for minor allele frequency >0.05 and Hardy-Weinberg equilibrium (HWE) (p < 0.000001) using SAS/Genetics, version 9.4 (Statistical Analysis System, SAS-Institute, Cary, NE, USA), 128,863 SNPs were left for the analysis.
The GWAS was performed with a general linear model. The analysis was run using TASSEL, version 3.0.164 [13]. The GLM explained for sex effects and the SNP genotypes. In addition, we tested several extended models employing up to five principal components (PCAs) to show the robustness of the outcome of the GWAS. All these models yielded the same genome-wide associated SNPs as the GLM model. Thus, adding principal components for cryptic data structure did not change the results of the GLM model. A plot of the first two PCAs for cases and controls employed for the GWAS is shown in Additional file 2. We applied the Bonferroni correction to determine the threshold for genome-wide significance in order to avoid false-positive associations. The Bonferroni correction set the threshold for significance at 0.05/128,863 = 3.88e−7 for a P-value of 0.05. A sex-stratified case-control analysis was performed using SAS/Genetics, version 9.4 (SAS). Homozygosity mapping was done in sliding windows with a minimum of 4 consecutive homozygous SNPs within a range of 50 kb using PLINK, version 1.07 [14].
A polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) for a sample of 77 PRT, 9 JRT from Germany and 179 controls from 20 different dog breeds was employed for genotyping the CAPN1:c.344G > A and KCNJ10:c.627C > G mutations. The design of primers was carried out using Primer 3 (http://bioinfo.ut.ee/primer3-0.4.0/) and the dog genome assembly CanFam3.1 (Additional file 3).
The gene model we used for KCNJ10 was based on the transcript XM_005640901.1 of the dog genome assembly CanFam3.1 (Additional file 4). This transcript includes three exons whereof only two exons (exon 2 and 3) are translated into a protein with 414 amino acids (aa). A previous study [7] used the transcript XM_545752.4 as gene model (denoted XM_545752.3 in previous reports [7, 11]) with two exons encoding 379 aa. Exon 2 and 3 of KCNJ10 and their adjacent genomic regions were sequenced in 10 PRT (cases PRT I-III and PRT IV-VIII and controls PRT IX and PRT X) and 3 JRT (cases JRT I-III) using an ABI 3500 Genetic Analyzer (Applied Biosystems by Life Technologies, Darmstadt, Germany) (Additional file 3). In total, 45 genetic variants were detected in these 13 dogs. Exon 1 could not be amplified because several prime pairs designed for this canine genomic region or primer pairs designed from other species including human, mouse and rat did not result in amplicons with the expected size nor the amplicons could be sequenced. The exon 1 region did not seem to be correctly annotated in the dog reference genome and thus, may prevent retrieval of possible genetic variants for KCNJ10 exon 1 in whole genome sequence data.
In addition, we tried to amplify the gene KCNJ9 (gene ID 100855823) annotated at 22,100,627–22,104,165 base pairs (bp) on dog chromosome (CFA) 38 (CanFam3.1). The record of this gene contained large gaps. We searched for canine expressed sequence tags (ESTs) using the canine coding sequence of KCNJ9 and the basic local alignment tool (blast) of NCBI (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The best hit with an E-value of 1e−77 and a total score of 293 (DN374672.1) of this search did not fit to the genomic region where KCNJ9 was annotated in the dog genome reference assembly. The same was found for hits with larger E-values. Therefore, we were not able to build a gene model for KCNJ9 or sequence this gene.
All KCNJ10 genetic variants detected in the 10 PRT and 3 JRT were further evaluated by sequencing KCNJ10 from one dog of 9 other breeds. These dogs had a normal phenotype in regard to SCA and included the following breeds: Akita, Bernese mountain dog, Dalmatian, German Drahthaar, German shepherd, Irish wolfhound, Shar Pei, Tibetan terrier and Briard. Analysis of sequencing results had been performed using Sequencher software, version 4.8 (Gene Codes, Ann Arbor, MI, USA). The evaluation study resulted in 16 private variants for PRT and JRT. We then filtered the 16 genetic variants for homozygous mutant genotypes in affected PRT and JRT. The single nucleotide variant (SNV) reported in a previous study [7] and only one other variant (KCNJ10:g.22141027insC) remained. Validation of this result was performed in further 17 PRT. We developed a test on a LI-COR 4300 DNA Analyzer (LI-COR Biosciences, Lincoln, NE, USA) using a 167-bp amplicon (Additional file 3). The forward primer was labelled with IRD700 at the 5′end. All samples of the 77 PRT and 9 JRT as well as 84 dogs from 40 other different breeds were used to generate the KCNJ10:g.22141027insC containing amplicon. The amplicons were size-fractionated using gel electrophoresis on an automated sequencer LI-COR 4300 DNA Analyzer on 6 % polyacrylamide denaturing gels. Allele sizes were scored against IRD700-labeled DNA ladders (Additional file 5). For bioinformatic analysis of the effects of the KCNJ10:g.22141027insC variant we used the complete fasta-sequence of KCNJ10 and employed the Regulatory RNA Motifs and Elements Finder (RegRNA) (http://regrna.mbc.nctu.edu.tw/html/about.html) [15] and the variant effect predictor (http://www.ensembl.org/Homo_sapiens/Tools/VEP?db=core).
Results
Genome-wide association study
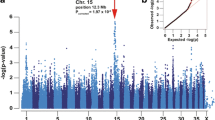
The GWAS using 15 cases and 29 controls identified three genome-wide significantly associated SNPs on dog chromosome 38 (CFA38) (Additional file 6) and a quantile-quantile plot (Q-Q plot) for expected versus observed –log10 P-values was calculated to control for population stratification (Additional file 7). All SNPs with –log10P-values >5 are located on CFA38 and were in HWE. The genomic inflation factor was equal to 1, indicative that data stratification had no effect on the outcomes of the GWAS. The significantly associated SNPs are located at 18.53, 25.21 and 25.31 Mb (CanFam2) and at 15.53, 22.19 and 22.30 Mb (CanFam3.1) on CFA38 with the strongest signal at 22.30 Mb (Praw = 5.14×10−9). The P-values for the SNPs with the highest associations after Bonferroni-correction were at 0.031 (BICF2G63072533), 0.036 (BICF2P378938) and 6.6×10−4 (BICF2P478530) (Additional file 8). In the associated interval, 9/15 cases and one control shared a homozygous region over 147 consecutive SNPs spanning 1893 kb at 23,387,952–25,280,713 bp (CanFam2) and at 20,388,358–22,267,778 (CanFam3.1). Inclusion of further two cases resulted in an even shorter homozygous interval with a size of 124 kb. However, for all other cases, an extended homozygosity region could not be determined (Additional file 9). This may indicate that hereditary ataxia had its origin in several founders or recombinations broke down the homozygous identical-by-descent region caused by a common founder to a very small region not detectable via the beadchip data.
A potential candidate gene in the associated and homozygous interval is KCNJ10 at 25,148,668–25,149,816 bp (CanFam2) and at 22,114,718–22,143,618 bp (CanFam3.1) [7, 11]. The two closest and significantly associated SNPs are 58,212 (BICF2P378938) and 149,816 (BICF2P478530) bp downstream to KCNJ10. An association was not evident for SNPs on CFA18 where CAPN1 is located. The gene KCNJ9 could not be verified using expressed sequence tags (ESTs) for searching this region. Other candidate genes could not be detected in the associated interval. Further members of the KCNJ-gene family were not considered because the GWAS did not indicate significant signals on locations outside CFA38.
Genotyping CAPN1 and KCNJ10 variants
CAPN1 genotyping results showed that all 77 PRT and 9 JRT were homozygous wild-type. Genotyping the KCNJ10:c.627C > G mutation revealed 38/77 PRT (49.4 %) with the wild-type, 22/77 (28.6 %) as heterozygous and 17/77 (22.1 %) as homozygous mutated. In 1/38 of the homozygous wild-type tested PRT, hereditary ataxia was clinically and histopathologically confirmed. Two other homozygous wild-type tested PRT were diagnosed to be affected after clinical examination and/or according to the report of the owner. Among JRT, only 1/9 dogs was homozygously affected and 7/9 heterozygous. Each one of the heterozygous and homozygous wild-type JRT were clinically affected (Table 1). In addition, 179 controls from 20 different dog breeds were genotyped for both mutations. All these dogs were homozygous wild-type for both mutations (Additional file 10).
Sequencing KCNJ10
In order to identify further mutations associated with hereditary ataxia explaining the cases with the homozygous KCNJ10 C/C genotype, exon 2 and 3 of KCNJ10 with their exon/intron or exon/UTR boundaries were sequenced in PRT and JRT. We included 3 affected PRT with the homozygous KCNJ10 C/C genotype and all 3 affected JRT with the different KCNJ10 genotypes. In addition, we sequenced 7 PRT (5 affected PRT and 2 unaffected) with the concordant KCNJ10 genotype. A total of 45 genetic variants could be identified including 41 single nucleotide variants (SNVs) and 4 indels (3 insertions and 1 deletion) (Additional file 11) whereof most of them were located in the 3′UTR (CanFam3.1) (Additional file 4). Sequencing further 9 dogs of other breeds (Additional file 12) and filtering showed 16 variants private for PRT and JRT (Additional file 13). A further filtering for homozygous mutant genotypes in affected PRT and JRT resulted in a 1-bp-insertion (KCNJ10:g.22141027insC) prevalent in these animals. This 1-bp-insertion was located within a stretch of 7 consecutive C bases (Additional file 5). Bioinformatic analysis using RegRNA indicated the KCNJ10:g.22141027insC affecting regulation of gene expression via a regulatory RNA motif or miRNA target site. A regulatory RNA motif (5′-GGGAGGGG-3′) was identified 6 bp downstream to the KCNJ10:g.22141027insC. The motif of the miRNA (hsa-miR-1180) target site contains the KCNJ10:g.22141027insC (5′-acACAACCCCCCCGACGCCGGGAg-3′). Genotyping a pedigree containing 17 PRT for both mutations, KCNJ10:g.22141027insC and KCNJ10:c.627C > G, showed segregation of both mutations with the affection status with exception of one animal (Additional file 14). Genotyping results of the KCNJ10:g.22141027insC are summarized in Table 2. Out of the 77 PRT, 34 (44.2 %) showed the wild-type, 26 (33.8 %) the heterozygous and 17 (22.1 %) the homozygous mutated genotype. In 2/26 of the heterozygous PRT, hereditary ataxia was clinically and histopathologically confirmed and 2/34 homozygous wild-type tested PRT were affected according to a clinical examination and/or according to the report of the owner. The genotyping results for the KCNJ10:g.22141027insC mutation of JRT showed two homozygous mutated genotypes among the affected individuals. A further affected individual had the wildtype genotype. Genotyping further 84 dogs of 40 other breed dogs revealed 59 (70.3 %) wildtype homozygous, 21 (25.0 %) heterozygous and 4 (4.8 %) homozygous mutated genotypes (Additional file 15).
Discussion
The results of the GWAS indicate that the associated genomic region on CFA38 may harbour the most likely candidate genes for hereditary ataxia in the sample under study. Thus, our study corroborated the previous candidate gene search using whole genome sequence data [7]. We could not detect samples with the CAPN1:c.344G > A mutation. This may indicate that this mutation may be rare in the German PRT and JRT populations. In agreement with a previous study [7], one histopathologically confirmed case of hereditary ataxia and two clinically affected cases carried neither the CAPN1:c.344G > A allele nor the homozygous KCNJ10:c.627C > G genotype. This may indicate that at least one further mutation may be involved in hereditary ataxia of PRT and JRT. As the largest proportion of affected dogs were concordant with the segregation of the KCNJ10:c.627C > G alleles the remaining unexplained cases may harbour a rare mutation. However, breeders have to be aware that selective breeding based on genetic tests for the KCNJ10:c.627C > G and CAPN1:c.344G > A mutations cannot completely preclude affected dogs of the breeds PRT or JRT despite of homozygous wild-type genotypes of the parents.
The present sequence analysis tagged a 1-bp-insertion associated with hereditary ataxia in PRT and JRT. This mutation may have an effect on regulation of KCNJ10 expression as indicated through bioinformatic results from RegRNA. However, the presence of this 1-bp-insertion in other breeds may require further research in phenotyping spinocerebellar ataxia associated with myokymia, seizures, or both. In conclusion, a few but not all cases of hereditary ataxia being heterozygous or homozygous for the wild-type allele of KCNJ10 could be explained by the KCNJ10:g.22141027insC mutation.
Conclusions
Our data confirmed the presence of the KCNJ10:c.627C > G mutation in PRT and JRT affected with hereditary ataxia. However, some cases of hereditary ataxia could not be explained through the KCNJ10:c.627C > G nor the CAPN1:c.344G > A mutation. Genetic testing using the KCNJ10:c.627C > G and the CAPN1:c.344G > A mutations deems not to be sufficient to capture all cases of hereditary ataxia in PRT and JRT. This lets us assume that at least one additional form of hereditary ataxia may segregate in the breeds PRT and JRT. The KCNJ10:g.22141027insC mutation was evident in a few cases but could not explain all not yet resolved cases. Further efforts are necessary to collect cases of hereditary ataxia not explained by the presently known mutations to unravel the responsible mutations in these cases of hereditary ataxia.
Abbreviations
- CFA:
-
Dog chromosome
- GWAS:
-
Genome-wide association study
- JRT:
-
Jack Russell Terrier
- KCNJ10 :
-
Potassium inwardly-rectifying channel, subfamily J, member 10
- LOA:
-
Late onset ataxia
- PRT:
-
Parson Russell Terrier
- SCA:
-
Spinocerebellar ataxia
- SNP:
-
Single nucleotide polymorphism
- SNV:
-
Single nucleotide variant
References
Hartley WJ, Palmer AC. Ataxia in Jack Russell Terriers. Acta Neuropathol (Berl). 1973;26(1):71–4.
Wessmann A, Goedde T, Fischer A, Wohlsein P, Hamann H, Distl O, Tipold A. Hereditary ataxia in the Jack Russell Terrier–clinical and genetic investigations. J Vet Intern Med. 2004;18(4):515–21.
Van Ham L, Bhatti S, Polis I, Fatzer R, Braund K, Thoonen H. ‘Continuous muscle fibre activity’ in six dogs with episodic myokymia, stiffness and collapse. Vet Rec. 2004;155(24):769–74.
Vanhaesebrouck AE, Van Soens I, Poncelet L, Duchateau L, Bhatti S, Polis I, Diels S, Van Ham L. Clinical and electrophysiological characterization of myokymia and neuromyotonia in Jack Russell Terriers. J Vet Intern Med. 2010;24(4):882–9.
Vanhaesebrouck AE, Bhatti SF, Franklin RJ, Van Ham L. Myokymia and neuromyotonia in veterinary medicine: A comparison with peripheral nerve hyperexcitability syndrome in humans. Vet J. 2013;197(2):153–62.
Bhatti SF, Vanhaesebrouck AE, Van Soens I, Martlé VA, Polis IE, Rusbridge C, Van Ham LM. Myokymia and neuromyotonia in 37 Jack Russell terriers. Vet J. 2011;189(3):284–8.
Gilliam D, O’Brien DP, Coates JR, Johnson GS, Johnson GC, Mhlanga-Mutangadura T, Hansen L, Taylor JF, Schnabel RD. A homozygous KCNJ10 mutation in Jack Russell Terriers and related breeds with spinocerebellar ataxia with myokymia, seizures, or both. J Vet Intern Med. 2014;28(3):871–7.
Urkasemsin G, Olby NJ. Canine Hereditary Ataxia. Vet Clin North Am Small Anim Pract. 2014;44(6):1075–89.
Björck G, Mair W, Olsson SE, Sourander P. Hereditary ataxia in Fox Terriers. Acta Neuropathologica (Berl). 1962; Suppl 1:45–48.
Rohdin C, Lüdtke L, Wohlsein P, Jäderlund KH. New aspects of hereditary ataxia in smooth-haired fox terriers. Vet Rec. 2010;166(18):557–60. doi:10.1136/vr.b4821.
Rohdin C, Gilliam D, O’Leary CA, O’Brien DP, Coates JR, Johnson GS, Jäderlund KH. A KCNJ10 mutation previously identified in the Russell group of terriers also occurs in Smooth-Haired Fox Terriers with hereditary ataxia and in related breeds. Acta Vet Scand. 2015;57:26. doi:10.1186/s13028-015-0115-1.
Forman OP, DeRisio L, Mellersh CS. Missense mutation in CAPN1 is associated with spinocerebellar ataxia in the Parson Russell Terrier dog breed. PLoS One. 2013;8(5):e64627. doi:10.1371/journal.pone.0064627.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–5.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Huang HY, Chien CH, Jen KH, Huang HD. RegRNA: an integrated web server for identifying regulatory RNA motifs and elements. Nucleic Acids Res. 2006;34(Web Server issue):W429–434.
Acknowledgements
The authors thank the dog breeders for providing EDTA-blood samples, pedigree data and veterinary diagnoses for hereditary ataxia. All dog owners gave informed consent to participate in this study.
Funding
ACG obtained internal funds of the University of Veterinary Medicine Hannover. The funders had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
All data and materials can be found in the tables and additional files.
Authors’ contributions
OD and AT conceived and designed the study. ACG, JM and OD performed the experiments and analysed the data. ACG, OD and AT coordinated the collection of samples. ACG, JM and OD drafted and finalized the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
All animal work has been conducted according to the national and international guidelines for animal welfare. The Lower Saxony state veterinary office at the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany, was the responsible Institutional Animal Care and Use Committee (IACUC) for this specific study. This specific study had been approved by the IACUC of Lower Saxony, the state veterinary office Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany (registration number 23-19c20/15). All owners of the dogs were informed on this specific project and all owners of the dogs gave written consent to participate with their dogs in this project.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Distribution of ataxia phenotypes for Parson Russell (PRT) and Jack Russell Terriers (JRT). (DOCX 15 kb)
Additional file 2:
Plot of the first (PCA1) versus the second principal component (PCA2) by hereditary ataxia affected Parson Russell Terriers (affected) and controls (non-affected) used for the genome-wide association study (GWAS). The black dots indicate controls and the orange dots affected PRT. Number of single nucleotide polymorphisms (SNPs) that informed PCAs was at 128,863 SNPs. Variance of SNPs explained by PCA1 and PCA2 was at 0.19 and 0.17, respectively. (DOC 69 kb)
Additional file 3:
Primer pair sequences, restriction enzymes, annealing temperature (AT) and amplicon size used for genotyping the mutations CAPN1:c.344G > A and KCNJ10:c.627C > G by PCR-RFLP are given. Primer pair sequences, annealing temperature (AT) and amplicon size of PCR for genotyping the mutation KCNJ10:g.22141027insC validated on a LI-COR 4300 DNA Analyzer as well as primer pair sequences and product-sizes for Sanger sequencing PCR-amplicons of exon 2 and 3 of KCNJ10 are shown. (DOC 57 kb)
Additional file 4:
Canine gene model of KCNJ10 and detected variants. The gene model was built using the Ensemble annotation (assembly CanFam3.1). (A) represents transcript XM_005640901.1 and (B) represents XM_545752.4. Translated exons are shown as black boxes and untranslated exons are shown as open boxes. Exon numbers are given above the boxes. Continuous lines indicate introns. Sizes of exons and introns are given below the boxes and lines. Positions of start codon and stop codon are also present. Locations and motifs of variants identified in our study are listed above the exons. Both the single nucleotide variant (SNV) reported by Gilliam et al. [7] (KCNJ10:c.627C > G) and the variant validated in our study (KCNJ10:g.22141027insC) are framed by a black open box. (PDF 41 kb)
Additional file 5:
(A) The genomic sequences of an unaffected and affected Parson Russell Terrier (PRT) are shown. The unaffected dog is homozygous for the wild-type allele. The sequence of the dog affected by hereditary ataxia shows a 1-base pair (bp) insertion in a seven-C stretch within exon 3 of the KCNJ10 gene. The seven-C stretch with and without the inserted base C is framed by a red open box. The homozygous wild-type and the homozygous mutant variant in the forward sequence as well as the heterozygous variant in both the forward and reverse sequence are shown. (B) Fragment length analysis on a 6 % polyacrylamide gel for evaluating the 1-bp insertion in KCNJ10 in hereditary ataxia affected Parson and Jack Russell Terriers. The homozygous wild-type (wt/wt) in two unaffected PRT are shown. The size of the normal PCR-product is 166 bp. The heterozygous genotype (wt/mut) in one unaffected and the homozygous genotype (mut/mut) for the mutant allele in two affected PRT are also present. (PDF 169 kb)
Additional file 6:
Manhattan plot of –log10 P-values of the genome-wide association study for hereditary ataxia in Parson Russell Terriers using a general model analysis. On the X-axis, the SNPs are given by dog chromosome number. The –log10P-values for each SNP genotype effect are plotted against the SNP position on each chromosome. Chromosomes are differentiated by colors. The color keys are given below the plot. The blue line indicates the threshold of the –log10 P-values for genome-wide significance after Bonferroni correction. (PDF 138 kb)
Additional file 7:
Q-Q-plot of expected –log10 P-values versus observed–log10 P-values from the general model analysis for hereditary ataxia in Parson Russell Terriers. Shown are all 128,863 SNPs included in the genome-wide association analysis with the grey line corresponding to the null hypothesis of no association. (PDF 56 kb)
Additional file 8:
Summary of results for the genome-wide association study using a general model analysis and sex-stratified case-control analysis for hereditary ataxia in Parson Russell Terriers. The SNP-ID, the position on dog chromosome (CFA) in base pairs (bp) according to the dog genome assembly CanFam2, minor allele, minor allele frequency (MAF) for all, affected (MAFA) and unaffected (MAFU) dogs (controls), variance explained by the single SNP (VSNP), −log10 P-values (−log10P) and Bonferroni-corrected -log10 P-values (−log10PBonf) of the general model analysis are given. Odds ratios (OR) with 95 % confidence intervals (CI) are from a sex-stratified case-control analysis for cases and controls. (DOC 42 kb)
Additional file 9:
Overview on the results of extended homozygosity mapping in Parson Russell Terriers (PRT) genotyped on the canine Illumina high density beadchip. The IDs of PRT, KCNJ10 c.627C > G genotype, the SNP-IDs and positions in base pairs (bp) for the start and end of the homozygosity region are given. The PRT-ID 2 indicates dogs which were used for sequencing in this study and PRT-ID 3 indicates the designation for PRT (P.n.) which are shown in the pedigree (see Additional file 12). KCNJ10 is located at 25,148,668–25,149,816 on CFA38 (CanFam2). (DOC 51 kb)
Additional file 10:
Distribution of genotyping results for the mutation CAPN1:c.344G > A and KCNJ10:c.627C > G in different dog breeds. Numbers of tested homozygous wild-type (wt/wt), heterozygous (wt/mut) and homozygous mutant (mut/mut) genotypes are given. (DOC 51 kb)
Additional file 11:
Identification of variants in the gene KCNJ10 in affected and unaffected Parson Russell Terriers (PRT) and Jack Russell Terriers (JRT). IDs of the variants, locations in the gene, accession numbers and the genotype for each variant are given. The single nucleotide variant reported by Gilliam et al. [7] (KCNJ10:c.627C > G) written in bold. Indel mutations are denoted as wt (wild-type) or mut (mutant). (DOC 108 kb)
Additional file 12:
Sequencing results of nine dogs of different breeds for evaluation of the variants detected in Parson Russell Terriers (PRT) and Jack Russell Terriers (JRT). IDs of the variants and the genotype for each variant are given. The single nucleotide variant reported by Gilliam et al. [7] (KCNJ10:c.627C > G) written in bold. Indel mutations are denoted as wt (wildtype) or mut (mutant). Indel mutations are denoted as wt (wild-type) or mut (mutant). (DOC 97 kb)
Additional file 13:
Variants in the gene KCNJ10 which are private for Parson Russell Terriers (PRT) and Jack Russell Terriers (JRT). IDs of the variants, locations in the gene, accession numbers and the genotype for each variant are given. Both the single nucleotide variant reported by Gilliam et al. [7] (KCNJ10:c.627C > G) and KCNJ10:g.22141027insC identified as further putative variant written in bold. Indel mutations are denoted as wt (wild-type) or mut (mutant). (DOC 64 kb)
Additional file 14:
The pedigree demonstrates the relationship of 17 Parson Russell Terriers (PRT) (pedigree number 1–17) selected from the sample of our study. Genotypes for both, the KNCJ10:g.22141027insC (first row) and KCNJ10:c.627C > G [7] (second row) variant are given for each of the 17 dogs. KNCJ10:g.22141027insC genotyping resulted in 5/17 wild-type (wt/wt), 9/17 heterozygous (wt/mut) and 3/17 homozygous mutant (mut/mut) PRT. KCNJ10:c.627C > G genotyping resulted in 9/17 wild-type (wt/wt), 5/17 heterozygous (wt/mut) and 3/17 homozygous mutant (mut/mut) PRT. (PDF 22 kb)
Additional file 15:
Distribution of genotyping results for the mutation KCNJ10:g.22141027insC in different dog breeds. Numbers of tested homozygous wild-type (wt/wt), heterozygous (wt/mut) and homozygous mutant (mut/mut) genotypes are given. (DOC 57 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gast, A.C., Metzger, J., Tipold, A. et al. Genome-wide association study for hereditary ataxia in the Parson Russell Terrier and DNA-testing for ataxia-associated mutations in the Parson and Jack Russell Terrier. BMC Vet Res 12, 225 (2016). https://doi.org/10.1186/s12917-016-0862-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-016-0862-x