
Abstract
Background
Avian pathogenic Escherichia coli (APEC) causes avian colibacillosis, which results in economic and welfare costs in the poultry industry worldwide. The pathogenesis of avian pathogenic E. coli strains is not well defined. Here, the function of an outer membrane protein encoded by the ireA gene of avian pathogenic E. coli strain DE205B was investigated.
Results
The ireA gene was distributed in 32.9 % (46/140) of tested E. coli strains, with high percentages in the phylogenetic ECOR groups B2 (58.8 %, 10/17) and D (55.9 %, 19/34). The gene expression level of ireA of APEC strain DE205B in high Fe M9 media was 1.8 times higher (P < 0.05) than that in low Fe M9 media. An ireA deletion mutant and complementary strain were constructed. Compared with the wild-type strain DE205B, the expression of most ferric uptake genes in the ireA deletion mutant were significantly upregulated (P < 0.05). The adhesion ability of the ireA deletion mutant to DF-1 cells was significantly decreased. The survival rate of ireA deletion mutant was reduced 21.17 % (P < 0.01), 25.42 (P < 0.05) and 70.0 % (P < 0.01) under alkali, high osmolarity, and low temperature (4 °C) conditions, respectively, compared with the wild-type strain.
Conclusions
The results suggested that the protein encoded by the iron-regulated gene ireA has roles in adhesion and stress resistance in avian pathogenic E. coli.
Similar content being viewed by others

Background
Avian pathogenic Escherichia coli (APEC), a subgroup of extra-intestinal pathogenic E. coli (ExPEC) causes avian colibacillosis and imposes economic losses on the poultry industry worldwide [1]. However, the pathogenesis of APEC is poorly understood. Many virulence genes have been studied to identify virulence factors in APEC, including those involved in adhesion, iron-regulation, toxin/cytotoxin production and serum resistance [2]. Iron is an essential element involved in important biological processes [3]. Biological activities in cells, such as peroxide reduction, nucleotide biosynthesis and electron transport, are facilitated by iron ions [4]. Extra-intestinal sites have low iron contents; therefore, ExPEC strains struggle to take up iron from the host during infection [5]. During natural infection, the initiation, progression and transmission of most bacterial infections depend on the ability of the invading pathogen to acquire iron from the complicated environment [6]. During iron acquisition, the cell must produce transmembrane receptors for siderophores that chelate iron ions [7]. There are various receptors that chelate iron ions encoded by bacterial genes, such as chuA, the SitABCD system, iron, iha, iutA, and ireA. Outer membrane protein ChuA participates in heme acquisition in enterohemorrhagic E. coli and uropathogenic E. coli (UPEC) strains, and is important for the pathogenicity of APEC [8, 9]. The SitABCD system, identified in the APEC strain MT512 by comparative genomic analysis, was reported to be associated with the pathogenicity of APEC [9, 10].
IreA was suggested to be involved in Fe acquisition and to act as an iron-regulated virulence gene in the blood- or urine-derived ExPEC E. coli isolated from humans [11]; however, its exact role in APEC strains remains unknown. Herein, an ireA deletion mutant was constructed to study the ireA gene function in the APEC strain DE205B.
Results
Prevalence of the ireA gene among E. coli Strains
As shown in Table 1, ireA was present in 32.9 % (46/140) of E. coli strains, with 19.0 % (12/63) in phylogenetic ECOR group A, 19.2 % (5/26) in B1, 58.8 % (10/17) in B2 and 55.9 % (19/34) in group D (Additional file 1: Table S1). Thus, the ireA gene was significantly more frequently distributed in the B2 and D groups than in the A and B1 groups.
Expression of the ireA gene
The ireA gene expression was tested by Immunoblotting. Western blotting was performed with anti-His serum, showing expected fusion protein bands for ireA (39 kDa) from strains DE205B. However, only fusion his protein (18 kDa) was detected from the blank plasmid control (Fig. 1). These results indicated that ireA was expressed under laboratory conditions.

Expression of ireA by western blotting. Expression of fusion ireA protein was detected by immunoblotting. Fusion ireA was detected in pET32a(+), while there is only his protein was detected in the blank plasmid. Lane M, protein marker; lane 1, fusion ireA; lane 2, fusion his
Gene expression of ireA in M9 media with different iron content
The relative gene expression of ireA in low Fe M9 media was 1.8 times higher than that in high Fe M9 media (P < 0.01) (Fig. 2).

Expression of ireA in M9 media. The first and second columns represent the relative gene expressions of ireA in M9 media with low and high Fe content, respectively. The gene expression levels showed significant differences in the two kinds of media (P < 0.01)
Growth curve
The CFU of the wild-type strain DE205B, mutant strain DE205BΔireA and the complementary strain DE205BCΔireA was monitored for 12 h. There was no significant difference between the growth curves of the wild-type and mutant strains (Fig. 3), which indicated that deletion of ireA had no effects on the growth of DE205B.

Growth curve of different strains. Growth curves of the wild-type strain DE205B, mutant strain DE205BΔireA and complementary strain DE205BCΔireA. Bacterial growth was estimated by plate counting as Colony Forming Units (CFU)
Expression variations of Ferric uptake system and adherence genes
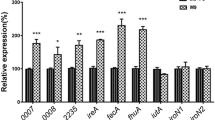
The expressions of fepC, feoB, chuT, fyuA, irp1, irp2, chuA and fepA were detected in the mutant and compared with their expressions in the wild-type strain DE205B. Most of the ferric uptake genes in the mutant strain DE205BΔireA, except irp1, irp2 and chuA, were significantly upregulated (P < 0.05) (Fig. 4). Irp1, irp2 and chuA were non-significantly upregulated. Furthermore, the expressions of the eight ferric uptake genes in the complementary strain DE205BCΔireA were restored to wild-type levels.

Expressions of Fe acquisition genes. Expressions of Fe acquisition genes of the wild-type strain DE205B, mutant strain DE205BΔireA and complementary strain DE205BCΔireA were tested by qPCR. The relative expression levels in the different mutant strains were calculated using the 2-△△Ct method
The expression of adherence genes yfcO, yfcQ, aufG, fmlD, fmlE, yadN and fimH showed no significant difference between the wild-type strain DE205B and the ireA mutant strain (Fig. 5).

Expressions of adhesion genes. Expressions of adhesion genes, including yfcO, yfcQ, aufG, fmlD, fmlE, yadN and fimH, of the wild-type strain DE205B, mutant strain DE205BΔireA and complementary strain DE205BCΔireA were tested by qPCR. The relative expression levels in the different mutant strains were calculated using the 2-△△Ct method
Adherence assays
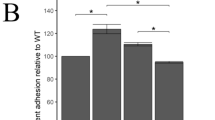
The number of mutant DE205BΔireA bacteria adhering to DF-1 cells was significantly decreased compared with the number of DE205B wild-type adherent cells (P < 0.05) (Fig. 6). The adhesion to DF-1 cells of complementary strain DE205BCΔireA was partly restored to wild-type levels.

Adherence assays. The adhesion abilities of the wild-type strain DE205B, mutant strain DE205BΔireA and complementary strain DE205BCΔireA to DF-1 cells were tested. The numbers of adherent cells were calculated by plate counting. The number of DE205BΔireA mutant bacteria adhering to DF-1 cells was significant decreased compared with the number of DE205B wild-type bacteria (P < 0.05)
Animal infection
The LD50 of the wild-type, deletion mutant and complementary strains were 1.74 × 105, 2.45 × 105, and 3.16 × 105, respectively (Table 2). The results showed that there was no significant difference between the LD50 of the wild-type and deletion mutant strains, indicating that the deletion of ireA gene has no obvious effect on the virulence of DE205B toward ducks.
Determination of resistance to environmental stress
Compared with the wild-type strain, the survival rate of the ireA deletion mutant was reduced by 21.17 % (P < 0.01) and 25.42 % (P < 0.05) under alkali and high osmolarity conditions, respectively (Fig. 7). The resistance to alkali and high osmolarity conditions of the complementary strain DE205BCΔireA was almost restored to wild-type levels. For the temperature challenge, compared to the wild type strain, the ireA deletion mutant was reduced 70.0 % (P < 0.01) under the low (4 °C) temperature (Fig. 8). The resistance to low temperature of the complementary strain DE205BCΔireAwas partly restored. The results showed that presence of the ireA gene increased the stress-resistance of APEC strain DE205B.

Resistance to alkaline and high osmolarity. The resistance to alkali (a) and high osmolarity (b) of wild-type strain DE205B, mutant strain DE205BΔireA and complementary strain DE205BCΔireA was tested by exposing the strains to alkaline and high osmolarity conditions and calculating their survival rates. Compared with the wild-type strain, the survival rate of the ireA deletion mutant was reduced 21.17 % (P < 0.01) and 25.42 (P < 0.05) under alkaline and high osmolarity conditions, respectively

Challenge under the low temperature of different strains. Each bacterial suspension with concentration of 107 CFU/ml (DE205B, DE205BΔireA or DE205BCΔireA) were incubated at 4 °C for 7days. The survival rates of wild-type and mutant strains were calculated by plate counting and compared using GraphPad Prism 5. The ireA deletion mutant was reduced 70.0 % (P < 0.01) under the low (4 °C) temperature compared to that of the wild type strain
Discussion
The ireA gene is an iron-regulated gene and is involved in iron acquisition in human pathogenic E. coli isolates and our study proved this protein functions in APEC. Additionally, we identified two new functions of ireA using the deletion mutant. In the present study, ireA was demonstrated to contribute to the adhesion to DF-1 cells. Moreover, the expressions of several adhesion genes were tested and the results showed no significant differences between wild-type and mutant strain, indicating that the ireA gene indeed plays a role in adhesion. Tarr et al., identified adhesin Iha from an O157:H7 strain of E. coli [12]. This adhesin shared high similarities with several identified or putative siderophore receptors. Siderophore receptor IrgA was reported to contribute to growth in the rabbit ileal loop model in vivo and to enhance virulence in an infant mouse model, suggesting a possible role in colonization [13, 14]. Here, we proved that iron-regulated gene ireA plays a role in the adhesion of APEC strains. The ireA gene also increased stress-resistance under alkali and high osmolarity conditions, as well as underlow temperature. Thus, the redundancy of siderophore receptors might reflect their multifunctional roles.
E. coli strains were reported to be classified into four main phylogenetic groups (A, B1, B2, and D) [15]. Virulent ExPEC strains mainly belong to phylogroup B2 and D, whereas most commensal strains belong to phylogroup A [16]. The ireA gene was distributed more frequently in the B2 (58.8 %) and D (55.9 %) groups than in the A (19 %) and B1 (19.2 %) groups, indicating that ireA might be associated with the virulence of the APEC strain DE205B. Thus, the result correlated with that Russo’s report: ireA was detected in 13 (26 %) of 50 random clinical isolates from patients and in none (0 %) of 14 fecal isolates, which presumably represented commensal strains [11]. Taken together, these results indicated that ireA might be a virulence gene in both human and avian ExPEC E. coli strains.
The expression level of ireA in DE205B was decreased in high Fe M9 media compared with that in low Fe media, indicating that ireA is involved in iron-regulation in this APEC strain. This result agreed with the report of Russo et al., who found that ireA was a iron-regulated virulence gene in the blood- or urine-derived isolates of ExPEC E. coli [11]. Most ferric uptake genes, such as fepC, feoB, chuT, fyuA and fepA were upregulated in the ireA deletion mutant strain. This might represent a compensatory function for ireA gene deletion. Fe acquisition is important for many microorganisms, especially for pathogens that grow in the host, which attempts to limit Fe availability. It is thought that pathogens harbor multiple Fe acquisition systems to ensure that Fe is gained from the host cells to provide a selective advantage. Alternatively, certain siderophores, and their cognate receptors, might be more active in certain environments, such as inside or outside the gastrointestinal tract [17, 18]. Moreover, multiple systems might represent ‘alternatives’ that protect against the disruption of one system caused by genomic rearrangements or mutations.
In the duck infection experiments, the LD50 showed no significant difference between the wild-type DE205B and the ireA deletion mutant strain. Thus, it seemed that ireA deletion had no obvious effect on the virulence of DE205B. However, DE205B has several Fe acquisition systems. We showed that most of the other Fe acquisition genes were upregulated in the ireA gene deletion mutant. Thus, the ireA gene might indeed contribute to the virulence of the APEC strain DE205B, while other Fe acquisition genes displayed compensatory functions when the ireA gene was deleted.
Conclusions
In summary, the ireA gene was mainly distributed in the more virulent phylogenetic ECOR group B and D. Compared with the wild-type strain, the adhesion and resistance to environmental stress of the ireA deletion mutant were significant decreased. This indicated that ireA is a Fe iron-regulated gene that aids adhesion and stress-resistance in the APEC strain DE205B.
Methods
Bacterial strains, plasmids and growth conditions
The APEC strain DE205B, which was characterized previously, was isolated from the brain of a duck with neurological symptoms in our previous work [19–22]. Its serotype is O2:K1, which is one of the predominant serotypes of APEC [9, 23]. All E. coli strains were grown in Luria-Bertani (LB) medium at 37 °C with shaking or on LB plates with 2 % agar. When necessary, the LB medium was supplemented with appropriate antibiotics: ampicillin (Amp; 100 μg/mL) and kanamycin (Kan; 50 μg/mL), unless otherwise specified. Information on the main strains and plasmids used in this study are listed in Table 3. M9 minimal medium was used to assess the expression of ireA, which was prepared as follows: an aliquot was mixed with200 ml of 5 × M9 salt solution (containingNa2PO4 · 7 H2O, 12.8 g; KH2PO4, 3.0 g; NaCl, 0.5 g; NH4Cl, 1.0 g;),2 ml of 1 M MgSO4, 20 ml of 20 % glucose solution, 0.1 ml of1 M CaCl2, and then dissolved in1000 ml double distilled water andfiltrated through a 0.22-μm membrane.
Prevalence of ireA among E. coli Strains
The distribution of ireA among 140 E. coli strains (Additional file 1: Table S1) maintained in our laboratory was tested using PCR. The PCR primers were designed using Primer Premier 5. The specific primers ireA-F and ireA-R are listed in Table 4. The PCR conditions were: 5 min at 95 °C for initial denaturing; 30 cycles of 1 min at 95 °C, 30 s at 50 °C and 1 min at 72 °C; followed by 10 min at 72 °C for extension. The DNA of DE205B and ddH2O were used as positive and negative templates, respectively. The PCR products were electrophoresed through 1 % agarose gels. Phylogenetic E. coli reference (ECOR) groups of single isolates of the 140 E. coli strains were determined using triplex PCR, as described previously [16, 24].
Expression of the ireA gene
The expression of the ireA gene was tested by fusion expression and western blotting as previously reported [25, 26]. The fusion fragment of ireA (including the ireA promoter and 579bp of ireA sequence) and His tag were inserted into plasmid pET32a(+). The fusion PCR primers P1 and P2 were listed in Table 4. Plasmid pET32a(+) without the fusion fragment was used as a blank control. For immunoblotting, protein samples were subjected to sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes (Amersham Pharmacia Biotech, Piscataway, NJ, USA), as described previously [27]. Anti-His serum was the primary antibody, horseradish peroxidase-conjugated goat anti-mouse IgG was the secondary antibody and 3,3′-diaminobenzidine was used as the substrate.
Regulation of ireA expression in M9 media
M9 minimal medium was used to assess the expression of ireA.M9 medium or M9 medium with Fe (0.1 mM Fe(NO3)3) were used as low and high Fe content media, respectively. DE205B was cultured in both M9 media to the mid-log phase and the expression of ireA was detected by quantitative real-time reverse transcription PCR(qRT-PCR). The real –time PCR primers were designed by PrimerQuest Tool IDT (http://sg.idtdna.com/primerquest/Home/Index). Briefly, total RNA was extracted from 1 ml of bacteria culture using the Trizol RNA isolation protocol (Invitrogen, Shanghai, China) and cDNA was amplified by reverse transcription according to the instructions of the primeScript RT reagent Kit (Takara). Quantitative real-time PCR (qPCR) was carried out using the ABI Prism 7300 and Sequence Detection System software version 1.4 (Applied Biosystems, Foster City, CA, USA), according to the instructions of the SYBR Premix Ex Taq (Takara, Dalian, China). QPCR primers for ireA (QireA-F and QireA-R) are listed in Table 4. DnaE was used as a reference gene. Assays were performed three times. The relative expressions of ireA in different media were calculated using the 2-△△Ct method [28]. Statistical analysis was performed using an unpaired t test in Graphpad Prism 5.0.
Construction of the ireA deletion mutant and complementary strain
An ireA knockout strain of DE205B was constructed using the lambda red recombinase system described by Datsenko and Wanner [29]. The specific primers ireAMu-F and ireAMu-R were designed to amplify the target gene ireA. The kanamycin resistance gene, which contained sequences homologous to the 5′ and 3′ ends of the target sequence, was amplified using plasmid pKD4 as a template. The PCR products were then transformed by electroporation into DE205B containing the lambda red recombinase expression plasmid pKD46. The transformed bacterial cells were first incubated at 30 °C for 2 h in super optimal broth with catabolite repression (SOC) broth, and then grown on LB agar containing kanamycin at 37 °C. Mutants were confirmed by PCR and sequenced using primers k1 and k2 (Table 4) in combination with primers ireA-1 and ireA-2 (Table 4) flanking the ireA region. To remove the kanamycin resistance gene, plasmid pCP20 was transformed into the mutant and a kanamycin sensitive mutant strain was selected. Finally, the ireA deletion mutant strain without kanamycin resistance was named as DE205BδireA (Additional file 2: Figure S1).
To construct the complementary strain, the ireA gene, including its putative promoters, was amplified using primers ireACo-F and ireACo-R (Table 4). The following amplification program were used: 5 min at 95 °C for initial denaturing; 35 cycles of 30 s at 95 °C, 30 s at 55 °C and 2.5 min at 72 °C; 10 min at 72 °C for extension. The PCR product of ireA gene was purified and subcloned into plasmid pSTV-28. The complementary strain DE205BCΔireA was generated by transforming vector pSTV-28-ireA into the deletion mutant (Additional file 3: Figure S2).
Growth curve
The growths of the DE205B wild-type and mutant strains were compared in LB medium at 37 °C over a course of 12 h, starting at 107 CFU/ml. Bacterial growth was estimated by plate counting as Colony Forming Units (CFU). Assays were performed three times.
The expression of the Ferric uptake system and adherence genes
The effect of ireA deletion on the regulation of the ferric uptake system, including fepC, feoB, chuT, fyuA, irp1, irp2, chuA and fepA was detected using qRT- PCR. Seveal adherence genes, including yfcO, yfcQ, aufG, fmlD, fmlE, yadN and fimH were also selected to test their expression levels. The wild-type DE205B and the ireA deletion mutant were cultured in LB to mid-log phase and total RNA was extracted from 1 ml of bacterial culture using the Trizol RNA isolation protocol (Gibco BRL, USA, cat. no.15596-026). cDNA was reverse transcribed and real-time PCR was carried out as described above. The qRT-PCR primers for the ferric uptake system and adherence genes are listed in Table 4. The real-time PCR primers were designed unsing PrimerQuest Tool IDT (http://sg.idtdna.com/primerquest/Home/Index). The relative expression levels of the genes were calculated using the 2-△△Ct method [28]. Assays were performed three times, and the statistical analysis was performed using an unpaired t test in Graphpad Prism 5.0. On the figures, error bars indicate the standard deviation.
Adherence assays
The adherence assay was performed as described previously [24]. Briefly, chicken embryo fibroblast (CEF) DF-1 cells were seeded at approximately 1 × 105 cells per well in 24-well tissue culture trays (TPP, Shanghai, China) and grown in Dulbecco’s modified Eagle medium (DMEM) with 10 % fetal bovine serum at 37 °C in a 5 % CO2 humidified atmosphere without antibiotics. DF-1 cells were washed once with DMEM and then inoculated with 500 ul of 2 × 107 CFU/ml bacteria per well for 2 h at 37 °C in the presence of 5 % CO2. The cells were washed, lysed with ddH2O, and the number of bacterial cells was calculated by plate counting. In all assays, wells only containing DF-1 cells were used as negative controls. The adherence assays were conducted three times. The statistical analysis was performed using an unpaired t test in Graphpad Prism 5.0.
Animal infections
Animal infections were performed as described previously [19, 20]. We purchased 7-day-old ducklings and young duck feeds from Anhui Poultry Farm (Anhui, China). Bacterial strains were cultured to the exponential phase, harvested, washed three times in PBS and then adjusted to the appropriate doses. Twenty-five 7-day-old ducks were inoculated intramuscularly 0.2 ml of each bacterial suspension (DE205B, DE205BΔireA or DE205BCΔireA) at four concentrations (5 × 108 CFU/ml, 5 × 107 CFU/ml, 5 × 106 CFU/ml, and 5 × 105 CFU/ml). Assays were performed three times. Seven ducks were used for each dose. Seven ducks were injected with PBS as a negative control. Death of the ducks was monitored for 7 days post infection. We calculated the LD50 of each strain using the method described by Spearman-Karber [30].
Determination of resistance to environmental stress
Resistance to environmental stress was tested for the wild-type and the mutant strain, as described by La Ragione et al. [31]. Bacteria were cultured in LB broth overnight and harvested by centrifugation. The cells were resuspended in PBS and adjusted to 107 CFU/ml in PBS. For alkali challenge, 100 μl of adjusted cells were mixed with 100 μl Tris buffer (1 M, pH10.0) and 800 μl ddH2O (final concentration, 100 mM, pH10.0) and incubated at 37 °C for 30 min. For high osmolarity endurance, cells were mixed with an equal volume of 4.8 M NaCl (final concentration, 2.4 M) and incubated at 37 °C for 1 h. Bacteria were exposed to PBS (pH 7.0) as a control.
Temperature challenge was performed as previously described with modifications [32], each bacterial suspension, at a concentration of 107 CFU/ml (DE205B, DE205BΔireA or DE205BCΔireA) was incubated at 4 °C for 7days. Assays were performed three times. The survival rates of wild-type and mutant strains were calculated by plate counting and compared using GraphPad Prism 5.
References
Dziva F, Hauser H, Connor TR, van Diemen PM, Prescott G, Langridge GC, Eckert S, Chaudhuri RR, Ewers C, Mellata M, et al. Sequencing and functional annotation of avian pathogenic Escherichia coli serogroup O78 strains reveal the evolution of E. coli lineages pathogenic for poultry via distinct mechanisms. Infect Immun. 2013;81(3):838–49.
Dissanayake DR, Octavia S, Lan R. Population structure and virulence content of avian pathogenic Escherichia coli isolated from outbreaks in Sri Lanka. Vet Microbiol. 2014;168(2–4):403–12.
Garenaux A, Houle S, Folch B, Dallaire G, Truesdell M, Lepine F, Doucet N, Dozois CM. Avian lipocalin expression in chickens following Escherichia coli infection and inhibition of avian pathogenic Escherichia coli growth by Ex-FABP. Vet Immunol Immunopathol. 2013;152(1–2):156–67.
Heinemann IU, Jahn M, Jahn D. The biochemistry of heme biosynthesis. Arch Biochem Biophys. 2008;474(2):238–51.
Gao Q, Wang X, Xu H, Xu Y, Ling J, Zhang D, Gao S, Liu X. Roles of iron acquisition systems in virulence of extraintestinal pathogenic Escherichia coli: salmochelin and aerobactin contribute more to virulence than heme in a chicken infection model. BMC Microbiol. 2012;12:143.
Kouse AB, Righetti F, Kortmann J, Narberhaus F, Murphy ER. RNA-mediated thermoregulation of iron-acquisition genes in Shigella dysenteriae and pathogenic Escherichia coli. PLoS One. 2013;8(5):e63781.
Pilarczyk-Zurek M, Chmielarczyk A, Gosiewski T, Tomusiak A, Adamski P, Zwolinska-Wcislo M, Mach T, Heczko PB, Strus M. Possible role of Escherichia coli in propagation and perpetuation of chronic inflammation in ulcerative colitis. BMC Gastroenterol. 2013;13:61.
Nagy G, Dobrindt U, Kupfer M, Emody L, Karch H, Hacker J. Expression of hemin receptor molecule ChuA is influenced by RfaH in uropathogenic Escherichia coli strain 536. Infect Immun. 2001;69(3):1924–8.
Li G, Laturnus C, Ewers C, Wieler LH. Identification of genes required for avian Escherichia coli septicemia by signature-tagged mutagenesis. Infect Immun. 2005;73(5):2818–27.
Schouler C, Koffmann F, Amory C, Leroy-Setrin S, Moulin-Schouleur M. Genomic subtraction for the identification of putative new virulence factors of an avian pathogenic Escherichia coli strain of O2 serogroup. Microbiology. 2004;150(Pt 9):2973–84.
Russo TA, Carlino UB, Johnson JR. Identification of a new iron-regulated virulence gene, ireA, in an extraintestinal pathogenic isolate of Escherichia coli. Infect Immun. 2001;69(10):6209–16.
Tarr PI, Bilge SS, Vary Jr JC, Jelacic S, Habeeb RL, Ward TR, Baylor MR, Besser TE. Iha: a novel Escherichia coli O157:H7 adherence-conferring molecule encoded on a recently acquired chromosomal island of conserved structure. Infect Immun. 2000;68(3):1400–7.
Goldberg MB, DiRita VJ, Calderwood SB. Identification of an iron-regulated virulence determinant in Vibrio cholerae, using TnphoA mutagenesis. Infect Immun. 1990;58(1):55–60.
Tashima KT, Carroll PA, Rogers MB, Calderwood SB. Relative importance of three iron-regulated outer membrane proteins for in vivo growth of Vibrio cholerae. Infect Immun. 1996;64(5):1756–61.
Herzer PJ, Inouye S, Inouye M, Whittam TS. Phylogenetic distribution of branched RNA-linked multicopy single-stranded DNA among natural isolates of Escherichia coli. J Bacteriol. 1990;172(11):6175–81.
Clermont O, Bonacorsi S, Bingen E. Rapid and simple determination of the Escherichia coli phylogenetic group. Appl Environ Microbiol. 2000;66(10):4555–8.
Bearden SW, Perry RD. The Yfe system of Yersinia pestis transports iron and manganese and is required for full virulence of plague. Mol Microbiol. 1999;32(2):403–14.
Tsolis RM, Baumler AJ, Heffron F, Stojiljkovic I. Contribution of TonB- and Feo-mediated iron uptake to growth of Salmonella typhimurium in the mouse. Infect Immun. 1996;64(11):4549–56.
Wang S, Niu C, Shi Z, Xia Y, Yaqoob M, Dai J, Lu C. Effects of ibeA deletion on virulence and biofilm formation of avian pathogenic Escherichia coli. Infect Immun. 2011;79(1):279–87.
Wang S, Xia Y, Dai J, Shi Z, Kou Y, Li H, Bao Y, Lu C. Novel roles for autotransporter adhesin AatA of avian pathogenic Escherichia coli: colonization during infection and cell aggregation. FEMS Immunol Med Microbiol. 2011;63(3):328–38.
Zhuge X, Wang S, Fan H, Pan Z, Ren J, Yi L, Meng Q, Yang X, Lu C, Dai J. Characterization and functional analysis of AatB, a novel autotransporter adhesin and virulence factor of avian pathogenic Escherichia coli. Infect Immun. 2013;81(7):2437–47.
Wang S, Shi Z, Xia Y, Li H, Kou Y, Bao Y, Dai J, Lu C. IbeB is involved in the invasion and pathogenicity of avian pathogenic Escherichia coli. Vet Microbiol. 2012;159(3–4):411–9.
Blanco JE, Blanco M, Mora A, Jansen WH, Garcia V, Vazquez ML, Blanco J. Serotypes of Escherichia coli isolated from septicaemic chickens in Galicia (northwest Spain). Vet Microbiol. 1998;61(3):229–35.
Dai J, Wang S, Guerlebeck D, Laturnus C, Guenther S, Shi Z, Lu C, Ewers C. Suppression subtractive hybridization identifies an autotransporter adhesin gene of E. coli IMT5155 specifically associated with avian pathogenic Escherichia coli (APEC). BMC Microbiol. 2010;10:236.
Urban JH, Vogel J. Translational control and target recognition by Escherichia coli small RNAs in vivo. Nucleic Acids Res. 2007;35(3):1018–37.
Wu Z, Wu C, Shao J, Zhu Z, Wang W, Zhang W, Tang M, Pei N, Fan H, Li J, et al. The Streptococcus suis transcriptional landscape reveals adaptation mechanisms in pig blood and cerebrospinal fluid. RNA. 2014;20(6):882–98.
Wang S, Bao Y, Meng Q, Xia Y, Zhao Y, Wang Y, Tang F, ZhuGe X, Yu S, Han X, et al. IbeR facilitates stress-resistance, invasion and pathogenicity of avian pathogenic Escherichia coli. PLoS One. 2015;10(3):e0119698.
Ali-Benali MA, Alary R, Joudrier P, Gautier MF. Comparative expression of five Lea Genes during wheat seed development and in response to abiotic stresses by real-time quantitative RT-PCR. Biochim Biophys Acta. 2005;1730(1):56–65.
Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97(12):6640–5.
Fung KP, Wong TW. Estimation of quantal dose response of drugs by the Spearman-Karber method: a computer program written in BASIC. J Singapore Paediatr Soc. 1989;31(3–4):129–32.
La Ragione RM, Sayers AR, Woodward MJ. The role of fimbriae and flagella in the colonization, invasion and persistence of Escherichia coli O78:K80 in the day-old-chick model. Epidemiol Infect. 2000;124(3):351–63.
McLeod A, Mage I, Heir E, Axelsson L, Holck AL. Effect of relevant environmental stresses on survival of enterohemorrhagic Escherichia coli in dry-fermented sausage. Int J Food Microbiol. 2016;229:15–23.
Acknowledgements
Authors are grateful to all of the staff at Key Laboratory Animal Bacteriology of Nanjing Agricultural University for helping conduct the experiments.
Funding
This work was supported by grants from the National Basic Research Program of China (2015CB554203), the Fund of Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Fundamental Research Funds for the Central Universities (KYZ201326), the Youth Foundation of the National Natural Science Foundation of China (No.31402213), the Natural Science Foundation of Jiangsu Province, China (No. BK20140686).
Availability of data and materials
All the data supporting our findings is contained within the manuscript.
Authors’ contributions
YL carried out the gene deletion and adhesion assays, as well as expression of ireA gene, JD carried out the tests of resistance to environmental stress, XZ, HW and LH carried out the animal infections, JR, LC, DL carried out the qPCR and gene prevelance tests. FT responsible for the growth curve, whole exprements design and draft of the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent to publish
Not applicable.
Ethics and consent to participate
Our experiments were conducted with the permission of the Ministry of Science and Technology of Jiangsu Province. The license number is SYXK(SU) 2010-0005. All efforts were made to minimize animal suffering.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Detailed information about the distribution of the ireA gene in APEC strains. (DOCX 21 kb)
Additional file 2: Figure S1.
Technical routes of construction of the ireA deletion mutant. (TIF 98 kb)
Additional file 3: Figure S2.
Technical routes of construction of the ireA complementary strain. (TIF 102 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, Y., Dai, J., Zhuge, X. et al. Iron-regulated gene ireA in avian pathogenic Escherichia coli participates in adhesion and stress-resistance. BMC Vet Res 12, 167 (2016). https://doi.org/10.1186/s12917-016-0800-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-016-0800-y