
Abstract
Background
Bartonellosis is an emerging zoonotic infection responsible for a variety of clinical syndromes in humans and animals. Members of the genus Bartonella exhibit high degrees of genetic diversity and ecologic plasticity. The infection is usually transmitted to animals and humans through blood-feeding arthropod vectors such as fleas, lice, ticks and sandflies. This study was conducted to investigate the prevalence of Bartonella species in 184 beef cattle, 40 dairy cattle, 40 sheep and 40 goats in eight animal farms across Peninsular Malaysia. Bartonella-specific PCR assays and sequence analysis of partial fragments of the citrate synthase gene were used for detection and identification of B. bovis. Isolation of B. bovis was attempted from PCR-positive blood samples. Molecular heterogeneity of the isolates was investigated based on sequence analysis of gltA, ITS, rpoB genes, ERIC-PCR, as well as using an established multilocus sequence typing (MLST) method. The carriage rate of B. bovis in ticks was also determined in this study.
Results
B. bovis was detected using Bartonella gltA-PCR assays from ten (4.5 %) of 224 cattle blood samples, of which three (1.3 %) were from beef cattle and seven (3.1 %) were from dairy cattle. None of the blood samples from the sheep and goats understudied were positive for B. bovis. Haemaphysalis bispinosa and Rhipicephalus (Boophilus) microplus were the predominant tick species identified in this study. B. bovis was detected from eight of 200 H. bispinosa ticks and none from the R. microplus ticks. Isolation of B. bovis was successful from all PCR-positive cattle blood samples, except one. Strain differentiation of B. bovis isolates was attempted based on sequence analysis of gltA, ITS, rpoB, and ERIC-PCR assay. B. bovis isolates were differentiated into six genotypes using the approach. The genetic heterogeneity of the isolates was confirmed using MLST method. Of the six MLST sequence types identified, five were designated new sequence types (ST23-27), while one (ST18) had been reported previously from Thailand isolates. All except one isolates were segregated into lineage II. A new lineage (IIa) is proposed for a single isolate obtained from a dairy cow.
Conclusions
The current study reported the first detection of B. bovis infection in the cattle and H. bispinosa ticks in Peninsular Malaysia. At least six genotypes of B. bovis were found circulating in the cattle understudied. New MLST sequence types were identified in Malaysian B. bovis isolates. Further study is necessary to explore the zoonotic potential of B. bovis and the vector compatibility of H. bispinosa ticks.
Similar content being viewed by others


Background
Bartonellae are small, Gram-negative fastidious bacteria which infect mammalian erythrocytes and endothelial cells. The organisms have been reported as the causative agents for a variety of clinical symptoms in human and animals [1, 2]. Transmission of the bacteria is usually through the bites of hematophagous arthropods such as fleas, sand flies, biting flies, lice and ticks [3–6]. Various bartonellae species have been associated with domestic and wild ruminants. The infections caused by bartonellae in the cattle are usually asymptomatic. Bartonella bovis, previously known as Bartonella weissii and originally isolated from domestic cats, has been reported as a pathogen which causes endocarditis and bacteremia in cattle [7–16].
The prevalence of B. bovis in cattle varied tremendously in different studies: USA (50–89 %), French Guyana (70 %), Georgia (57 %), Taiwan (42.4 %), France (36 %), Italy (24 %), Guatemala (21 %), West Africa (20 %), Thailand (10 %), Poland (6.8 %), Japan (0 %) and Kenya (0 %) [8–16]. Cattle ticks (Rhipicephalus microplus) and flies have been described as the potential vectors for transmission of B. bovis [3, 15]. In a recent study, multiple factors such as distribution and abundance of specific arthropods, and environmental factors (geographic characters, landscape, etc.) have been postulated to have some effect on the prevalence of B. bovis [14]. Other than the isolation of B. melophagi from sheep ked, little is known about the prevalence of Bartonella spp. in sheep and goats [17].
Information on the prevalence, genetic variation and arthropod vector is important for formulation of strategies for prevention and control of B. bovis infections. Several molecular approaches including sequence analysis of the Bartonella citrate synthase gene (gltA) [18], internal transcribed spacer of 12S-23S rRNA (ITS) [19], and beta subunit of the RNA polymerase gene (rpoB) fragments [20], enterobacterial repetitive intergenic consensus (ERIC)-PCR [21], and PCR-restriction fragment length polymorphism [18] have been used for genotyping of bartonellae. Recently, a multi-locus sequence typing (MLST) scheme has been developed to compare the genetic divergence of B. bovis [14]. The MLST data suggested genetic variations among 28 isolates from different geographical regions and a total of 22 sequence types and three lineages (I, II, and III) had been identified.
Data on the prevalence and genetic diversity of B. bovis in the domestic and wild animals in the Southeast Asia is scarce. So far, B. bovis infections have only been reported from cattle and water buffaloes from Thailand [14]. There has not been any report on the association of B. bovis with any arthropod in the region. This study was designed to determine the prevalence and molecular heterogeneity of Bartonella spp. in the cattle, sheep and goats from eight farms across Peninsular Malaysia and to investigate the presence of bartonellae in the ticks collected from the farms. Isolation and strain differentiation of B. bovis were also attempted.
Methods
Study sites and sample collection
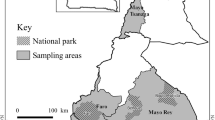
Written approval (JPV/PSTT/100-8/1) for animal blood sampling and assessment of tick samples was obtained from the Director, Department of Veterinary Services, Ministry of Agriculture and Agro-Based Industry, Malaysia who owns the animals understudied. Sampling was carried out by farm workers according to standard veterinary care and practice. Table 1 shows the locations of the farms selected in this study. Animal blood sampling was conducted from February to September 2013 in six cattle farms, a goat and a sheep farm at different regions of Peninsular Malaysia. Approximately 1–3 mL whole blood samples were collected from the animals via jugular vein in EDTA-coated tubes and transported on ice to the laboratory. Blood samples were stored at -20 °C prior to processing.
Ticks were collected from animals subjected for blood collection whenever possible. The ear, eyes, flank, abdomen, tail and perineal regions of the animals were examined for ticks. Ticks were identified to the genus level according to Walker et al. [22] and Geevarghese and Mishra [23], and preserved in -80 °C freezer prior to processing. For molecular identification of the ticks, the tick 16S rRNA gene was amplified and sequenced [24].
DNA extraction
DNA was extracted from 200 μL of animal blood samples using QIAamp DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Ticks were first thawed, washed in 5 % sodium hypochlorite and 70 % ethanol and rinsed in sterile distilled water prior to homogenization using surgical blades [25]. Each tick homogenate was then subjected to DNA extraction using a QIAamp DNA mini kit (Qiagen, Hilden, Germany).
Molecular detection and data analysis of Bartonella spp. from animal blood samples and tick samples
PCR assay targeting bartonellae citrate synthase gene (gltA) was used for detection of Bartonella DNA in the animal blood and tick samples [18]. All PCR assays were performed in a final volume of 20 μL containing 2 μL of DNA template, 1X ExPrime Taq DNA polymerase (GENET BIO, South Korea) and 0.2 μM of each primer, in a Veriti thermal cycler (Applied Biosystems, USA). Positive control derived from plasmid carrying the amplified gltA gene from Bartonella tribocorum and negative control (sterile distilled water) were included in each PCR run. A rodent-borne strain of Bartonella elizabethae (BeUM) was used as positive control for other PCR assays. PCR products were separated on a 1.5 % agarose gel at 100 V for 45 min and visualized using a UV transilluminator (G-Box, Syngene, UK). Amplicons were purified using GeneAll Expin™ Combo GP (GeneAll, South Korea) as described by the manufacturer. Sequencing was performed with a Big Dye Terminator Cycle Sequencing kit, version 3.1 (Applied Biosystems, USA) on an ABI PRISM 377 Genetic Analyzer (Applied Biosystems, USA), using forward and reverse primers.
Sequence assembly and alignment were performed using BioEdit Sequence Alignment Editor Software (Version 7.0.5.3). The resulting sequences were compared with known Bartonella sequences deposited in the GenBank database using the Basic Local Alignment Search Tool (BLAST) program (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Isolation of Bartonella spp
Isolation of B. bovis was attempted using PCR-positive blood samples. Each blood sample (100 μL) was inoculated in duplicate onto fresh Columbia agar plates supplemented with 5 % sheep blood (Isolab, Malaysia). The agar plates were incubated at 37 °C in 5 % CO2 for four weeks. Following incubation, isolates were subcultured for Gram staining, and DNA was extracted using a QIAamp DNA mini kit (Qiagen, Hilden, Germany). Confirmation of the identity of the isolates was carried out by using gltA-PCR followed by sequence analysis.
Strain differentiation B. bovis isolates
B. bovis isolates were differentiated based on sequence analysis of gltA (278 bp) [18], ITS (301–322 bp) [19], rpoB (671 bp) [20], and ERIC-PCR assay [21]. Enterobacterial repetitive intergenic consensus (ERIC)-PCR was performed by using ERIC 1R and ERIC 2 primers as described previously [26]. The PCR reaction mixture contained 2 μL of DNA template, 0.2 μM of each primer, 1 X PCR buffer, 0.2 mM dNTP mix, 1.5 mM magnesium chloride (MgCl2) and 1U of GoTaq @ Flexi DNA polymerase (Promega Corp, USA). Amplification was performed according to Dehio et al. [21] and the amplified fragments were separated by electrophoresis at 60 V in a 2 % agarose gel for 4 h.
MLST analysis
Sequence comparison for eight loci (16S rRNA, ftsZ, groEL, nuoG, ribC, rpoB, ssrA and ITS) were performed for each B. bovis isolate as described by Bai et al. [14]. A dendrogram was constructed based on the concatenated sequences of the eight loci using the neighbour-joining method of the MEGA 6.0 software and bootstrap analysis with 1,000 resamplings [27].
Results
A total of 304 blood samples collected from 184 beef cattle, 40 dairy cattle, 40 sheep and 40 goats were subjected to PCR detection for B. bovis DNA in this study (Table 1). The prevalence of B. bovis ranged from zero to 17.5 % across the eight animal farms. B. bovis was detected from ten (10/224; 4.5 %) of 224 cattle blood samples, of which three (3/224; 1.3 %) were from beef cattle and seven (7/224; 3.1 %) were from dairy cattle. None of the blood samples from the sheep and goats understudied were positive for B. bovis. The PCR-positive blood samples were derived from three beef cattle (one Nellore and two Yellow cattle cross Kedah-Kelantan breeds) in two farms (Farms 1 and 6) and seven dairy cattle (Mafriwal breed) in Farm 8 (Table 1). The age of the infected cattle ranged from one to four years old. No overt clinical signs were observed from those cattle with positive PCR findings for B. bovis.
Mixed populations of R. microplus and H. bispinosa were detected in the cattle in this study. The sheep were mainly infested by H. bispinosa. Of 270 ticks examined in this study, 70 and 200 ticks were identified as R. microplus and H. bispinosa, respectively. Sequence analysis of the 16S rRNA gene amplified from B. bovis-positive ticks revealed 100 % identity (200 bp) with that of H. bispinosa reported in India (GenBank: KC853418- KC853420) [28]. Eight (4.0 %) H. bispinosa ticks collected from B. bovis-infected cattle in two farms (Farms 1 and 6) were positive using Bartonella gltA PCR assay. These included one male tick and seven fully engorged female ticks. None of the R. microplus tick was positive for bartonellae.
B. bovis was successfully isolated from nine cattle blood samples in this study (Table 2). A total of one, two and six isolates were cultured from the blood samples collected from cattle in Farms 1, 6 and 8, respectively. Primary cultures showed the growth of tiny, discrete, and greyish colonies after 5–7 days of incubation. A low level of bacteremia (≈20 colony forming unit/mL) was suspected as only one to two single colonies were obtained from each blood sample. The isolates were confirmed as B. bovis by using gltA-PCR assay followed by sequence analysis.
The isolates were first differentiated based on the nucleotide variation of gltA, ITS, rpoB and ERIC-PCR assay (Table 2). The sequences derived from this study have been deposited in the GenBank database: B1-B2 (gltA, GenBank accession no.: KP230412-KP230413), S1-S3 (ITS, GenBank accession no.: KP230415-KP230417), and R1-R5 (rpoB, GenBank accession no.: KP230418-KP230422). Two gltA sequence types (B1-B2), demonstrating 100 % (n = 8) and 99 % (n = 1) similarities with B. bovis strain 91-4 (GenBank accession no.: AF293394), were identified in this study. The amplified ITS gene from nine isolates was differentiated into three sequence types (S1-S3). The 322 bp of the sequence type S1 (five isolates) and S2 (two isolates) showed 99 % homology with B. bovis strain 91-4 (GenBank accession no.: AY116638). Meanwhile, the 301 bp of the sequence type S3 were aligned 100 % (292/292) to that of B. bovis strain B38216 from the cattle in Guatemala (GenBank accession no.: KF218233, [14]). The rpoB sequences obtained from nine isolates were differentiated into five sequence types (R1-R5). Sequence type R1 was identical to that of B. bovis strain B25099 from cattle in Thailand (GenBank accession no.: KF218220) [14]. Sequence type R2, R3 and R4 showed the closest similarity (99 %) to that of B. bovis strain B32674 from a water buffalo in Thailand (GenBank accession no.: KF218223) [14]. The sequence type R5 identified in an isolate (F8-6) in Farm 8 was identical to that of a B. bovis isolate (N04-927) which caused bovine endocarditis in France (GenBank accession no.: EF432062) [8].
Figure 1 shows the fingerprinting profiles generated from the ERIC-PCR assay. A total of four to seven DNA fragments with the size ranging from 210 bp to 2 kb were generated from each isolate. Based on the fingerprinting profiles in Fig. 1, the isolates were differentiated into three genotypes (E1-E3). Table 2 summarizes the genotyping results of B. bovis isolates based on the combined sequence analysis of gltA, ITS, rpoB and ERIC-PCR assay. A total of six genotypes of B. bovis were identified from nine isolates in this study. Interestingly, six isolates from a single farm (Farm 8) were differentiated to five genotypes (Table 2, Fig. 1). The approach grouped the isolates F1-1 and F8-1 in Genotype 1, and the isolates F6-1, F8-2 and F8-3 in Genotype 3. The remaining isolates, i.e., F6-2, F8-4, F8-5 and F8-6, were differentiated to Genotype 2, 4, 5 and 6, respectively. Further differentiation of B. bovis from blood and tick samples based on the sequence analysis of multiple genes was not possible due to the failure in amplifying the target genes, probably owing to the low amounts of DNA in the samples.

ERIC-PCR fingerprinting profiles of nine B. bovis isolates. Lane M1, 100 bp ladder; Lane M2,1 kb ladder; Lane 1: F6-1 (E1); Lane 2: F6-2 (E1); Lane 3: F1-1 (E2); Lane 4: F8-4(E1); Lane 5: F8-1 (E2); Lane 6: F8-2 (E1); Lane 7: F8-3 (E1); Lane 8: F8-6 (E3) and Lane 9: F8-5 (E1)
Six sequence types were identified from the MLST analysis in this study (Table 3), of which five (designated as ST23-27) have not been described before. New sequences obtained from the MLST analysis in this study have been submitted to the GenBank database with the accession numbers KR733181, KR733182-KR733184, KR733185-KR733186, KR733187-KR733189, KR733190-KR733191, KR733192-KR733195, KR733196, KR733197-KR733121 for ftsZ, gltA, groEL, nuoG, ribC, rpoB, ssrA and ITS genes, respectively.
Three isolates (F8-2, F8-3 and F6-1) in this study were identified as ST18 which had been reported in B. bovis isolated from cattle in Thailand (B25093). ST23 was represented by two isolates (F1-1 and F8-1) in this study. Other STs (ST24-27) in this study were represented by a single isolate. One of the isolates (F8-6) with unique PCR fingerprint profile (E3) exhibited sequence variation in all the gene fragments except for ssrA gene. The dendrogram constructed based on the concatenated sequences of eight loci demonstrated the segregation of eight B. bovis isolates into lineage II (Fig. 2). The isolate F8-6 (ST27) was placed at a single branch adjacent to those isolates from the lineage II, with a 81 % bootstrap value. The isolate is proposed under a new lineage (designated as lineage IIa).

Phylogenetic placement of Malaysian B. bovis isolates based on the concatenated sequences of ftsZ, gltA, groEL, nuoG, ribC, rpoB, ssrA, and ITS genes. Reference sequences were retrieved from Bai et al. [14] for comparison. Bootstrap analysis was performed with 1000 replications. Numbers in brackets are GenBank accession numbers. Scale bar indicates the nucleotide substitutions per sites
Discussion
This study reports for the first time the molecular detection and isolation of B. bovis from cattle in Peninsular Malaysia. The prevalence (4.5 %) of the B. bovis in the cattle, as determined by direct amplification from blood samples in this study was low when compared to those of cattle (10 %) and water buffaloes (6.8 %) reported from Thailand, a neighbouring country of Malaysia [14]. A high prevalence (42.4 %) of B. bovis has been reported in beef cattle from Taiwan [15]. In this study, the prevalence of B. bovis infections was higher (7/40; 17.5 %) in dairy cattle, as compared to the beef cattle (3/204; 1.6 %). In a Japanese study, B. bovis was not detected from 305 cattle investigated across five prefectures [14]. The causes for the variation in the prevalence of B. bovis across the same geographical region are still not clear [14]. Factors such as the carriage rate of B. bovis in the ectoparasites, environmental exposure, animal breed, susceptibilities and husbandry practices are probably important. Although cattle examined in this study appeared healthy, B. bovis infection has been associated with endocarditis in previous investigations. Maillard et al. reported B. bovis infection in 9.1 % of 22 cows with endocarditis [7]. Additionally, B. bovis had been isolated from the heart of an Angus cow which succumbed to sudden death without any clinical sign [29]. As endocarditis caused by fastidious haemotropic bacteria is not frequently accompanied by clinical manifestations [30], direct amplification of B. bovis DNA from blood samples by using PCR method is important for surveillance of the infection in the animals.
R. microplus is a common tick in cattle while H. bispinosa tick has been reported to parasitize both cattle and goats [28]. B. bovis has been detected from 15.7 % of R. microplus ticks in Taiwan [15]. The cattle in this study were infested by both R. microplus and H. bispinosa, while the sheep were infested mainly by H. bispinosa. Although Farm 8 had the highest prevalence of B. bovis, ticks were not available for testing as the animals had undergone deticking prior to our sampling study. To the best of our knowledge, this is the first report of the detection of B. bovis in H. bispinosa ticks. Haemaphysalis genus is the second largest tick genus in the family Ixodidae [31] which has been associated with various tickborne pathogens including Bartonella [32], Theileria, Babesia, Rickettsia, Ehrlichia, Anaplasma [33], and spotted fever group rickettsiae [34]. Bartonella spp. other than B. bovis have been reported in H. longicornis in ticks from Korea [32] and China [35].
In this study, although B. bovis has been detected from both infected cattle (F6-2) and H. bispinosa ticks feeding on the same host, this does not necessarily demonstrate B. bovis infection in the ticks as the DNA detected might have originated from the blood ingested by ticks. Further characterization and experimental transmission studies are needed to demonstrate vector competency of the H. bispinosa for B. bovis transmission.
Sequence analysis of gltA, ITS and rpoB genes and ERIC-PCR assay was initially attempted for strain differentiation of B. bovis in this study. Among the three genes, most sequence variation was observed in the rpoB gene. Sequence analysis of gltA and ITS region were less discriminative due to their shorter fragments. ERIC-PCR assay was rapid and cost-effective but less discriminative compared to the results obtained based on the sequence analysis of the rpoB gene. However, when the results of the sequence typing of gltA, ITS, rpoB and ERIC-PCR were combined (as shown in Table 2), six genotypes were identified in the nine isolates. Although the discrimination power of this approach was similar with MLST analysis, further validation with more isolates is necessary.
In an effort to reveal the genetic diversity of B. bovis, Bai et al. reported the segregation of 22 STs into three lineages (I-III) from 28 strains originated from different continents [14]. The typing of nine B. bovis isolates understudied into six STs confirms the high genetic diversity of B. bovis, particularly for those isolated from dairy cattle in Farm 8, with the identification of five STs (Table 2). Specific host association had been demonstrated previously with isolates from lineages I and II in cattle and lineage III in water buffaloes [14]. It was hypothesized that lineage I was associated with the cattle of ‘taurine’ lineage while lineage II was associated with the cattle of ‘zebu’ lineage. The crossing lineages of B. bovis isolated from the cattle from Guatemala (lineage I and II) had been attributed to a mixed breed of cattle. Interestingly, in this study, B. bovis was isolated with a higher rate in the dairy cattle (Mafriwal breed). The tropical breed has been developed by crossbreeding between Sahiwal (Bos indicus) and Friesian (Bos taurus) cattle to gain higher weight and milk production [36]. The finding of new STs and proposal of a new lineage for B. bovis isolated from the crossbreed cattle are interesting and warrants further investigation.
Conclusions
The detection of B. bovis in cattle blood and H. bispinosa tick samples from six farms across Peninsular Malaysia was demonstrated in this study. In line with previous studies, B. bovis was genetically diverse, as evidenced by the identification of six genotypes based on molecular methods. MLST data shows the identification of new sequence variants of B. bovis which are distinct from those reported in other geographical regions. A new lineage (IIa) is proposed for a single isolate of B. bovis obtained from a dairy cow. The vectorial potential of H. bispinosa ticks of B. bovis between cattle warrants further investigation. Additionally, the role of B. bovis as a zoonotic infectious agent should be further explored, in view of the fact that humans may have direct exposure to B. bovis during handling of infected animals (slaughtering and evisceration process) and through bites of hematophagous arthropods in the farm.
Availability of supporting data
The data set(s) supporting the results of this article is included within the article. The sequences derived from this study have been deposited in the GenBank database: gltA (GenBank accession no.:KP230412-KP230414), ITS (GenBank accession no.: KP230415-KP230417), and rpoB (GenBank accession no.: KP230418-KP230422). New sequence types from the MLST analysis: KR733181, KR733182-KR733184, KR733185-KR733186, KR733187-KR733189, KR733190-KR733191, KR733192-KR733195, KR733196, KR733197-KR733121 for ftsZ, gltA, groEL, nuoG, ribC, rpoB, ssrA and ITS, respectively.
Abbreviations
- ERIC-PCR:
-
Enterobacterial repetitive intergenic consensus polymerase chain reaction
- MLST:
-
Multilocus Sequence Typing
References
Anderson BE, Neuman MA. Bartonella spp. as emerging human pathogens. Clin Microbiol Rev. 1997;10(2):203–19.
Breitschwerdt EB, Maggi RG, Chomel BB, Lappin MR. Bartonellosis: an emerging infectious disease of zoonotic importance to animals and human beings. J Vet Emerg Crit Care (San Antonio, Tex : 2001). 2010;20(1):8–30.
Tsai YL, Chang CC, Chuang ST, Chomel BB. Bartonella species and their ectoparasites: selective host adaptation or strain selection between the vector and the mammalian host? Comp Immunol Microbiol Infect Dis. 2011;34(4):299–314.
Drali R, Sangare AK, Boutellis A, Angelakis E, Veracx A, Socolovschi C, et al. Bartonella quintana in body lice from scalp hair of homeless persons, France. Emerg Infect Dis. 2014;20(5):907–8.
Janecek E, Mietze A, Goethe R, Schnieder T, Strube C. Bartonella spp. infection rate and B. grahamii in ticks. Emerg Infect Dis. 2012;18(10):1689–90.
Chung CY, Kasten RW, Paff SM, Van Horn BA, Vayssier-Taussat M, Boulouis HJ, et al. Bartonella spp. DNA associated with biting flies from California. Emerg Infect Dis. 2004;10(7):1311–3.
Maillard R, Petit E, Chomel B, Lacroux C, Schelcher F, Vayssier-Taussat M, et al. Endocarditis in cattle caused by Bartonella bovis. Emerg Infect Dis. 2007;13(9):1383–5.
Welc-Faleciak R, Grono K. The first cases of Bartonella bovis infection in cattle from Central Europe. Vet Microbiol. 2013;162(2–4):954–6.
Martini M, Menandro ML, Mondin A, Pasotto D, Mazzariol S, Lauzi S, et al. Detection of Bartonella bovis in a cattle herd in Italy. Vet Rec. 2008;162(2):58–9.
Raoult D, La Scola B, Kelly PJ, Davoust B, Gomez J. Bartonella bovis in cattle in Africa. Vet Microbiol. 2005;105(2):155–6.
Saisongkorh W, Rolain JM, Suputtamongkol Y, Raoult D. Emerging Bartonella in humans and animals in Asia and Australia. J Med Assoc Thai. 2009;92(5):707–31.
Bermond D, Boulouis HJ, Heller R, Van Laere G, Monteil H, Chomel BB, et al. Bartonella bovis Bermond et al. sp. nov. and Bartonella capreoli sp. nov., isolated from European ruminants. Int J Syst Evol Microbiol. 2002;52(Pt 2):383–90.
Cherry NA, Maggi RG, Cannedy AL, Breitschwerdt EB. PCR detection of Bartonella bovis and Bartonella henselae in the blood of beef cattle. Vet Microbiol. 2009;135(3–4):308–12.
Bai Y, Malania L, Alvarez Castillo D, Moran D, Boonmar S, Chanlun A, et al. Global distribution of Bartonella infections in domestic bovine and characterization of Bartonella bovis strains using multi-locus sequence typing. PLoS One. 2013;8(11), e80894.
Tsai YL, Chomel BB, Chang CC, Kass PH, Conrad PA, Chuang ST. Bartonella and Babesia infections in cattle and their ticks in Taiwan. Comp Immunol Microbiol Infect Dis. 2011;34(2):179–87.
Breitschwerdt EB, Sontakke S, Cannedy A, Hancock SI, Bradley JM. Infection with Bartonella weissii and detection of Nanobacterium antigens in a North Carolina beef herd. J Clin Microbiol. 2001;39(3):879–82.
Kumsa B, Parola P, Raoult D, Socolovschi C. Bartonella melophagi in Melophagus ovinus (sheep ked) collected from sheep in northern Oromia, Ethiopia. Comp Immunol Microbiol Infect Dis. 2014;37(1):69–76.
Norman AF, Regnery R, Jameson P, Greene C, Krause DC. Differentiation of Bartonella-like isolates at the species level by PCR-restriction fragment length polymorphism in the citrate synthase gene. J Clin Microbiol. 1995;33(7):1797–803.
Maggi RG, Breitschwerdt EB. Potential limitations of the 16S-23S rRNA intergenic region for molecular detection of Bartonella species. J Clin Microbiol. 2005;43(3):1171–6.
Renesto P, Gouvernet J, Drancourt M, Roux V, Raoult D. Use of rpoB gene analysis for detection and identification of Bartonella species. J Clin Microbiol. 2001;39(2):430–7.
Dehio C, Lanz C, Pohl R, Behrens P, Bermond D, Piemont Y, et al. Bartonella schoenbuchii sp. nov., isolated from the blood of wild roe deer. Int J Syst Evol Microbiol. 2001;51(Pt 4):1557–65.
Walker AR, Bouattour A, Camicas J-L, Estrada-Pena A, Horak IG, Latif AA, et al. Ticks of domestic animals in Africa: a guide to identification of species. Bioscience Reports: Edinburgh Scotland, U.K.; 2003.
Geevarghese G, Mishra AC: Haemaphysalis Ticks of India: Elsevier Science, London; 2011
Black WC, Piesman J. Phylogeny of hard- and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc Natl Acad Sci U S A. 1994;91(21):10034–8.
Duh D, Punda-Polic V, Avsic-Zupanc T, Bouyer D, Walker DH, Popov VL, et al. Rickettsia hoogstraalii sp. nov., isolated from hard- and soft-bodied ticks. Int J Syst Evol Microbiol. 2010;60(Pt 4):977–84.
Versalovic J, Koeuth T, Lupski JR. Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Res. 1991;19(24):6823–31.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis, version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Brahma RK, Dixit V, Sangwan AK, Doley R. Identification and characterization of Rhipicephalus (Boophilus) microplus and Haemaphysalis bispinosa ticks (Acari: Ixodidae) of North East India by ITS2 and 16S rDNA sequences and morphological analysis. Exp Appl Acarol. 2014;62(2):253–65.
Erol E, Jackson C, Bai Y, Sells S, Locke S, Kosoy M. Bartonella bovis isolated from a cow with endocarditis. J Vet Diagn Invest. 2013;25(2):288–90.
Buczinski S, Belanger AM. Bovine tricuspid endocarditis as a cause of increased serum concentration of cardiac troponins. Can Vet J. 2010;51(2):195–7.
Horak IG, Camicas JL, Keirans JE. The Argasidae, Ixodidae and Nuttalliellidae (Acari: Ixodida): a world list of valid tick names. Exp Appl Acarol. 2002;28(1–4):27–54.
Kim CM, Kim JY, Yi YH, Lee MJ, Cho MR, Shah DH, et al. Detection of Bartonella species from ticks, mites and small mammals in Korea. J Vet Sci. 2005;6(4):327–34.
Chen Z, Liu Q, Liu JQ, Xu BL, Lv S, Xia S, et al. Tick-borne pathogens and associated co-infections in ticks collected from domestic animals in central China. Parasit Vectors. 2014;7(1):237.
Tijsse-Klasen E, Hansford KM, Jahfari S, Phipps P, Sprong H, Medlock JM. Spotted fever group rickettsiae in Dermacentor reticulatus and Haemaphysalis punctata ticks in the UK. Parasit Vectors. 2013;6:212.
Sun J, Liu Q, Lu L, Ding G, Guo J, Fu G, et al. Coinfection with four genera of bacteria (Borrelia, Bartonella, Anaplasma, and Ehrlichia) in Haemaphysalis longicornis and Ixodes sinensis ticks from China. Vector Borne Zoonotic Dis (Larchmont, NY). 2008;8(6):791–5.
Panandam JM, Raymond AK. Development of the Mafriwal dairy cattle of Malaysia. Available online: http://agtr.ilri.cgiar.org/casestudy/jothi/Jothi.htm.
Acknowledgments
The authors would like to thank Dato’ Dr. Ibrahim Che Embung, Dr. Abu Hassan Muhammad Ali, Dr. Arman Kison and Mr. Chang Kum Wah from the Department of Veterinary Services, Ministry of Agriculture and Agro-Based Industry, Malaysia, and managers and workers of each farm for their collaborative efforts. Our appreciation is also extended to Dr. Low Van Lun, Mr. Tan Tiong Kai, Ms Asha Devi and Ms Meeta Devi from University of Malaya for their assistance in sample collection. This project was funded by High Impact Research-MOHE Grant [E000013-20001 (subprogramme-4)], University Malaya Research Grant (RP013-2012A) and Postgraduate Research Fund (PG006-2013B) from University of Malaya, Kuala Lumpur, Malaysia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
KKL carried out the tick’s identification and DNA extraction, molecular detection, isolation, MLST, data analysis and wrote the manuscript. KFX performed the DNA extraction for the cattle blood and cattle ticks, ERIC-PCR and MLST. HNQZ approved and TQ provided assistance for sample collection from eight farms in this study. TST initiated and designed the study, supervised the laboratory works, data analysis and wrote the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Kho, KL., Koh, FX., Jaafar, T. et al. Prevalence and molecular heterogeneity of Bartonella bovis in cattle and Haemaphysalis bispinosa ticks in Peninsular Malaysia. BMC Vet Res 11, 153 (2015). https://doi.org/10.1186/s12917-015-0470-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-015-0470-1