
Abstract
Background
Multiple myeloma (MM) is a severely debilitating and fatal B-cell neoplastic disease. The discovery of disease-associated proteins with causal genetic evidence offers a chance to uncover novel therapeutic targets.
Methods
First, we comprehensively investigated the causal association between 2994 proteins and MM through two-sample mendelian randomization (MR) analysis using summary-level data from public genome-wide association studies of plasma proteome (N = 3301 healthy individuals) and MM (598 cases and 180,756 controls). Sensitivity analyses were performed for these identified causal proteins. Furthermore, we pursued the exploration of enriched biological pathways, prioritized the therapeutic proteins, and evaluated their druggability using the KEGG pathway analysis, MR-Bayesian model averaging analysis, and cross-reference with current databases, respectively.
Results
We identified 13 proteins causally associated with MM risk (false discovery rate corrected P < 0.05). Six proteins were positively associated with the risk of MM, including nicotinamide phosphoribosyl transferase (NAMPT; OR [95% CI]: 1.35 [1.18, 1.55]), tyrosine kinase with immunoglobulin-like and EGF-like domains 1 (TIE1; 1.14 [1.06, 1.22]), neutrophil cytosol factor 2 (NCF2; 1.27 [1.12, 1.44]), carbonyl reductase 1, cAMP-specific 3',5'-cyclic phosphodiesterase 4D (PDE4D), platelet-activating factor acetylhydrolase IB subunit beta (PAFAH1B2). Seven proteins were inversely associated with MM, which referred to suppressor of cytokine signaling 3 (SOCS3; 0.90 [0.86, 0.94]), Fc-gamma receptor III-B (FCGR3B; 0.75 [0.65,0.86]), glypican-1 (GPC1; 0.69 [0.58,0.83]), follistatin-related protein 1, protein tyrosine phosphatase non-receptor type 4 (PTPN4), granzyme B, complement C1q subcomponent subunit C (C1QC). Three of the causal proteins, SOCS3, FCGR3B, and NCF2, were enriched in the osteoclast differentiation pathway in KEGG enrichment analyses while GPC1 (marginal inclusion probability (MIP):0.993; model averaged causal effects (MACE): − 0.349), NAMPT (MIP:0.433; MACE: − 0.113), and NCF2 (MIP:0.324; MACE:0.066) ranked among the top three MM-associated proteins according to MR-BMA analyses. Furthermore, therapeutics targeting four proteins are currently under evaluation, five are druggable and four are future breakthrough points.
Conclusions
Our analysis revealed a set of 13 novel proteins, including six risk and seven protective proteins, causally linked to MM risk. The discovery of these MM-associated proteins opens up the possibility for identifying novel therapeutic targets, further advancing the integration of genome and proteome data for drug development.
Similar content being viewed by others

Background
Multiple myeloma (MM) is a hematologic neoplasm caused by the malignant proliferation of clonal plasma cells. In 2019, more than 155,688 people were diagnosed with multiple myeloma worldwide, and approximately 100,000 deaths are attributed to MM per year [1]. Various pharmacological strategies have been developed against MM, including proteasome inhibitors, immunomodulatory agents, and alkylating agents, which have successfully increased patient survivorship [2]. However, most patients still experience relapse and resultant mortality due to drug resistance; therefore, the 5-year survival rate of MM patients in high-risk populations remains 50% or lower [3]. Hence, it is imperative to find novel therapeutic targets for the development of new anti-multiple myeloma agents.
Proteins are versatile biologically active compounds involved in the regulation of multiple cellular and physiological functions. A new generation of proteomics technologies has enabled the identification of ectopic protein expressions and further exploration of potential biomarkers and therapeutic targets for cancer [4]. For example, by employing a quantitative proteomics approach, Chen et al. recognized proteinase inhibitor 9 (SERPINB9) as a promising novel therapeutic target for bortezomib-resistant recurrent and relapsed MM [5]. More recently, a proteomic profiling analysis revealed that cyclin-dependent kinase 6 (CDK6) upregulation is a targetable resistance mechanism for lenalidomide, highlighting the expanding importance of proteomic research in MM [6]. However, most such studies were restricted to small sample sizes and/or limited protein species. And it is important to note that the causal relevance of associations from these nonrandomized observational studies remains largely unresolved due to their susceptibility to confounders or reverse causation.
Mendelian randomization (MR) is a popular approach for causal inferences by using genetic variants as instrumental variables (IVs) that mimic a lifetime randomized controlled trial [7]. It exploits the natural random allocation of genetic variants at conception, so results from MR are much less likely to be biased by reverse causation or residual confounding. With the development of genome-wide association studies (GWASs) on human plasma proteome, an optimization framework by integrating genomic and proteomic databases for biomarker discovery has emerged [8]. In particular, MR studies leveraging protein quantitative trait loci from variants have contributed to elucidating novel targets for breast cancer [9], lung cancer [10], and ovarian cancer [11], which suggests that the analytical method is empirically validated and reliable.
We therefore applied a proteome-wide MR analysis by combing the high-throughput proteomes with genetic data to assess the causal effects of the circulating proteins on the risk of MM. Furthermore, to explore the clinical utility of these proteins, we branch out the existing research and give out a three-step parallel approach: (i) revealing their roles in the etiology of MM; (ii) disentangling the prioritization of these proteins; and (iii) evaluating the druggability of potential target proteins.
Methods
Study design
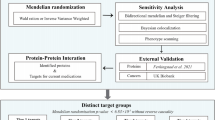
The flow diagram summarizing the methodology of the study is depicted in Fig. 1 and the detailed information of the data source is displayed in Additional file 1: Table. S1. Firstly, we leveraged the two-sample MR method to explore the causal relationships between 2994 plasma proteome and the risk of MM using summary-level data from publicly GWASs of plasma proteome in INTERVAL study (N = 3301) [12] and MM in FinnGen consortium (598 cases and 180,756 controls) [13]. Three assumptions are required for the MR method: (1) the single nucleotide polymorphism (SNP) selected as the genetic instrument is robustly associated with the exposure (“relevance”); (2) the SNP is not associated with confounders (“exchangeability”); and (3) SNP affect the outcome only through the exposure (“exclusion restriction”) [14]. False discovery rate (FDR) correction was used to account for multiple testing. Second, we further explore the enriched pathways, priority of therapeutic targets for MM, and druggability in these causal proteins. To be specific, the Kyoto encyclopedia of genes and genome (KEGG) pathway analysis was performed to identify significantly enriched pathways in the causal proteins and MR-Bayesian model averaging (MR-BMA) was leveraged to rank causal proteins by their marginal score. To evaluate clinical development activity of candidate drugs targeting the candidate proteins, we comprehensively queried an updated list of druggable genes [15], the ChEMBL [16] database, and a clinical trials registry website.

A flow chart of the study design and a schematic illustration of two-sample MR. BMI, body mass index; FDR, false discovery rate; GSMR, generalized summary-data-based Mendelian randomization; KEGG, Kyoto encyclopedia of genes and genome; MM, multiple myeloma; MR-BMA, MR-Bayesian model averaging; SNPs, single-nucleotide polymorphisms
Plasma proteome GWAS data
The publicly available proteome GWAS dataset was derived from summary statistics of 2994 blood proteomes in 3301 healthy individuals of European ancestry from INTERVAL study [12]. For selection of individuals, Sun et al. randomly selected two non-overlapping sub-cohorts of 2731 and 831 from INTERVAL study and 3301 participants remained for analysis after genetic quality control. In the published GWAS study, relative concentrations of plasma proteins were measured by a multiplexed, aptamer-based approach (SOMAscan assay) and the reliability of protein measurements was validated using several subsequent experiments and replication of known associations with non-genetic factors. After quality control process, 3283 SOMA aptamers (SOMAmers) were left and mapped to 2994 unique plasma proteins in the final GWAS. In each sub-cohort, rank-inverse normalized protein residuals from linear regression of log-transformed protein levels on age, sex, duration between blood draw and processing, and the first three principal components were used as phenotypes and an additive genetic model was used to test genetic associations between 10.6 million imputed SNPs and levels of 2994 plasma proteins. Lastly, association results from the two sub-cohorts were combined using fixed-effects meta-analysis.
MM GWAS data
GWAS data of MM used for primary analysis was extracted from summary statistics of 181,354 individuals of European ancestry from the FinnGen consortium (R5 release, https://finngen.gitbook.io/documentation/v/r5/) [13]. The FinnGen Study is a Finnish, nationwide GWAS meta-analysis of 9 biobanks, which was non-overlapped with the samples of plasma proteome GWAS. The biobanks have been linked with longitudinal digital health record data from national health registries. The GWAS of MM in the FinnGen Study included 598 cases and 180,756 individuals without cancers as controls. The MM was defined according to the international classification of diseases-10 (ICD-10) and ICD-9 in the record of hospital discharge registry and cause of death registry. The genetic associations between SNPs and MM were adjusted for age, sex, 10 principal components, and genotyping batch.
Another summary-level data on MM for sensitivity analysis was derived from a GWAS of 405,018 individuals of European ancestry from UK Biobank (552 cases and 404,466 controls) [17, 18]. MM was defined by electronic health records-derived ICD billing codes 204.4. Scalable and accurate implementation of generalized mixed model (SAIGE) with adjustment for genetic relatedness, sex, birth year, and the first 4 principal components was performed to test the genetic associations. There was also no overlap since samples of MM and plasma proteins were obtained from different cohorts.
Selection of genetic instruments
To satisfy the assumptions of MR (Fig. 1), a set of criteria was drawn up. First, SNPs used as genetic instruments for each plasma protein were selected by a genome-wide significant level (P < 5 × 10–8). If the number of SNPs for the protein is less than 3, we adopted a suggestive genome-wide P-value threshold (P < 5 × 10–6) to identify enough SNPs (at least 3) in common between proteins and MM. Second, SNPs with linkage disequilibrium (LD) R2 > 0.001 within 10 Mb were excluded to ensure the independence of the SNPs. Third, SNPs with F-statistics < 10 were excluded to minimize instrument bias [19]. Fourth, we examined whether any of these SNPs were associated with confounders (age at recruitment, BMI, alcohol consumption, smoking, and physical activity) and outcomes at a P-value of Bonferroni level (0.05/number of SNPs) using the publicly available GWAS summary data. The associations of these SNPs with alcoholic drinks per week and smoking initiation were extracted from GWAS and sequencing consortium of alcohol and nicotine use (GSCAN) consortium [20]. Summary-level data of BMI and physical activity were derived from a meta-analysis of GWASs (~ 700,000 individuals) from genetic investigation of anthropometric traits (GIANT) consortium and UK Biobank and a GWAS (377,234 individuals) from UK Biobank, respectively [21, 22]. Given MM always occurs in old age, older populations have the competing risk of mortality and MM, which could create survival bias [23]. To alleviate the bias, we excluded the SNPs associated with survival which were proxied by age at recruitment, as described previously [24]. The associations of these SNPs with age at recruitment were derived from the MR-Base platform, including 361,194 individuals from UK Biobank [25]. Detailed information regarding these GWAS datasets is displayed in Additional file 1: Table. S1. Since no SNP were selected as genetic instruments for MM using the genome-wide significant threshold (P < 5 × 10–8), we also applied a less stringent threshold of 5 × 10–6 to obtain more SNPs for MM, as described before [24]. The remaining steps for the selection of genetic instruments for MM adhere to the aforementioned criteria.
Statistical analysis
Power analysis
A priori statistical power was calculated by a web application (https://shiny.cnsgenomics.com/mRnd/) [26]. Assuming an alpha level of 5%, and a proportion of MM cases (1%), our analyses had a power of > 80% to detect a causal effect of OR = 1.30, when the protein explained by the SNPs (R2) is at least 5%.
Two-sample MR of plasma proteins on MM and sensitivity analyses
If a genetic instrument (SNP) was missing from the MM GWAS, the SNP was replaced by a proxy SNP in high LD R2 > 0.80 using LDlink (https://ldlink.nci.nih.gov/) [27]. Inverse-variance weighted (IVW) with random-effects method was leveraged to estimate causal effects of 2994 plasma proteins on the risk of MM. Random-effects IVW method was performed to take into account potential heterogeneity [28]. To account for multiple testing across 2994 plasma proteins analyses, an FDR-corrected P-value threshold of < 0.05 was established, which we used as a heuristic to define evidence for the association between protein and the risk of MM.
We conducted a series of sensitivity analyses to assess the robustness of our findings. First, we performed additional MR methods on our significant results from the IVW analyses, including MR-Egger, weighted median, and generalized summary-data-based MR (GSMR). MR-Egger was used to assess the presence of pleiotropic effects on MM, and the intercept distinct from zero provides evidence for pleiotropic effects [29]. An I2GX was calculated to quantify the suitability of MR-Egger method and an I2GX less than 0.9 indicates the presence of considerable risk of bias in the MR-Egger analysis [30]. Weighted median provided a robust result although half of genetic instruments are invalid or pleiotropic [31]. GSMR used the heterogeneity in dependent instruments (HEIDI)-outlier method to exclude outlier or heterogenous genetic instruments and accounts for LD among SNPs using the reference dataset for LD estimation [32]. In this study, the genotype data of Europeans from phase 3 of the 1000 Genomes project was used as the reference dataset. Additionally, Steiger filtering method was performed to identify whether there are reverse causal SNPs (i.e., those explaining more variance in the outcome than the exposure) [33]. The reverse causal SNPs violate the MR assumption of “exclusion restriction”. Second, we repeated the primary analysis using summary-level data of MM from UK Biobank and combined the primary results and results of UK Biobank using fixed-effects meta-analysis. Third, we leveraged two-sample MR method to assess whether clinically diagnosed MM showed evidence of causally impacting levels of significant proteins, rather than vice versa. Finally, where applicable, we performed two-sample MR using only SNPs in the cis-region of the gene encoding the protein (defined as within ± 1Mb window from the gene) to investigate whether the significant associations identified in our study were being driven by cis-regulator SNPs. Wald ratio method was used for one SNP and IVW method was conducted for more than one SNP.
KEGG pathway analysis
KEGG is an open and widely used database integrating information on genomes, biological pathways, diseases, and drugs. KEGG pathway analysis was performed to mine pathways enriched in the list of significant proteins. To account for multiple testing, the FDR corrected P-value on the pathway less than 0.05 was significant.
MR-BMA method
MR-BMA is a novel multivariable MR paradigm that ranks risk factors by the probability of that risk factor being a causal determinant of the outcome and selects causal risk factors for outcomes in a Bayesian framework from a high-dimensional set of related and potentially highly correlated candidate risk factors [34]. To overcome the high correlation of plasma proteins and “measured pleiotropy” caused by other significant proteins, MR-BMA was leveraged to identify the dominating proteins over the others and assess which candidate causal proteins should be prioritized for MM, as described before [34,35,36]. We integrated the significant protein-associated SNPs (according to the aforementioned criteria) as instrumental variables in MR-BMA because we wanted to disentangle their causal roles. In MR-BMA, the multivariable MR analysis using weighted regression was undertaken in multiple combinations of proteins, including all single protein, all pairs of proteins, and all triples. The goodness-of-fit of the regression model was assessed by posterior probability of that combination being the true causal proteins. Then, a score was assigned to each protein that was calculated by adding the posterior probabilities of models including that protein, which is called marginal inclusion probability (MIP) and represented the probability of that protein being a causal determinant of MM. Model-averaged causal effect (MACE) for each protein was calculated and represent conservative estimates of the direct causal effect of a protein on the MM averaged across these models. Empirical P-values for MIP of each protein are calculated using a permutation method, with adjustment for multiple testing via the false-discovery rate (FDR) correction. Subsequently, Cochran’s Q-statistic was used to identify outlying SNPs and Cook’s distance was used to identify influential SNPs in the visited MR-BMA models (posterior probability > 0.02). Finally, we re-run the MR-BMA analysis omitting influential SNPs and outliers as our primary analysis, as suggested previously [34, 35].
Evaluation of druggability
We evaluated the druggability of the candidate target proteins by querying a list of druggable genes [15], ChEMBL database (release 27) [16], and https://www.ClinicalTrials.gov website; 4479 druggable genes were shown in the list and assembled in three tiers, including targets of approved drugs and drugs in clinical development (tier 1), proteins closely related to drug targets or with associated drug-like compounds (tier 2), and extracellular proteins and members of key drug-target families (tier 3) [15]. ChEMBL is a large, open-access bioactivity database (https://www.ebi.ac.uk/chembl), and we retrieved the ChEMBL to obtain information on compound name, molecule type, action type, and clinical development activity of the targeted proteins. Additionally, we searched https://www.ClinicalTrials.gov to obtain the name and clinical phase of protein targeted drug, if applicable.
All analyses were two-sided and conducted using TwoSampleMR (version 0.5.6), GSMR (version 1.0.9) and clusterProfiler (version 3.14.3) packages in R software (version 3.6.3). R-code for MR-BMA was sourced from https://github.com/verena-zuber/demo_AMD.
Results
Causal effect of plasma proteins on MM
Of the 2994 proteins being studied, the primary MR analysis showed causal relationships between 13 plasma proteins and the risk of MM (PFDR < 0.05; Fig. 2). According to the criteria of genetic instrument selection, we selected 3 to 16 SNPs to genetically proxied 13 proteins (Additional file 1: Table. S2). Of note, the suppressor of cytokine signaling 3 (SOCS3) had the most significant MR result (OR [95% CI]: 0.90 [0.86, 0.94]; PFDR < 0.001; Figs. 2 and 3). Except for SOCS3, primary MR analysis also showed genetically predicted higher levels of six proteins were associated with lower risk of MM, including protein tyrosine phosphatase non-receptor type 4 (PTPN4; 0.87 [0.82, 0.92]; PFDR < 0.001), granzyme B (GZMB; 0.90 [0.86, 0.94]; PFDR = 0.003), follistatin-related protein 1 (FSTL1; 0.68 [0.57, 0.81]; PFDR = 0.007), glypican 1 (GPC1; 0.69 [0.58,0.83]; PFDR = 0.017), Fc-gamma receptor III-B (FCGR3B; 0.75 [0.65,0.86]; PFDR = 0.024), and complement C1q subcomponent subunit C (C1QC; 0.93 [0.89, 0.96]; PFDR = 0.043; Fig. 3 and Additional file 1: Table. S3). Genetically predicted higher levels of six proteins were associated with a higher risk of MM, including nicotinamide phosphoribosyl transferase (NAMPT; 1.35 [1.18, 1.55]; PFDR = 0.005), Tyrosine kinase with immunoglobulin-like and EGF-like domains 1 (TIE1; 1.14 [1.06, 1.22]; PFDR = 0.043), carbonyl reductase 1 (CBR1; 1.46 [1.26, 1.69]; PFDR < 0.001), cAMP-specific 3',5'-cyclic phosphodiesterase 4D (PDE4D; 1.29 [1.16, 1.43]; PFDR = 0.002), platelet-activating factor acetylhydrolase IB subunit beta (PAFAH1B2; 1.43 [1.27, 1.61]; PFDR < 0.001), and neutrophil cytosol factor 2 (NCF2; 1.27 [1.12, 1.44]; PFDR = 0.040; Fig. 3 and Additional file 1: Table. S3).

Volcano plot showing effects of human plasma proteins on the risk of multiple myeloma. Data are expressed as odds ratios (OR) estimated by the inverse variance-weighted (IVW) method. The red dots represent the plasma proteins was significant positively associated with the risk of multiple myeloma (PFDR < 0.05). The blue dots represent the plasma proteins significant was inversely associated with the risk of multiple myeloma (PFDR < 0.05). The black dashed line represents the association threshold of FDR corrected P-value < 0.05. C1QC, complement C1q subcomponent subunit C; CBR1, carbonyl reductase 1; FCGR3B, Fc-gamma receptor III-B; FSTL1, follistatin-related protein 1; FDR, false discovery rate; GPC1, glypican 1; GZMB, granzyme B; NAMPT, nicotinamide phosphoribosyl transferase; NCF2, neutrophil cytosol factor 2; PAFAH1B2, platelet-activating factor acetyl hydrolase IB subunit beta; PDE4D, cAMP-specific 3',5'-cyclic phosphodiesterase 4D; PTPN4, protein tyrosine phosphatase non-receptor type 4; SOCS3, suppressor of cytokine signaling 3; TIE1, Tyrosine kinase with immunoglobulin-like and EGF-like domains 1; SNP, single nucleotide polymorphism

Mendelian randomization results of causal risk proteins on the risk of multiple myeloma. FDR P-value indicates the P-value was adjusted for multiple testing using false-discovery rate (FDR) method. C1QC, complement C1q subcomponent subunit C; CBR1, carbonyl reductase 1; FCGR3B, Fc-gamma receptor III-B; FSTL1, follistatin-related protein 1; FDR, false discovery rate; GPC1, glypican 1; GZMB, granzyme B; NAMPT, nicotinamide phosphoribosyl transferase; NCF2, neutrophil cytosol factor 2; PAFAH1B2, platelet-activating factor acetyl hydrolase IB subunit beta; PDE4D, cAMP-specific 3',5'-cyclic phosphodiesterase 4D; PTPN4, protein tyrosine phosphatase non-receptor type 4; SOCS3, suppressor of cytokine signaling 3; TIE1, Tyrosine kinase with immunoglobulin-like and EGF-like domains 1; SNP, single nucleotide polymorphism
Pathway analysis
A KEGG pathway analysis was conducted. Three of 13 causal proteins were significantly enriched in the pathway of osteoclast differentiation (hsa04380; PFDR = 0.020), including SOCS3, FCGR3B, and NCF2.
MR-BMA analysis to rank causal proteins on MM
We applied MR-BMA in 13 causal proteins to disentangle their causal roles. We integrated the genetic instruments of 13 proteins and pruned the SNPs at an LD threshold of r2 < 0.001, leaving 75 SNPs for MR-BMA analysis (Additional file 1: Table. S4). After initially performing the MR-BMA method, ten models with posterior probability larger than 0.02 were selected to calculate Cochran’s Q statistic and Cook’s distance for each SNP. An influential SNP (rs10919543) was identified (Additional file 1: Table. S5) and no outliers were identified (Additional file 1: Table. S6). After removing the influential SNP, MR-BMA analysis was repeated and the top 10 models ranked according to their posterior probability were provided in Additional file 1: Table. S7. GPC1 (MIP:0.993; MACE: − 0.349; PFDR = 0.013), NAMPT (MIP: 0.433; MACE: − 0.113; PFDR = 0.022), and NCF2 (MIP: 0.324; MACE: 0.066; PFDR = 0.022) are ranked in top-three among the 13 proteins (Table 1). It is noted that MR-BMA is designed to rank the causal importance of the 13 proteins. As such, when the PFDR is greater than 0.05, it does not imply the absence of causality between those proteins and MM risk. Instead, it indicates that the rank of the subsequent 10 proteins remains uncertain.
Druggability and clinical-phase drug for candidate protein targets
We comprehensively searched a list of druggable genes [15], the ChEMBL (release 27) database [16], and the clinical trial registry website to evaluate the druggability and drug development of the 13 candidate proteins. We categorized candidate targets into three statuses, including approved, in development (in the phase of clinical trials), and druggable (listed as druggable targets). Notably, NAMPT-targeted drug DAPORINAD is entering phase I/II trials for cutaneous T-cell Lymphoma, melanoma, and B-cell chronic lymphocytic leukemia (Table 2). Additionally, NAMPT-target drugs TEGLARINAD, ATG-019, and KPT-9274 were all in the phase I trial for non-Hodgkin’s lymphoma or acute myeloid leukemia (Table 2). FCGR3B-targeted drug IMGATUZUMAB was currently being evaluated in clinical trials for colorectal neoplasms, head and neck neoplasms, and non-small-cell lung and PED4D inhibitor was currently being evaluated in clinical trials for Alzheimer’s disease, fragile X syndrome, and depression (Table 2). Although no ongoing trials for FSTL1, CBR1, GZMB, GPC1, and C1QC, they are potential druggable targets (Table 2). NCF2, PTPN4, SOCS3, and PAFAH1B2 are not currently listed as potential drug targeted according to the druggable gene list, ChEMBL database, and clinical trial registry website. However, enforced expression of SOCS3 is proposed as a potential treatment for triple-negative breast cancer and hepatocellular carcinoma [37, 38].
Sensitivity analysis
To further increase robustness of the findings, we performed a set of sensitivity analyses. First, GSMR method showed consistent results with primary MR analysis (Additional file 1: Table. S3). Although MR-Egger and weighted methods showed no evidence of the associations between certain proteins and MM, the directions of the effects were consistent with the primary results, indicating the primary results were not substantially altered (Additional file 1: Table. S3). Moreover, I2GX for MR-Egger estimates of NAMPT, FSTL1, CBR1, PTPN4, PAFAH1B2, and GPC1 is less than 90%, indicating MR-Egger estimates for the effect of six proteins on MM to be biased. MR-Egger intercept test indicated no evidence of horizontal pleiotropy (all P-values for intercept > 0.05; Additional file 1: Table. S3). Furthermore, GSMR method detected no HEIDI-outliers, indicating no evidence of pleiotropy (Additional file 1: Table. S3). In addition, all IVs showed the correct causal direction in the Steiger filtering analysis (i.e., IVs explain more variance in the exposure than the outcome; Additional file 1: Table. S2). Second, MR-IVW analysis of 13 proteins on MM was performed using the GWAS of MM from UK Biobank. With the exception of FSTL1, the effect directions of the other 12 proteins on MM in the UK Biobank were consistent with the primary results obtained from FinnGen consortium (Additional file 1: Table S8). Similarly, fixed-effects meta-analysis showed combined effects of 12 proteins on MM was significant and consistent with the primary results (P < 0.05), except for FSTL1 (P = 0.579; Additional file 1: Table. S8). Third, we investigated the causal relationships between MM and 13 proteins. According to the criteria of genetic instrument selection, we selected 8 SNPs for MM (Additional file 1: Table. S9). IVW showed no evidence of relationships between genetic predisposition to MM and 13 proteins, and MR-Egger, weighted median, and GSMR also showed concordant results (Additional file 1: Table. S10). Finally, we performed MR analyses using only cis-SNPs from all significant proteins. Out of 13 proteins, only six proteins have at least one cis-SNP and could therefore be analyzed. The results showed CBR1, GZMB, GPC1, C1QC, and FCGR3B were causally associated with the risk of MM (Additional file 1: Table. S11). All other significant associations might be driven by trans-SNPs.
Discussion
In the present study, a pipeline composed of analytical techniques was utilized to analyze 2994 circulating proteins in relation to MM. The primary two-sample MR analysis revealed that 13 proteins were causally correlated with MM risk, with 6 showing positive associations (NAMPT, TIE1, CBR1, PDE4D, PAFAH1B2, and NCF2) and 7 showing inverse associations (FSTL1, PTPN4, SOCS3, GZMB, GPC1, C1QC, and FCGR3B). These included association with MM has been implicated elsewhere, such as NAMPT [39], PDE4D [40], PAFAH1B2 [41], SOCS3 [42], and GZMB [43]. The next step was the KEGG enrichment analysis, which showed that three of the causally associated proteins, SOCS3, FCGR3B, and NCF2, were enriched in the osteoclast differentiation pathway. Subsequently, MR-BMA analysis indicated that NAMPT, GPC1, and NCF2 ranked among the top three MM-associated proteins. At last, a list of 4 in-development protein-targeted drugs and 5 druggable proteins supported the incorporation of genomics and proteomics in the drug development programs again. Taken together, these findings exemplify the utility of genetic analysis in identifying both known and novel loci and pathways with causal implications for MM.
The etiology of MM is intricate, encompassing the dysfunction of multiple genes and signaling pathways as well as the abnormal regulation of cellular processes. Multiple lines of evidence have underscored a bidirectional prosurvival regulatory loop exists between osteoclasts (OCs) and MM cells in the bone marrow microenvironment [44]. On one hand, OCs exert immunomodulatory effects via upregulating various inhibitory checkpoint molecules and immune-suppressive cytokines, contributing to the immunosuppressive microenvironment in MM [45]. On the other hand, MM cells drive OCs formation and activation while hampering OCs generation and function. This cascade leads to bone resorption, impedes osteoblast activity, ultimately resulting in bone destruction and osteoporosis [46]. Therefore, numerous studies are exploring signaling molecules in OCs differentiation as potential therapeutic avenues for MM treatment, with denosumab serving as an illustrative example due to its effective capability in delaying and mitigating bone-related events [47, 48]. Our results consistently point to an enrichment of proteins associated with OCs differentiation, specifically SOCS3, FCGR3B, and NCF2, in MM. SOCS3, as a member of the suppressor family of cytokine signaling, acts to inhibit the activation of the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway. By negatively regulating the central JAK-STAT pathway, SOCS3 can further orchestrate bone cell growth, differentiation, and maintenance [49, 50]. Similarly, FCGR3B is the only inhibitory member of the FcγR immunomodulator family. Recent evidence suggests that cross-regulation of immunoreceptor tyrosine-based activation motif and Fc-γ receptors could promote the suppression of spleen tyrosine kinase activation, thus leading to the inhibition of osteoclast differentiation [51]. Finally, the gene encoding NCF2 also encodes the niacinamide adenine dinucleotide phosphate oxidase complex, thereby indirectly inducing osteoclast differentiation [52]. Collectively, our results lend support to the causal roles of these proteins and corroborate the significance of osteoclast differentiation in the etiology of MM.
Furthermore, given the interrelated nature of protein characteristics, a MR-BMA analysis was conducted to identify the priority causal proteins. It should be noted that the primary objective of this methodology is to detect causal risk factors among a high-dimensional set of candidates rather than to unbiasedly estimate the magnitude of their causative effects [34]. So, our results highlight the need for prioritization of NAMPT, GPC1, and NCF2 as they may be more proximal to the occurrence of MM. In detail, NAMPT serves as a rate-limiting enzyme in the salvage pathway of nicotinic acid dinucleotide synthesis [53]. In line with our findings, a recent study found that OT-82 exhibits a potent effect on MM, which can be attributed to its ability to induce cell death through the inhibition of NAMPT [54]. In addition, NAMPT is currently in clinical trials for hematological malignancies such as lymphoma, non-Hodgkin’s lymphoma, and acute myeloid leukemia. For NCF2, early studies specified that NCF2 is overexpressed in gastric cancer and promotes the progression of gastric cancer by activating the NF-kB signaling pathway [55]. Recently, there is emerging evidence that high expression of NCF2 is associated with poor prognosis in patients suffering from acute myeloid leukemia [56]. So, with its involvement in osteoclast differentiation as mentioned above, NCF2 is poised to play a vital role in the underlying mechanisms of MM. For GPC1, the available literature provides conflicting information on the role of GPC1. The previous perception of GPC1 as a biomarker for prostate cancer has been challenged by recent findings that reveal its complex, paradoxical role in the regulation of prostate cancer cell proliferation and migration [57]. Thus, despite being considered as a potential target for cancer therapy in some solid tumors, the actual application of targeting GPC1 has not been realized. Furthermore, new evidence suggests that GPC1 expression in bone marrow-derived stromal cells exerts inhibitory effects on cancer cells, making GPC1 a promising target for the development of anti-cancer therapies targeting fibroblast cells [58]. Nonetheless, the involvement of these proteins in MM may be substantial and merits further research attention.
In addition to the above proteins, the significance of other proteins in MM should not be disregarded. According to our druggable list, there are ongoing efforts to develop drugs that specifically target TIE1 and PDE4D. TIE1 is a tyrosine kinase receptor expressed by endothelial and hematopoietic cells and is functionally involved in major vascular diseases like atherosclerosis and tumor angiogenesis [59]. Despite the lack of precise information on clinical applications, numerous studies view TIE1 inhibitors as a potential therapeutic approach for antiangiogenic treatment [59, 60]. PDE4D, a primary cAMP-hydrolyzing enzyme in cells, is also a promising drug target. Studies have demonstrated therapeutic benefits of PDE4D inhibitors in the treatment of Alzheimer’s disease, Huntington’s disease, schizophrenia, and depression [61]. However, recent studies have indicated that targeting PDE4D can be used for the treatment of ER positive breast cancer [62], prostate cancer [63], or hepatocellular carcinoma [64]. Future research could explore the potential of PDE4D inhibitors for the treatment of MM. Furthermore, our findings indicate that CBR1, FSTL1, C1QC, and GZMB possess potential for pharmacological and clinical utilization and may be targeted through the use of small molecules or antibodies. The association of CBR1 with cancer has been extensively studied, especially with the recent discovery of its high expression in Philadelphia-like B-line acute lymphoblastic leukemia [65]. Likewise, it has been demonstrated that FSTL1 can suppress the proliferation of nicotine-induced lung cancer cells [66], and C1QC has proven valuable for the diagnosis of skin cutaneous melanoma with improved overall survival [67]. Furthermore, GZMB, as a crucial component in natural killer cells, has made a significant contribution to the treatment of MM [43]. Finally, the limited research on PAFAH1B2 or PTPN4 hinders the acquisition of extensive knowledge on their effects on MM. Nonetheless, PAFAH1B2 expression has been reported as a prognostic marker for MM in validation analysis [41] while PTPN4 has been found to serve as an upstream therapeutic target in the treatment of prostate cancer [68], indicating the potential for the two proteins as future research entry points.
Our study has several advantages. We innovatively explored a prospective way to intervene with circulating proteins to lower MM risk by studying the enriched pathway, the priority of therapeutic targets, and druggability of the potential causal proteins. Benefiting from the large-scale and non-overlapped GWASs data of proteome and MM, we could incorporate more functional proteins into our study and obtain more powerful MR estimates. Further, during the actual execution, a state-of-art method MR-BMA was conducted to probe the prioritized proteins and existing databases were comprehensively searched to depict the druggability profile of target proteins. We gained an advantage by applying a suggestive genome-wide P-value threshold (5 × 10–6) during the selection of genetic instruments, enabling the inclusion of a broader range of analyzable candidate proteins compared to the conventional standard of 5 × 10–8 [24, 69]. Lastly, the bidirectional MR analysis adds strength to the robustness of our findings by indicating that reverse causation is unlikely to have influenced the observed associations.
Several limitations need to be considered when interpreting our findings. First, our study was conducted using overall MM without specifying disease subtypes characterized by the immunoglobulin. Given the etiologic and prognostic heterogeneity within each subtype symptom, it is desirable to identify subtype-specific causal proteins. However, such analyses are currently constrained by the limited availability of genetic data underlying each MM subtype. Second, the causal estimation of several proteins on MM was not fully confirmed by MR-Egger method and weighted median method. However, it is important to highlight that the direction of estimates mostly aligns with the primary MR method (i.e., IVW method), and additional assessments such as Steiger filtering, GSMR HEIDI-outlier test, MR-Egger intercept test, and Cochran’s Q test did not reveal any evidence of invalid SNPs. Consequently, despite the lack of corroboration, these collective results serve to reinforce the robustness of our findings. These results still enhance the robustness of our findings. Third, MR is not perfectly analogous to a randomized controlled trial (RCT). Therefore, effects of potential causal proteins on MM derived from MR analyses may differ in magnitude from those anticipated in an RCT, and should be interpreted as life-course effects. But that does not contradict our intention to employ MR as an expedited approach to complement clinical trials and enhance their reliability. Fourth, the non-linear effects of some proteins on MM risk cannot be excluded. It is intriguing to consider the possibility that a protein may impact MM risk at extremely low or high levels, but detecting such effects in practical clinical settings can be challenging. Fifth, it is possible that the null effects of certain proteins on MM we observed may have been a consequence of inadequate statistical power due to the power of 80% to detect an OR of at least 1.30, considering R2 = 5%, while the proportion of variance explained by SNPs for certain proteins is less than 5% [12]. In addition, the only protein with a difference between the FinnGen Consortium and the UK Biobank dataset, FSTL1, should also be further investigated in a larger MM cohort. Thus, replication in larger studies of MM would be worthwhile. Finally, our analysis was confined to European ethnicity, and race issues frequently lead to the underuse of treatment and unintended interruptions in MM treatment [70]; thus, we need to be careful in generalizability of our findings to other ethnic groups.
Conclusions
To summarize, our analysis identified six risk proteins (NAMPT, TIE1, CBR1, PDE4D, PAFAH1B2, NCF2) and seven protective proteins (FSTL1, PTPN4, SOCS3, GZMB, GPC1, C1QC, FCGR3B) that are causally associated with MM risk. Additionally, we shed light on the role of these proteins in MM, prioritized the identified proteins, and evaluated their feasibility for drug development. These findings hold promise for advancing the integration of genome and proteome data to discover new drug targets for the treatment of MM. Further studies were warranted to explore the mechanisms through which the potentially causal proteins influence the risk of MM.
Availability of data and materials
GWAS data of plasma proteome are publicly available to researchers at http://www.phpc.cam.ac.uk/ceu/proteins/. GWAS data of MM of individuals from FinnGen was derived at https://www.finngen.fi/en/access_results and GWAS data of MM of individuals from UK Biobank was derived at https://pheweb.org/UKB-SAIGE/pheno/204.4. The sources for downloading GWAS data of confounders were presented in Additional file 1: Table. S1. Further information is available from the corresponding author upon request.
Abbreviations
- C1QC:
-
Complement C1q subcomponent subunit C
- CDK6:
-
Cyclin-dependent kinase 6
- CBR1:
-
Carbonyl reductase 1
- CI:
-
Confidence interval
- FCGR3B:
-
Fc-gamma receptor III-B
- FDR:
-
False discovery rate
- FSTL1:
-
Follistatin-related protein 1
- GPC1:
-
Glypican 1
- GIANT:
-
Genetic investigation of anthropometric traits
- GSCAN:
-
GWAS and sequencing consortium of alcohol
- GSMR:
-
Generalized summary-data-based Mendelian randomization
- GWASs:
-
Genome-wide association studies
- GZMB:
-
Granzyme B
- HEIDI:
-
Heterogeneity in dependent instruments
- ICD:
-
International Classification of Diseases
- IVs:
-
Instrumental variables
- IVW:
-
Inverse–variance weighted
- JAK-STAT:
-
Janus kinase-signal transducer and activator of transcription
- KEGG:
-
Kyoto encyclopedia of genes and genome
- LMM:
-
Liner mixed model
- LD:
-
Linkage disequilibrium
- MACE:
-
Model averaged causal effects
- MIP:
-
Marginal inclusion probability
- MM:
-
Multiple myeloma
- MR:
-
Mendelian randomization
- MR-BMA:
-
Mendelian randomization-Bayesian model averaging
- NAMPT:
-
Nicotinamide phosphoribosyl transferase
- NCF2:
-
Neutrophil cytosol factor 2
- OCs:
-
Osteoclasts
- OR:
-
Odds ratio
- PAFAH1B2:
-
Platelet-activating factor acetylhydrolase IB subunit beta
- PDE4D:
-
CAMP-specific 3',5'-cyclic phosphodiesterase 4D
- PTPN4:
-
Protein tyrosine phosphatase non-receptor type 4
- SERPINB9:
-
Proteinase inhibitor 9
- SNPs:
-
Single nucleotide polymorphisms
- SOCS3:
-
Suppressor of cytokine signaling 3
- TIE1:
-
Tyrosine kinase with immunoglobulin-like and EGF-like domains 1
References
Cowan AJ, Green DJ, Kwok M, Lee S, Coffey DG, Holmberg LA, Tuazon S, Gopal AK, Libby EN. Diagnosis and management of multiple myeloma: a review. JAMA. 2022;327(5):464–77.
Palumbo A, Bringhen S, Ludwig H, Dimopoulos MA, Bladé J, Mateos MV, Rosiñol L, Boccadoro M, Cavo M, Lokhorst H, et al. Personalized therapy in multiple myeloma according to patient age and vulnerability: a report of the European Myeloma Network (EMN). Blood. 2011;118(17):4519–29.
Shah N, Chari A, Scott E, Mezzi K, Usmani SZ. B-cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia. 2020;34(4):985–1005.
Cai X-W, Yu W-W, Yu W, Zhang Q, Feng W, Liu M-N, Sun M-H, Xiang J-Q, Zhang Y-W, Fu X-L. Tissue-based quantitative proteomics to screen and identify the potential biomarkers for early recurrence/metastasis of esophageal squamous cell carcinoma. Cancer Med. 2018;7(6):2504–17.
Chen Y, Quan L, Jia C, Guo Y, Wang X, Zhang Y, Jin Y, Liu A. Proteomics-based approach reveals the involvement of SERPINB9 in recurrent and relapsed multiple myeloma. J Proteome Res. 2021;20(5):2673–86.
Ng YLD, Ramberger E, Bohl SR, Dolnik A, Steinebach C, Conrad T, Müller S, Popp O, Kull M, Haji M, et al. Proteomic profiling reveals CDK6 upregulation as a targetable resistance mechanism for lenalidomide in multiple myeloma. Nat Commun. 2022;13(1):1009.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–65.
Henry A, Gordillo-Marañón M, Finan C, Schmidt AF, Ferreira JP, Karra R, Sundström J, Lind L, Ärnlöv J, Zannad F, et al. Therapeutic targets for heart failure identified using proteomics and mendelian randomization. Circulation. 2022;145(16):1205–17.
Shu X, Zhou Q, Sun X, Flesaker M, Guo X, Long J, Robson ME, Shu X-O, Zheng W, Bernstein JL. Associations between circulating proteins and risk of breast cancer by intrinsic subtypes: a Mendelian randomisation analysis. Br J Cancer. 2022;127(8):1507–14.
Yang Z, Yu R, Deng W, Wang W. Genetic evidence for the causal association between programmed death-ligand 1 and lung cancer. J Cancer Res Clin Oncol. 2021;147(11):3279–88.
Considine DPC, Jia G, Shu X, Schildkraut JM, Pharoah PDP, Zheng W, Kar SP. Genetically predicted circulating protein biomarkers and ovarian cancer risk. Gynecol Oncol. 2021;160(2):506–13.
Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558(7708):73–9.
Kurki MI, Karjalainen J, Palta P, Sipilä TP, Kristiansson K, Donner KM, Reeve MP, Laivuori H, Aavikko M, Kaunisto MA, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–18.
Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63.
Finan C, Gaulton A, Kruger FA, Lumbers RT, Shah T, Engmann J, Galver L, Kelley R, Karlsson A, Santos R, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med. 2017;9(383):eaag1166.
Mendez D, Gaulton A, Bento AP, Chambers J, De Veij M, Félix E, Magariños MP, Mosquera JF, Mutowo P, Nowotka M, et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47(D1):D930–40.
Gagliano Taliun SA, VandeHaar P, Boughton AP, Welch RP, Taliun D, Schmidt EM, Zhou W, Nielsen JB, Willer CJ, Lee S, et al. Exploring and visualizing large-scale genetic associations by using PheWeb. Nat Genet. 2020;52(6):550–2.
Zhou W, Zhao Z, Nielsen JB, Fritsche LG, LeFaive J, Gagliano Taliun SA, Bi W, Gabrielsen ME, Daly MJ, Neale BM, et al. Scalable generalized linear mixed model for region-based association tests in large biobanks and cohorts. Nat Genet. 2020;52(6):634–9.
Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–64.
Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, Datta G, Davila-Velderrain J, McGuire D, Tian C, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51(2):237–44.
Klimentidis YC, Raichlen DA, Bea J, Garcia DO, Wineinger NE, Mandarino LJ, Alexander GE, Chen Z, Going SB. Genome-wide association study of habitual physical activity in over 377,000 UK Biobank participants identifies multiple variants including CADM2 and APOE. Int J Obes (Lond). 2018;42(6):1161–76.
Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, Visscher PM, et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet. 2018;27(20):3641–9.
Smit RAJ, Trompet S, Dekkers OM, Jukema JW, le Cessie S. Survival bias in Mendelian randomization studies: a threat to causal inference. Epidemiology. 2019;30(6):813–6.
Wang Q, Shi Q, Lu J, Wang Z, Hou J. Causal relationships between inflammatory factors and multiple myeloma: a bidirectional Mendelian randomization study. Int J Cancer. 2022;151(10):1750–9.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408.
Brion M-JA, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2013;42(5):1497–501.
Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31(21):3555–7.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat Med. 2017;36(11):1783–802.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45(6):1961–74.
Bowden J, Smith GD, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14.
Zhu Z, Zheng Z, Zhang F, Wu Y, Trzaskowski M, Maier R, Robinson MR, McGrath JJ, Visscher PM, Wray NR, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun. 2018;9(1):224.
Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13(11):e1007081.
Zuber V, Colijn JM, Klaver C, Burgess S. Selecting likely causal risk factors from high-throughput experiments using multivariable Mendelian randomization. Nat Commun. 2020;11(1):29.
Zuber V, Gill D, Ala-Korpela M, Langenberg C, Butterworth A, Bottolo L, Burgess S. High-throughput multivariable Mendelian randomization analysis prioritizes apolipoprotein B as key lipid risk factor for coronary artery disease. Int J Epidemiol. 2021;50(3):893–901.
Julian TH, Cooper-Knock J, MacGregor S, Guo H, Aslam T, Sanderson E, Black GCM, Sergouniotis PI. Phenome-wide Mendelian randomisation analysis identifies causal factors for age-related macular degeneration. Elife. 2023;12:e82546.
Kim G, Ouzounova M, Quraishi AA, Davis A, Tawakkol N, Clouthier SG, Malik F, Paulson AK, D’Angelo RC, Korkaya S, et al. SOCS3-mediated regulation of inflammatory cytokines in PTEN and p53 inactivated triple negative breast cancer model. Oncogene. 2015;34(6):671–80.
Liu Z-K, Li C, Zhang R-Y, Wei D, Shang Y-K, Yong Y-L, Kong L-M, Zheng N-S, Liu K, Lu M, et al. EYA2 suppresses the progression of hepatocellular carcinoma via SOCS3-mediated blockade of JAK/STAT signaling. Mol Cancer. 2021;20(1):79.
Cea M, Cagnetta A, Fulciniti M, Tai Y-T, Hideshima T, Chauhan D, Roccaro A, Sacco A, Calimeri T, Cottini F, et al. Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition. Blood. 2012;120(17):3519–29.
Rickles RJ, Pierce LT, Giordano TP, Tam WF, McMillin DW, Delmore J, Laubach JP, Borisy AA, Richardson PG, Lee MS. Adenosine A2A receptor agonists and PDE inhibitors: a synergistic multitarget mechanism discovered through systematic combination screening in B-cell malignancies. Blood. 2010;116(4):593–602.
Kumari R, Majumder MM, Lievonen J, Silvennoinen R, Anttila P, Nupponen NN, Lehmann F, Heckman CA. Prognostic significance of esterase gene expression in multiple myeloma. Br J Cancer. 2021;124(8):1428–36.
Liang P, Cheng SH, Cheng CK, Lau KM, Lin SY, Chow EYD, Chan NPH, Ip RKL, Wong RSM, Ng MHL. Platelet factor 4 induces cell apoptosis by inhibition of STAT3 via up-regulation of SOCS3 expression in multiple myeloma. Haematologica. 2013;98(2):288–95.
Zhong Y, Meng F, Zhang W, Li B, van Hest JCM, Zhong Z. CD44-targeted vesicles encapsulating granzyme B as artificial killer cells for potent inhibition of human multiple myeloma in mice. J Control Release. 2020;320:421–30.
Raimondo S, Saieva L, Vicario E, Pucci M, Toscani D, Manno M, Raccosta S, Giuliani N, Alessandro R. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J Hematol Oncol. 2019;12(1):2.
Fu J, Li S, Ma H, Yang J, Pagnotti GM, Brown LM, Weiss SJ, Mapara MY, Lentzsch S. The checkpoint inhibitor PD-1H/VISTA controls osteoclast-mediated multiple myeloma bone disease. Nat Commun. 2023;14(1):4271.
Westhrin M, Kovcic V, Zhang Z, Moen SH, Nedal TMV, Bondt A, Holst S, Misund K, Buene G, Sundan A, et al. Monoclonal immunoglobulins promote bone loss in multiple myeloma. Blood. 2020;136(23):2656–66.
Terpos E, Zamagni E, Lentzsch S, Drake MT, García-Sanz R, Abildgaard N, Ntanasis-Stathopoulos I, Schjesvold F, de la Rubia J, Kyriakou C, et al. Treatment of multiple myeloma-related bone disease: recommendations from the Bone Working Group of the International Myeloma Working Group. Lancet Oncol. 2021;22(3):e119–30.
Hussain M, Khan F, Al Hadidi S. The use of bone-modifying agents in multiple myeloma. Blood Rev. 2023;57:100999.
Mahony R, Ahmed S, Diskin C, Stevenson NJ. SOCS3 revisited: a broad regulator of disease, now ready for therapeutic use? Cell Mol Life Sci. 2016;73(17):3323–36.
Shao F, Pang X, Baeg GH. Targeting the JAK/STAT signaling pathway for breast cancer. Curr Med Chem. 2021;28(25):5137–51.
Shanmugarajan S, Beeson CC, Reddy SV. Osteoclast inhibitory peptide-1 binding to the Fc gammaRIIB inhibits osteoclast differentiation. Endocrinology. 2010;151(9):4389–99.
Lee NK, Choi YG, Baik JY, Han SY, Jeong D-W, Bae YS, Kim N, Lee SY. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106(3):852–9.
Venkateshaiah SU, Khan S, Ling W, Bam R, Li X, van Rhee F, Usmani S, Barlogie B, Epstein J, Yaccoby S. NAMPT/PBEF1 enzymatic activity is indispensable for myeloma cell growth and osteoclast activity. Exp Hematol. 2013;41(6):547–557.e2.
Korotchkina L, Kazyulkin D, Komarov PG, Polinsky A, Andrianova EL, Joshi S, Gupta M, Vujcic S, Kononov E, Toshkov I, et al. OT-82, a novel anticancer drug candidate that targets the strong dependence of hematological malignancies on NAD biosynthesis. Leukemia. 2020;34(7):1828–39.
Zhang J-X, Chen Z-H, Chen D-L, Tian X-P, Wang C-Y, Zhou Z-W, Gao Y, Xu Y, Chen C, Zheng Z-S, et al. LINC01410-miR-532-NCF2-NF-kB feedback loop promotes gastric cancer angiogenesis and metastasis. Oncogene. 2018;37(20):2660–75.
Paolillo R, Boulanger M, Gâtel P, Gabellier L, De Toledo M, Tempé D, Hallal R, Akl D, Moreaux J, Baik H, et al. The NADPH oxidase NOX2 is a marker of adverse prognosis involved in chemoresistance of acute myeloid leukemias. Haematologica. 2022;107(11):2562–75.
Quach ND, Kaur SP, Eggert MW, Ingram L, Ghosh D, Sheth S, Nagy T, Dawson MR, Arnold RD, Cummings BS. Paradoxical role of glypican-1 in prostate cancer cell and tumor growth. Sci Rep. 2019;9(1):11478.
Kaur SP, Verma A, Lee HK, Barnett LM, Somanath PR, Cummings BS. Inhibition of glypican-1 expression induces an activated fibroblast phenotype in a human bone marrow-derived stromal cell-line. Sci Rep. 2021;11(1):9262.
La Porta S, Roth L, Singhal M, Mogler C, Spegg C, Schieb B, Qu X, Adams RH, Baldwin HS, Savant S, et al. Endothelial Tie1-mediated angiogenesis and vascular abnormalization promote tumor progression and metastasis. J Clin Invest. 2018;128(2):834–45.
D’Amico G, Korhonen EA, Anisimov A, Zarkada G, Holopainen T, Hägerling R, Kiefer F, Eklund L, Sormunen R, Elamaa H, et al. Tie1 deletion inhibits tumor growth and improves angiopoietin antagonist therapy. J Clin Invest. 2014;124(2):824–34.
Cardinale A, Fusco FR. Inhibition of phosphodiesterases as a strategy to achieve neuroprotection in Huntington’s disease. CNS Neurosci Ther. 2018;24(4):319–28.
Mishra RR, Belder N, Ansari SA, Kayhan M, Bal H, Raza U, Ersan PG, Tokat ÜM, Eyüpoğlu E, Saatci Ö, et al. Reactivation of cAMP pathway by PDE4D inhibition represents a novel druggable axis for overcoming tamoxifen resistance in ER-positive breast cancer. Clin Cancer Res. 2018;24(8):1987–2001.
Rahrmann EP, Collier LS, Knutson TP, Doyal ME, Kuslak SL, Green LE, Malinowski RL, Roethe L, Akagi K, Waknitz M, et al. Identification of PDE4D as a proliferation promoting factor in prostate cancer using a Sleeping Beauty transposon-based somatic mutagenesis screen. Cancer Res. 2009;69(10):4388–97.
Ren H, Chen Y, Ao Z, Cheng Q, Yang X, Tao H, Zhao L, Shen A, Li P, Fu Q. PDE4D binds and interacts with YAP to cooperatively promote HCC progression. Cancer Lett. 2022;541:215749.
Gupta DG, Varma N, Kumar A, Naseem S, Sachdeva MUS, Sreedharanunni S, Binota J, Bose P, Khadwal A, Malhotra P, et al. Genomic and proteomic characterization of Philadelphia-like B-lineage acute lymphoblastic leukemia: a report of Indian patients. Cancer. 2023;129(8):1217–26.
Chiou J, Su C-Y, Jan Y-H, Yang C-J, Huang M-S, Yu Y-L, Hsiao M. Decrease of FSTL1-BMP4-Smad signaling predicts poor prognosis in lung adenocarcinoma but not in squamous cell carcinoma. Sci Rep. 2017;7(1):9830.
Yang H, Che D, Gu Y, Cao D. Prognostic and immune-related value of complement C1Q (C1QA, C1QB, and C1QC) in skin cutaneous melanoma. Front Genet. 2022;13:940306.
Gan J, Liu S, Zhang Y, He L, Bai L, Liao R, Zhao J, Guo M, Jiang W, Li J, et al. MicroRNA-375 is a therapeutic target for castration-resistant prostate cancer through the PTPN4/STAT3 axis. Exp Mol Med. 2022;54(8):1290–305.
Palmos AB, Millischer V, Menon DK, Nicholson TR, Taams LS, Michael B, Sunderland G, Griffiths MJ, Hübel C, Breen G. Proteome-wide Mendelian randomization identifies causal links between blood proteins and severe COVID-19. PLoS Genet. 2022;18(3):e1010042.
Joshi H, Lin S, Fei KZ, Renteria AS, Jacobs H, Mazumdar M, Jagannath S, Bickell NA. Multiple myeloma, race, insurance and treatment. Cancer Epidemiol. 2021;73:101974.
Acknowledgements
We appreciate a lot for the funding of the Ningbo Science and Technology Bureau (No.2023J204 and No. 2022J053). We also appreciate all investigators for sharing the GWAS datasets.
Funding
This work was supported by Ningbo Science and Technology Bureau (No.2023J204 and No. 2022J053).
Author information
Authors and Affiliations
Contributions
JH and QW contributed to conceptualize the study. QW, JH, and QS contributed to design the study. QW, ZW, and JL contributed to the data curation. QW, QS, ZW, and JH contributed to the methodology development. QW, QS, and ZW contributed to write the plan of analysis. QW, QS, ZW, and JL conducted statistical analyses. QW, QS, ZW, and JL helped in the validation and performed sensitivity analyses. JH supervised the process of the project. QW, QS, JH, ZW, and JL interpreted the data and wrote the original draft of the manuscript. JH, QW, JL, ZW, and QS critically revised the paper. JH is the guarantor of this study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All studies included in cited genome-wide association studies had approved by a relevant review board. All participants had provided the inform consent. The GWAS of proteome was approved by National Research Ethics Service (11/EE/0538). The GWASs of multiple myeloma in FinnGen consortium and UK Biobank were approved by the Coordinating Ethics Committee of the Hospital District of Helsinki and Uusimaa (HUS/990/2017) and North West Multi-center Research Ethics Committee (11/NW/0382), respectively.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Detailed information for data source of confounders used in the current study. Table S2. Genetic variants for Mendelian randomization analysis of the associations between causal risk proteins and multiple myeloma. Table S3. Mendelian randomization results of causal risk proteins on multiple myeloma. Table S4. Information of genetic variants for MR-BMA analysis. Table S5. Test of influential genetic variants in MR-BMA. Table S6. Test of outlying genetic variants in MR-BMA. Table S7. Top 10 models ranked by the model posterior probability after model diagnostics. Table S8. Meta-analysis for the effect of genetically predicted causal risk proteins on the risk of MM from FinnGen and UK Biobank. Table S9. Genetic variants for Mendelian randomisation analysis of the associations between multiple myeloma and significant proteins. Table S10. Mendelian randomization results of multiple myeloma on significant proteins. Table S11. Mendelian randomization of blood proteins on multiple myeloma using only variants in the cis-region of the gene encoding the blood proteins.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Q., Shi, Q., Wang, Z. et al. Integrating plasma proteomes with genome-wide association data for causal protein identification in multiple myeloma. BMC Med 21, 377 (2023). https://doi.org/10.1186/s12916-023-03086-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-023-03086-0