
Abstract
Background
Offspring born to women with pregestational type 1 diabetes (T1DM) are exposed to an intrauterine hyperglycemic milieu and has an increased risk of metabolic disease later in life. In this present study, we hypothesize that in utero exposure to T1DM alters offspring DNA methylation and gene expression, thereby altering their risk of future disease.
Methods
Follow-up study using data from the Epigenetic, Genetic and Environmental Effects on Growth, Metabolism and Cognitive Functions in Offspring of Women with Type 1 Diabetes (EPICOM) collected between 2012 and 2013.
Setting
Exploratory sub-study using data from the nationwide EPICOM study.
Participants
Adolescent offspring born to women with T1DM (n=20) and controls (n=20) matched on age, sex, and postal code.
Main outcome measures
This study investigates DNA methylation using the 450K-Illumina Infinium assay and RNA expression (RNA sequencing) of leucocytes from peripheral blood samples.
Results
We identified 9 hypomethylated and 5 hypermethylated positions (p < 0.005, |ΔM-value| > 1) and 38 up- and 1 downregulated genes (p < 0.005, log2FC ≥ 0.3) in adolescent offspring born to women with T1DM compared to controls. None of these findings remained significant after correction for multiple testing. However, we identified differences in gene co-expression networks, which could be of biological significance, using weighted gene correlation network analysis. Interestingly, one of these modules was significantly associated with offspring born to women with T1DM.
Functional enrichment analysis, using the identified changes in methylation and gene expression as input, revealed enrichment in disease ontologies related to diabetes, carbohydrate and glucose metabolism, pathways including MAPK1/MAPK3 and MAPK family signaling, and genes related to T1DM, obesity, atherosclerosis, and vascular pathologies. Lastly, by integrating the DNA methylation and RNA expression data, we identified six genes where relevant methylation changes corresponded with RNA expression (CIITA, TPM1, PXN, ST8SIA1, LIPA, DAXX).
Conclusions
These findings suggest the possibility for intrauterine exposure to maternal T1DM to impact later in life methylation and gene expression in the offspring, a profile that may be linked to the increased risk of vascular and metabolic disease later in life.
Similar content being viewed by others


Background
The “Developmental Origins of Health And Disease” hypothesis describes how an adverse early life environment may lead to increased risk of adult disease [1]. Offspring born to women with pregestational type 1 diabetes (T1DM) are exposed to a hyperglycemic intrauterine milieu and recent studies have shown an increased risk of developing type 2 diabetes (T2DM), cardiovascular disease, and obesity [2,3,4]. The pathophysiological mechanisms behind this increased risk in the offspring are unclear, and whether a link to intrauterine T1DM exposure exists remains to be elucidated. Epigenetic modifications have been proposed as a possible mechanism by which maternal diabetes induces long-term effects [5]. However, our knowledge on epigenetic programming of offspring born to women with T1DM is still limited.
Methylation of cytosine is the best-studied epigenetic mark, with the addition of a methyl group to the C5 position of the cytosine-ring, and generally occurs on cytosines followed by a guanine (CpG). Genomic regions with an overrepresentation of CpG sites are termed CpG islands and are found at 60–70% of human gene promoters [6]. Hypermethylation of CpG islands is involved in transcriptional repression, thus, changes in DNA methylation adds level of regulation to the DNA molecule without changing the DNA sequence and via this mechanism DNA transcription can be altered [7]. Even though CpG sites are more sparse in gene bodies than in promotors, studies have described CpG hypermethylation in gene bodies to positively correlate with RNA expression [8]. Changes in DNA methylation is well known to be involved in cell differentiation and embryogenesis and is considered to be part of developmental biology [9]. Besides CpG methylation, epigenetic alterations also include histone modifications and non-coding RNA [5].
Recent studies in cord blood and peripheral blood have linked especially maternal overweight but also, to a lesser degree, maternal gestational diabetes (GDM) to offspring epigenetic alterations [10,11,12,13]. Few studies have evaluated the impact of maternal T1DM on offspring epigenetic changes, and included studies exploring an adult cohort including both offspring born to women with gestational diabetes and T1DM, using muscle, adipose tissue, or preadipocytes, described increased expression of microRNA-15a and microRNA-15b in skeletal muscle tissue and decreased leptin promotor methylation in subcutaneous adipocytes in offspring of T1DM women [12, 14,15,16,17]. Only one study by Gaultier et al. explored the more genome-wide DNA methylation profile using Illumina Human Methylation 27 Beadchip in describing an association between intrauterine exposure to maternal T1DM and kidney function in the adult offspring [18].
In this study, we hypothesized that in utero exposure to maternal T1DM alters global DNA methylation and RNA expression and thereby alters the offspring’s risk of later disease development.
Methods
The EPICOM cohort
This study is a part of the EPICOM (Environmental Versus Genetic and Epigenetic Influences on Growth, Metabolism and Cognitive Functions in Offspring of Women with Type 1 Diabetes) study (ClinicalTrials.gov registration no. NCT01559181). From 1992 to 1999, pregnancies in women with pregestational T1DM were prospectively reported to a registry managed by the Danish Diabetes Association. Information regarding diabetes status and pregnancy outcome was reported to the registry by local obstetricians at eight hospitals in Denmark, who were responsible for antenatal care and delivery for pregnant women with T1DM. Coverage of cases from the reporting centers spanned from 75 to 93%, as described by Jensen et al. [19]. The Danish Diabetes Association Register consists of 1326 records of children born to women with T1DM. Only children born after 24 completed gestational weeks were registered. As part of the EPICOM study, we invited one child per mother (the oldest) to participate in a clinical study concerning metabolic risk. Controls were recruited from the background population and were matched by sex, age, and postal number. Details from this study have previously been described by Vlachova et al. [20].
Sample inclusion
Among the oldest participants of the EPICOM cohort, we selected 20 offspring of women with pregestational T1DM (index children) and their 20 matched controls (controls) as our exploratory cohort of DNA methylation and RNA expression (Table 1). Besides fulfilling the matching criteria for inclusion in the EPICOM cohort, we made sure that our exploratory cohort did not differ in body mass index (BMI), glucose tolerance, HbA1c, cholesterol, or triglyceride levels and that the index child and their control were age matched. The index/control pairs were chosen representing the pairs with the smallest age difference, living in the same postal code, gender matched, and not suffering from any chronical illnesses. DNA methylation and RNA expression profiling were performed on leucocytes isolated from peripheral blood samples.
Clinical examination
All measurements except from height were performed three times, and the mean value was used for the analyses. We measured height to the nearest 0.1 cm and weight (in kilograms) to the nearest 0.1 kg. Waist circumference was measured using a tape measuring to the nearest 0.5 cm midway between arcus costae and crista iliaca after exhalation and hip circumference corresponds to the widest measure around the hips. Preeclampsia was defined as blood pressure > 140/90 mmHg and proteinuria 2+ on a urine protein test strip (equal to 1.0 g/l).
Oral glucose tolerance test (OGTT)
We performed a standard 2-h OGTT by using a glucose load of 1.75 g/kg body weight up to a total of 75 g. Plasma glucose was measured at 0 and 120 min and serum insulin at 0 min (fasting).
Insulin sensitivity was evaluated by HOMA-IR and calculated using the original equation [21].
Biochemical analyses
Glucose was measured in venous plasma with a hexokinase-glucose-6-phosphate dehydrogenase assay (Abbott Diagnostics, Abbott Park, Illinois, USA). Serum insulin was measured by ELISA using dual-monoclonal antibodies (ALPCO Diagnostics, Salem, New Hampshire, USA). Lipids were measured by enzymatic calorimetric analysis, end-up reaction (Abbott). Analyses of maternal HbA1c between 1993 and 1999 were measured with local assays. Correction was made to a common standard (normal range of standard assay, 0.044–0.064) by multiplying the HbA1c value with a correction factor as previously described (mean of the reference values for a standard assay divided by the mean of the reference values for the given assay) [19].
Statistics
Continuous variables with symmetric distribution are presented as means and standard deviation (SD), continuous variable with skewed distribution as medians, and interquartile range (IQR) or range. Comparison of groups was performed using Student’s T test, Wilcoxon rank-sum test, chi-square, or Fisher’s exact test. Statistical analyses were done in STATA 13.1.
DNA isolation and the 450K-Illumina Infinium assay
EDTA-treated peripheral blood samples were collected from the participants when they participated in the EPICOM study and were stored as EDTA treated at −80 °C until use. Genomic DNA was extracted from peripheral blood using QIAmp Mini Kit (Qiagen, Germany). For each sample, 1 μg of genomic DNA was bisulfite-converted using Zymo EZ DNA Methylation Kit according to the manufacturer’s recommendations. DNA methylation level was measured using the 450K-Illumina Infinium assay (Illumina, Inc.) at Aros Applied Biotechnology A/S.
(Pre-)processing of the 450K-Illumina Infinium assay data
All analyses were performed in R statistics, version 3.6.1, and R package minfi (version 1.32.0) was used for normalization, analysis, and visualization [22]. Detection p-values were calculated to identify failed positions with a p-value cut-off > 0.01. Probes were removed if they failed in more than 20% of the samples (n = 350). No samples were identified as failed, as the proportion of failed probes did not exceed 1% for any single sample. Density bean plots were used to identify outliers, and minfi’s inbuilt function was used to evaluate data with respect to extreme methylation outliers (> 3 SD away from the median). We performed background normalization and control normalization implementing the pre-processing choices of Genome Studio. Next, we applied subset-quantile-within-array-normalization correcting for technical differences between Infinium type I and II assay design allowing both within-array and between-sample normalization [23]. Cross-reactive probes (n = 29.541), probes with SNPs documented in C or G of the target (n = 18.284), and probes on sex chromosomes (n = 12,312) were excluded, leaving 415,009 probes. Methylation values were calculated as M-values (logit [beta]) (Equation (I)) [24].
Multidimensional scaling plots were evaluated to identify clusters of samples behaving differently than expected. Finally, the probes were annotated to the human genome version 19 using the enhanced Illumina annotation method developed by Price et al. [25].
Estimated differential cell counts
We adjusted for differences in cell proportions using minfi’s estimateCellCounts that implements Houseman et al.’s regression calibration algorithm [26]. This algorithm returns relative proportions of CD4+ and CD8+ T-cells, natural killer cells, monocytes, granulocytes, and B-cells in each sample.
Identifying differentially methylated positions (DMPs)
To identify positions where methylation is associated with intrauterine exposure to maternal T1DM, we fitted a linear model, which utilizes a generalized least squares model (lmFit of R package limma) allowing for missing values [27]. Significance was evaluated using F-test. The sample variances were estimated using an empirical Bayes approach with shrinkage towards the means. A Benjamini-Hochberg FDR below 0.05 was considered significant. We applied the model both without and with adjustment for estimated relative cell proportions (CD4+ and CD8+ T-cells, natural killer cells, monocytes, granulocytes, and B-cells).
Identifying differentially methylated regions (DMRs)
We used DMRcate to identify any autosomal DMRs [28]. DMRcate identifies and ranks the most differentially methylated regions across the genome based on kernel smoothing of the differential methylation signal. The model performs well on small sample sizes and builds on the well-established Limma package, allowing us to incorporate estimated cell proportions as covariates. A FDR below 0.05 with a |ΔM-value| < 1 was considered significant.
RNA sequencing (RNA-Seq) sample preparation
Blood samples were drawn using PAXgene Blood RNA Tubes and placed 2 h at room temperature, sequentially stored overnight at −20° before being stored at −80° until analysis.
RNA-Seq library construction and sequencing
Whole transcriptome, strand-specific RNA-Seq libraries were prepared from total RNA using the Ribo-Zero Globin technology (Illumina, Inc.) for depletion of rRNA and globin mRNA followed by library preparation using the ScriptSeq technology (Illumina, Inc.). Depletion and library preparation were automated on a Sciclone NGS (Caliper, Perkin Elmer) liquid handling robot. The total RNA (1.7 μg per sample) was subjected to Baseline-ZERO DNase prior to depletion. Total RNA was purified using Agencourt RNAClean XP Beads before and after DNase treatment followed by on-chip electrophoresis on a LabChip GX (Caliper, Perkin Elmer) and by UV measurements on a NanoQuant (Tecan). Cytoplasmic and mitochondrial rRNA as well as globin mRNA were removed from 400 ng DNAse-treated total RNA using the Ribo-Zero Globin Gold Kit (Human/Mouse/Rat, Illumina, Inc.) following the manufacturer’s instructions, and quality of the depleted RNA was estimated on a LabChip GX (Caliper, Perkin Elmer). Synthesis of strand-specific RNA-Seq libraries were conducted using the ScriptSeq v2 kit (Illumina, Inc.) following the recommended procedure, and the qualities of the RNA-Seq libraries were estimated by on-chip electrophoresis (HS Chip, LabChip GX, Caliper, Perkin Elmer) of a 1 μL sample. The DNA concentrations of the libraries were estimated using the KAPA Library Quantification Kit (Kapa Biosystems). The RNA-Seq libraries were multiplexed paired-end sequenced on a NextSeq 500 (75 + 6 + 75 bp) using a high-output flowcell.
RNA-Seq analysis
Paired de-multiplexed fastq files were generated using bclfastq (v.2.20 Illumina) and initial quality control was performed using FastQC. Adapter trimming was conducted using the GATK ReadAdaptorTrimmer tool followed by mapping to the human genome (hg19) in addition to transcripts from databases on non-coding RNAs (mirbase, mitranscriptome, rfam, snornabase, tjumirna, and trnascanse) using Bowtie and then further analyzed using Tophat, and Cufflinks and HTSeq-count (union method) [29, 30]. HTSeq-count (union method) was applied to produce raw counts which were then submitted for differential expression analysis in R using edgeR [31]. All non-informative features were filtered out by removing features with less than one count per million (CPM) in 39 samples removing 98,334 features, leaving 13,842 for downstream analysis. A generalized linear model was fitted, and p-values and log fold changes (Log2FC) were retrieved from the individual comparisons of index vs. controls. A Benjamini-Hochberg FDR below 0.05 was considered significant.
Weighted gene correlation network analysis (WGCNA)
Weighted gene correlation network analysis (WGCNA, v1.70.3) was applied to identify co-expressed genes from the RNA-Seq data, and how these co-expressed gene modules associated with the cohort group-parameter or clinical traits. Outlier samples were detected using hierarchical sample clustering, removing one female index sample. A signed co-expression network was constructed using a one-step approach, calculating adjacency choosing an appropriate soft thresholding power with approximate scale-free topology. Gene clustering was performed on the signed Topology Overlap Matrix by hierarchical clustering, identifying modules via the blockwiseModules function with a minModuleSize of 30 and a mergeCutHeight of 0.25. The module eigengenes were calculated via the moduleEigengenes function, and eigengene significance and corresponding p-value were obtained for each module-trait association. Intramodular connectivity and gene significance was extracted for each module of interest and hub genes were identified using the chooseTopHubInEachModule function.
Functional enrichment analysis
To investigate possible biological functions of the RNA expression changes in our cohort, we performed gene set enrichment analysis (GSEA) using Clusterprofiler [32]. The input genes were ranked based on the magnitude of changes in the index vs. controls comparison (log2FC). Disease associations were identified by disease ontology (DO) enrichment analysis, while Gene Ontology Biological Processes (GOBP) and REACTOME were used for functional and pathway enrichment. The top-enriched terms for each enrichment analysis were sorted according to p-value and presented as barplots. Furthermore, gene-concept networks that depict linkages of the genes and biological terms (DO, GOBP, and REACTOME terms) were made by evoking the cnetplot function of Clusterprofiler.
For methylation data, GOBP and REACTOME enrichment analysis were performed on all differentially methylated positions (p < 0.005) using the methylGSA package for R [33]. The probes were annotated to the human genome version 19 as previously described.
Correlation between DNA methylation and gene expression
DNA methylation has been shown to play an important role in modulating gene expression. Therefore, we correlated changes in methylation and gene expression of the gene closest to the methylation site. Shared differentially expressed genes (DEGs) and DMPs (closest gene to methylation site) between index and controls (p < 0.05) were plotted in a scatter plot based on log2FC and ΔM-value. In the second part of the analysis, we used Spearman’s rank correlation to analyze the correlation between the specific methylation level (M-value) and RNA expression level (normalized counts) for shared genes. We considered the correlations to be interesting (either negative or positive) if p < 0.1.
Analysis software
Statistics was done using R 3.6.1 (R Foundation for Statistical Computing, Vienna, Austria) with Bioconductor 3.9 [34]. DNA methylation data was analyzsed using the minfi [22], DMRcate [28], and Limma [35] package 1.32.0, and RNA-Seq data using edgeR [31]. Functional gene enrichment analysis was carried out using the R packages Clusterprofiler and MethylGSA [32, 33]. Graphics were made using basic R functions ggbio, Gviz, DEseq, DEXSeq, and ggplot2. The package knitr was used for data documentation.
Results
Through medical records and the original Diabetes Association registry, we retrieved information regarding pregnancy and birth for the majority of both index and controls (Table 1). The index and controls included in the exploratory cohort were similar on most parameters including BMI, OGTT fasting plasma glucose, OGTT 120 min plasma glucose, fasting insulin, HOMA-IR, HbA1c, systolic and diastolic blood pressure, and birth weight, although significant differences were present in some of these parameters in the larger EPICOM group [20]. Gestational age at birth was significantly different for the intrauterine T1DM exposed children compared to the controls (at that time it was common practice to induce labor ~2–3 weeks prior to the expected delivery date in women with diabetes) (gestational age 259 (range=217–278) days vs. 278 (258–301) days, p <0.001), as was delivery by caesarean section (T1DM offspring 68% vs. 28% in the control group, p=0.01). Also, maternal age at birth, maternal pre-pregnancy BMI, risk of polyhydramnios, and parity were similar between the women with T1DM and the mothers of the controls. For most of the women with T1DM, we also had knowledge of their glycemic regulation pregestationally and during the pregnancy as well as average daily amount of insulin, hypertension status, and level of albuminuria. For the RNA-Seq data, one sample from the control cohort was missing. This however did not change the overall phenotypic results.
Differentially methylated positions and regions
Performing whole-blood genome-wide DNA methylation analysis using the 450K-Illumina Infinium assay, we identified 13,867 DMPs with a p < 0.05 between index and controls. To reduce the number of false positives, a criterion of p < 0.005 and |ΔM-value| > 1 was applied, resulting in 14 DMPs (9 hypomethylated and 5 hypermethylated) (Fig. 1a and Additional file 1: Table. T1) However, after correction for multiple testing, no single DMP reached the threshold of a Benjamini-Hochberg false detection rate (FDR) (< 0.05). DMRs defined as methylation in groups of nearby positions have been proposed to be involved in transcriptional regulation. Therefore, we extended our analysis to the regional level. Applying a p < 0.005, 37 DMRs were found (Additional file 2: Table T2). However, no DMRs were identified applying a FDR < 0.05.

Differences in DNA methylation and gene expression between type 1 diabetes-exposed offspring and matched controls. a Volcano plot of −log10 p-value against delta-M-values of differentially methylated positions. Red dots denote DMPs between type 1 diabetes-exposed offspring and matched controls with a p-value < 0.005 and an absolute |ΔM-value| >1. The horizontal line represents p-value = 0.005. b Volcano plot of −log10 p-value against log fold change of differentially expressed coding genes. Red dots denote differentially expressed coding genes with a p < 0.005 and a log2FC ≥ 0.3. The horizontal line represents p = 0.005. c–e Functional enrichment analysis of the gene expression changes observed in type 1 diabetes-exposed offspring compared to matched controls. The differentially expressed coding genes were used as input for gene set enrichment analysis, to identify enriched disease phenotypes (DO) (c), biological processes (GOBP) (d), and biological pathways (REACTOME) (e). The top-enriched terms were sorted according to p-value (vertical black line denotes p = 0.05). A network plot (f, g) depicted the genes that were involved in the significant DO terms (f) and REACTOME terms (g)
RNA expression
Subsequently, we analyzed gene expression by RNA-Seq to test if any DEGs, coding or non-coding RNA, were present when comparing our two cohort groups (index vs. controls). We identified 39 coding DEGs (38 upregulated and 1 downregulated) (Table 2, Fig. 1b and Additional file 3: Fig. F1) (p < 0.005 and a log2FC ≥ 0.3). In addition, we also found 20 non-coding DEGs (7 upregulated tags and 13 downregulated tags) (p < 0.005 and a log2FC ≥ 0.3) (Additional file 4: Table T3 online). No DEGs reached an FDR < 0.05.
Functional enrichment analysis
To gain mechanistic insight into functional importance of the coding DEGs found above, we used enrichment analysis to associate these changes to disease ontology (DO), biological processes (GOBP), and biological pathways (REACTOME). We observed enrichment in disease ontologies relating to acquired metabolic disease, glucose metabolism disease, diabetes mellitus, carbohydrate metabolism disease, disease of metabolism, and clustering around genes including CXCL5, EGF, and SPARC (Fig. 1c–g). For biological processes and pathways, the analysis revealed enrichment relating to striated muscle tissue development, muscle tissue development, growth, and developmental growth and biological pathways including ECM proteoglycans, RAF/MAP kinase cascade, MAPK1/MAPK3 signaling, and MAPK family signaling (Fig. 1c–g and Additional file 5: Fig. F2 online).
Functional enrichment analysis was also carried out with identified DMPs (p < 0.005) as input. Again, enrichment in disease ontologies and biological pathways relating to carbohydrate metabolism, glucose transportation, and pathways including glucagon-like peptide-1 (GLP1) regulated insulin secretion were in the top-enriched terms (Additional file 6: Table T4 and Additional file 7: Table T5 online).
Weighted gene correlation network analysis
As we were unable to robustly identify changes at the single-gene level, we used WGCNA to detect modules of correlated genes, and to test if any of these modules could be related to our two cohort groups and the associated clinical traits. Based on co-expression similarities, the genes were split into 25 modules (Additional file 8: Fig. F3). The eigengenes of these modules, representing a summary of the expression of all genes within the module, were correlated with group status, index or control, and the clinical and paraclinical measurements. The modules that were significantly associated to our two cohort groups, sex or clinical traits, were selected (Fig. 2a). The strongest association observed was a negative correlation of the salmon module with the gender female (sex, cor = −0.97, p = 4e−23). This module consisted of 153 genes, and as expected, almost exclusively came from the sex chromosomes (21 from the X chromosome and 13 from the Y chromosome). Other female-specific traits like increased hip circumference and fat percentage were also negatively correlated with eigengene expression in this module, while male traits like increased height, waist-hip ratio, and lean body mass were positively correlated. Interestingly, we also observed that the greenyellow module, consisting of 201 genes, was positively correlated with the T1DM offspring group (cor = 0.48, p = 0.002) and unaffected by sex. We used a signed network structure to create our modules, and thus, a general upregulation of all genes within this module was expected for the index group. This assumption was supported by the presence of 22 out of the 38 upregulated DEGs in the greenyellow module. The association of the individual genes to the index group revealed that a subset of the module genes was more strongly correlated with the index group (e.g., CLU, SDPR, BEND2, EGF, STON2, TFPI, SPARC, TUBB1) (Fig. 2b). Furthermore, the greenyellow module was negatively correlated with offspring systolic blood pressure at follow-up (cor = −0.35, p = 0.03) (Fig. 2c), gestational age at birth (cor = −0.52, p = 7e−04) and the 5-min Apgar score (−0.53, p = 7e−04). Maternal BMI was not correlated to any modules; however, maternal age was positively correlated to the lightgreen module, which was also positively correlated to increased offspring weight and length. The presence of preeclampsia was not significantly associated to the greenyellow group module (T1DM offspring). However, we observed that preeclampsia was positively correlated to the magenta, darkred, and turquoise modules and negatively correlated to the tan, blue, brown, and darkgreen module. Interestingly, almost the exact same correlations were observed for fat percentage. Thus, presence of preeclampsia may predispose to an increased fat percentage later in life.

Weighted gene correlation network analysis (WGCNA) of all expressed genes. The genes were sorted into modules, with each module appointed a color, based on co-expression patters. a The eigengene of each module was correlated to the measured clinical traits. The correlation values and p-values were depicted for each correlation, which were colored based on positive (red) or negative (blue) correlation. b The module-membership vs gene significance for group (index vs control) for all genes within the greenyellow module. c The module-membership vs gene significance systolic blood pressure for all genes within the greenyellow module. d–f Functional enrichment analysis of the genes within the greenyellow module, to identify enriched biological processes (GOBP) (d) and biological pathways (REACTOME) (e) and disease phenotypes (DO) (f)
All genes from the greenyellow module were used as input for functional enrichment analysis to associate these changes to disease ontology (DO), biological processes (GOBP), and biological pathways (REACTOME) (Fig. 2d–f). Enrichment in ontologies and pathways relate to platelet degranulation/activation, blood coagulation, smooth muscle cell contraction, and fibrin clot formation. Within disease ontologies, we observed enrichment in terms relating to coagulation disease, atherosclerosis, thrombosis, myocardial infarction, and hemorrhagic disease.
Correlation between DNA methylation and gene expression
DNA methylation has been shown to play a regulatory role in mediating gene expression, and if located within a gene promotor, higher levels of methylation usually repress transcriptional expression [8]. To further explore the consequences of altered DNA methylation patterns, we therefore compared the DNA methylation data with our gene expression data (Fig. 3 and Additional file 9: Table T6). First, we visualized correlations between changes at the group level. That is, the mean change in methylation level plotted against the mean change in gene expression of the gene annotated to the methylation site. DEGs overlapping with DMPs (annotated to closest gene to the methylation site), between T1DM exposed offspring and controls (p-value < 0.05), were plotted in a scatter plot based on log2FC and ΔM-value.

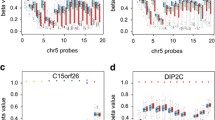
Correlated changes in DNA methylation and gene expression in type 1 diabetes-exposed offspring compared to matched controls. Scatter plot depicting mean methylation difference (delta-M) versus mean gene expression change (log2FC), of shared DEGs (p < 0.05) and DMPs (closest gene to methylation site, p < 0.05). Upper-left (red): Increased methylation corresponding to a decreased gene expression. Lower-right (blue): Decreased methylation corresponding to increased gene expression. Upper-right: Increased methylation corresponding to increased gene expression
This analysis revealed that 93 DMPs annotated to 76 DEGs showed a pattern where DNA hypomethylation was associated with an increased RNA expression (Fig. 3, lower-right (blue)). Fourteen DMPs annotated to 12 DEGs were found to show a pattern were DNA hypermethylation was associated with decreased RNA expression (Fig. 3, upper-left (red)). Two hundred one DMPs annotated to 129 genes showed a pattern where DNA hypermethylation was associated with increased RNA expression. As the second step of our correlation analysis, we performed a Spearman rank correlation for each DMP-DEG pair, using each subject’s methylation level at the involved DMPs and expression level (normalized counts) for the corresponding RNA expression data as input. Here, our analysis showed that 2 DMPs and their corresponding genes had significant inverse correlation between DNA hypomethylation and increased RNA expression (p < 0.1) (CIITA and TPM1, Table 3). DNA hypermethylation of CpG sites localized within the gene body has been described as being associated with increased RNA expression, and in our analysis, we found 4 annotated DMPs localized within the gene body and their corresponding genes displaying positive correlation between DNA methylation and RNA expression (p < 0.1) (PXN, ST8SIA1, LIPA, and DAXX, Table 3).
Discussion
This exploratory study suggests the existence of epigenetic marks (e.g., DNA methylation) that possibly influence the long-term health of offspring born to women with pregestational T1DM and elucidate intuitive pathways to be involved. Combining DNA methylation with gene expression data in offspring exposed to T1DM in utero is novel and provides new insight into the consequences of subtle epigenetic changes on gene expression and disease. Although none of the DMPs and DEGs remained significant after correction for multiple testing, our WGCNA indicated that subtle alterations in gene expression networks were present and may have a biological significance.
We identified 14 DMPs (p < 0.005 and |ΔM-value| > 1), 9 being hypomethylated and 5 hypermethylated, between T1DM exposed offspring and their matched unexposed controls. We also sought to identify any overall differences in RNA expression between index and controls and were able to identify a set of 39 predominantly upregulated genes (p < 0.005 and a log2FC ≥ 0.3). This set of upregulated genes was further validated by WGCNA. Here, 25 modules of co-expressed genes were identified, and one of these modules was positively correlated with the index group. A significant part of the genes in this module overlapped with the identified DEGs. Interestingly, this module was also negatively correlated to offspring systolic blood pressure at follow-up, gestational age at birth, and Apgar score at 5 min. As gestational age and Apgar score were generally lower for the index group, this seemed reasonable. Surprisingly, increased eigengene expression in this module, as observed in the index group, correlated with lower systolic blood pressure. From the enrichment analysis of the genes in this module, pathways and disease ontologies related to platelet activation and coagulation were among the top-enriched. Thus, vasodilation may be a consequence of the gene expression changes observed, and over time, this may lead to a malfunctioning vasculature resulting in some of the long-term pathologies observed in offspring born to women with T1DM [36].
The functional enrichment analysis of the DEGs showed clustering around the genes CXCL5, EGF, and SPARC, which are associated with diabetes, obesity, and insulin secretion and enriched in disease ontology terms of metabolic disease, diabetes, and glucose metabolism disease. These findings seem to fit the hypothesis of altered intrauterine programming of offspring born to women with T1DM [37,38,39]. The functional enrichment analysis points towards muscle tissue development and developmental growth to be associated with intrauterine exposure to T1DM and that the MAPK signaling pathway could play a role in the pathogenesis. The link between intrauterine exposure to T1DM and later in life alterations in skeletal muscle has previously been described by Houshmand-Oeregaard et al., and even though no studies have described a link between intrauterine exposure to diabetes and later in life alterations in the MAPK pathway, the central placement of MAPK in insulin signaling makes it a plausible object for pathogenetic changes [14, 40].
Our access to both DNA methylation data and gene expression data made it possible to explore the impact of epigenetic changes (e.g., DNA methylation) induced by intrauterine T1DM exposure on gene expression. We chose to perform a two-step analysis where we first explored the overall correlation between DMPs and DEGs at the group level. During step 2, we studied the individual correlation between ΔM-value at the involved DMP and log2FC for the corresponding DEG. No studies within the area of fetal programming as a consequence of maternal T1DM have to our knowledge performed a similar linkage study between DNA methylation and RNA expression data. However, in cancer studies, a similar methodology has been used [41]. Applying this method provided us with six genes where changes in DNA methylation between offspring exposed to maternal T1DM and controls correlated with relevant changes in gene expression (Table 3). PXN, which is localized at chromosome 12, has been linked to progression of T1DM and LIPA at chromosome 10 to the composition of lipoproteins [42, 43]. Mutations in TPM1 at chromosome 15 is associated with both the development of the metabolic syndrome and cardiac hypertrophy [44, 45]. CIITA is associated with MHC class II (MHCII) expression and MHCII antigen presentation in adipocytes and is reported to trigger early adipose inflammation and insulin resistance as well as inducing changes to energy expenditure in skeletal muscle [46, 47].
In our study, we chose to match our participants so that they did not overtly differ in phenotypic appearance. This approach resulted in a very homogenous group where the major difference was in T1DM exposure status. The matching was an attempt to exclude any phenotypic differences to be the cause of differences in methylation status. However, a consequence of the matching could, in theory, result in the observed changes in methylation and gene expression as being protective towards the development of later in life metabolic disease, as none of the participants in the exploratory cohort had overt metabolic disease in contrast to the participants in the full EPICOM study [20]. Also, it is well known that gestational age is associated with DNA methylation, and as gestational age differed between our two cohorts, this could have influenced our findings [48].
Epigenetic changes in relation to intrauterine exposure to either nutrition or maternal diabetes has previously been described in animal studies but the results have been difficult to apply to human studies [49, 50]. In recent years, methylation analyses of cord blood have shown promising results, but the use of cord blood methylation status as a biomarker of long-term offspring risk of disease development has yet to be fully described [11, 51].
Here we used leucocytes from peripheral blood from both T1DM exposed and non-exposed offspring. The most likely target tissue for the pathogenesis of long-term consequences of being born to a mother with T1DM may, however, be pancreatic β-cells, and muscle and fat tissue. These three tissues are not as readily available, and it is debated whether peripheral blood leucocytes reflect global epigenetic changes induced by maternal pregestational T1DM. Studies of tissue-specific differentially methylated regions have shown conservation of DNA methylation patterns across different tissues [52, 53]. However, replication of our findings in target tissues would further strengthen our hypothesis. In our study, we did not have access to specific white cell counts for each participant. Instead, we used the EstimateCellCounts algorithm from the minfi package, which enables us to minimize confounding related to cell composition, a strength compared to prior studies within the same area.
The samples used in our study were collected at a mean age of ~18 years. This provides time to develop phenotypic characteristics. However, it also provides the possibility that any epigenetic changes have occurred during the time period from birth to follow-up. In the EPICOM study, we did not have access to cord blood samples and it is therefore not possible for us to state that the epigenetic findings in our study have been present since birth. Lifestyle and diet in families where the mother has T1DM could be different compared to families without diabetes and this could affect the epigenetic findings [54, 55].
When interpreting our finding, one must consider the fact that no single DMP reached the FDR-adjusted value of 0.05. This is probably a consequence of our modest sample size in the exploratory cohort and an obvious limitation of our study. Instead, we used the same pragmatic threshold for DMPs as West et al. providing an opportunity to compare findings in related studies [56]. Our limited sample size also hindered a robust exploration of maternal preeclampsia or being born either small or large for gestational age and later in life epigenetic and transcriptomic alterations. Interestingly, the WGCNA analysis did indicate a correlation between the presence of preeclampsia and an increased fat percentage later in life. This, however, was not associated to the T1DM group.
The strength of this study is that it was performed on a group of prospectively studied offspring born to women with T1DM. For both diabetes-exposed offspring and their matched controls, we have extensive knowledge of intrauterine exposure, birth, neonatal period, and current health status. Our women with T1DM are well characterized, and no women with type 2 diabetes (T2DM) or gestational diabetes (GDM) were included in our cohort. This enables us to explore associations between both fetal and long-term consequences of being born to mother with T1DM and epigenetic and transcriptomic alterations. However, access to a control group of offspring of mothers with GDM or T2DM would have benefitted our study and rendered possible a study of which epigenetic and transcriptomic alterations is induced by maternal hyperglycemia and which is induced by e.g., maternal pre-pregnancy overweight.
Conclusions
Our data indicate intrauterine exposure to maternal T1DM to impact later in life methylation and gene expression in the offspring, a profile that may be linked to the increased risk of metabolic and vascular disease. However, comprehensive follow-up studies using a genome-wide approach and including relevant target tissue are needed.
Availability of data and materials
The dataset supporting the conclusions of this article is available in the bmcEGA MOMA repository (https://ega-archive.org/dacs/EGAC00001000145) and will be available upon request within the General Data Protection Regulation.
Abbreviations
- BMI:
-
Body mass index
- DEGs:
-
Differentially expressed genes
- DMPs:
-
Differentially methylated positions
- DMRs:
-
Differentially methylated regions
- DO:
-
Disease ontology
- EPICOM:
-
Epigenetic, Genetic and Environmental Effects on Growth, Metabolism and Cognitive Functions in Offspring of Women with Type 1 Diabetes
- FDR:
-
False detection rate
- GDM:
-
Gestational diabetes
- GLP1:
-
Glucagon-like peptide-1
- GOBP:
-
Gene Ontology Biological Processes
- GSEA:
-
Gene set enrichment analysis
- IQR:
-
Interquartile range
- OGTT:
-
Oral glucose tolerance test
- SD:
-
Standard deviation
- T1DM:
-
Type 1 diabetes
- T2DM:
-
Type 2 diabetes
- WGCNA:
-
Weighted gene correlation network analysis
References
Hochberg Z, Feil R, Constancia M, Fraga M, Junien C, Carel JC, et al. Child health, developmental plasticity, and epigenetic programming. Endocr Rev. 2011;32:159–224.
Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, et al. High prevalence of type 2 diabetes and pre-diabetes in adult offspring of women with gestational diabetes mellitus or type 1 diabetes. Diabetes Care. 2008;31:340–36.
Rijpert M, Evers I, de Vroede MAMJ, de Valk HW, Heijnen CJ, Visser GHA. Risk factors for childhood overweight in offspring of type 1 diabetic women with adequate glycemic control during pregnancy. Diabetes Care. 2009;32:2099–104.
Manderson JG, Mullan B, Patterson CC, Hadden DR, Traub AI, McCance DR. Cardiovascular and metabolic abnormalities in the offspring of diabetic pregnancy. Diabetologia. 2002;45:991–6.
Hjort L, Novakovic B, Grunnet LG, Maple-Brown L, Damm P, Desoye G, et al. Diabetes in pregnancy and epigenetic mechanisms—how the first 9 months from conception might affect the child’s epigenome and later risk of disease. Lancet Diabetes Endocrinol. 2019;7:796–806.
Illingworth RS, Bird AP. CpG islands--’a rough guide’. FEBS Lett. 2009;583:1713–20.
Szyf M, Bick J. DNA methylation: a mechanism for embedding early life experiences in the genome. Child Dev. 2013;84:49–57.
Jones PA. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92.
Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y, et al. The DNA methylation landscape of human early embryos. Nature. 2014;511:606–10.
Opsahl JO, Moen GH, Qvigstad E, Böttcher Y, Birkeland KI, Sommer C. Epigenetic signatures associated with maternal body mass index or gestational weight gain: a systematic review. J Dev Orig Health Dis. 2020. https://doi.org/10.1017/S2040174420000811.
Jönsson J, Renault KM, García-Calzón S, Perfilyev A, Estampador AC, Nørgaard K, et al. Lifestyle Intervention in Pregnant Women With Obesity Impacts Cord Blood DNA Methylation, Which Associates With Body Composition in the Offspring. Diabetes. 2021;70(4):854–66. https://doi.org/10.2337/db20-0487. Epub 2021 Jan 11. PMID: 33431374; PMCID: PMC7980200.
Kelstrup L, Hjort L, Houshmand-Oeregaard A, Clausen TD, Ninna S. Gene expression and DNA methylation of PPARGC1A in muscle and adipose tissue from adult offspring of women with diabetes in pregnancy. Diabetes. 2016;3:1–41.
Hjort L, Martino D, Grunnet LG, Naeem H, Maksimovic J. Gestational diabetes and maternal obesity are associated with epigenome-wide methylation changes in children. JCI insight. 2018;3:e122572.
Houshmand-Oeregaard A, Schrölkamp M, Kelstrup L, Hansen NS, Hjort L, Thuesen ACB, et al. Increased expression of microRNA-15a and microRNA-15b in skeletal muscle from adult offspring of women with diabetes in pregnancy. Hum Mol Genet. 2018;27:1763–71.
Hansen NS, Strasko KS, Hjort L, Kelstrup L, Houshmand-ØRegaard A, Schrölkamp M, et al. Fetal hyperglycemia changes human preadipocyte function in adult life. J Clin Endocrinol Metab. 2017;102:1141–50.
Houshmand-Oeregaard A, Hansen NS, Hjort L, Kelstrup L, Broholm C, Mathiesen ER, et al. Differential adipokine DNA methylation and gene expression in subcutaneous adipose tissue from adult offspring of women with diabetes in pregnancy. Clin Epigenetics. 2017;9:1–12.
Houshmand-Oeregaard A, Hjort L, Kelstrup L, Hansen NS, Broholm C, Gillberg L, et al. DNA methylation and gene expression of TXNIP in adult offspring of women with diabetes in pregnancy. PLoS One. 2017;12:1–18.
Gautier JF, Porcher R, Abi Khalil C, Bellili-Munoz N, Fetita LS, Travert F, et al. Kidney Dysfunction in Adult Offspring Exposed In Utero to Type 1 Diabetes Is Associated with Alterations in Genome-Wide DNA Methylation. PLoS One. 2015;10(8):e0134654. https://doi.org/10.1371/journal.pone.0134654. PMID: 26258530; PMCID: PMC4530883.
Jensen DM, Damm P, Moelsted-Pedersen L, Ovesen P, Westergaard JG, Moeller M, et al. Outcomes in type 1 diabetic pregnancies: a nationwide, population-based study. Diabetes Care. 2004;27:2819–23.
Vlachová Z, Bytoft B, Knorr S, Clausen TD, Jensen RB, Mathiesen ER, et al. Increased metabolic risk in adolescent offspring of mothers with type 1 diabetes: the EPICOM study. Diabetologia. 2015;58:1454–63.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9.
Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13:R44.
Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587.
Price ME, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, et al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics and Chromatin. 2013;6:1–15.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. https://doi.org/10.1186/1471-2105-13-86. PMID: 22568884; PMCID: PMC3532182.
Smyth GK. limma: Linear Models for Microarray Data. In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Statistics for Biology and Health. New York: Springer; 2005. https://doi.org/10.1007/0-387-29362-0_23.
Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, Lord RV, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. https://doi.org/10.1186/1756-8935-8-6. PMID: 25972926; PMCID: PMC4429355.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78.
Anders S, Pyl PT, Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. https://doi.org/10.1093/bioinformatics/btp616.
Yu G, Wang LG, Han Y, He QY. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omi A J Integr Biol. 2012;16:284–7.
Ren X, Kuan PF. methylGSA: a Bioconductor package and Shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics. 2019;35:1958–9.
Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. https://doi.org/10.1186/gb-2004-5-10-r80.
Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S, Smyth GK. Bioinformatics and computational biology solutions using R and Biocunductor; 2015. p. 397–420.
Yu Y, Arah OA, Liew Z, Cnattingius S, Olsen J, Sørensen HT, et al. Maternal diabetes during pregnancy and early onset of cardiovascular disease in offspring: population based cohort study with 40 years of follow-up. BMJ. 2019;367 Cvd:1–4.
Chavey C, Fajas L. CXCL5 drives obesity to diabetes, and further. Aging (Albany NY). 2009;1:674–7.
Maachi H, Fergusson G, Ethier M, Brill GN, Katz LS, Honig LB, et al. HB-EGF signaling is required for glucose-induced pancreatic β-cell proliferation in rats. Diabetes. 2020;69:369–80.
Kos K, Wilding JPH. SPARC: A key player in the pathologies associated with obesity and diabetes. Nat Rev Endocrinol. 2010;6:225–35.
King GL, Park K, Li Q. Selective insulin resistance and the development of cardiovascular diseases in diabetes: The 2015 Edwin Bierman Award Lecture. Diabetes. 2016;65:1462–71.
Diéz-Villanueva A, Sanz-Pamplona R, Carreras-Torres R, Moratalla-Navarro F, Henar Alonso M, Paré-Brunet L, et al. DNA methylation events in transcription factors and gene expression changes in colon cancer. Epigenomics. 2020;12:1593–610.
Prashanth G, Vastrad B, Tengli A, Vastrad C, Kotturshetti I. Identification of hub genes related to the progression of type 1 diabetes by computational analysis. BMC Endocr Disord. 2021;21:1–65.
Arnaboldi L, Ossoli A, Giorgio E, Pisciotta L, Lucchi T, Grigore L, et al. LIPA gene mutations affect the composition of lipoproteins: enrichment in ACAT-derived cholesteryl esters. Atherosclerosis. 2020;297(2019):8–15.
Savill SA, Leitch HF, Harvey JN, Thomas TH. Inflammatory adipokines decrease expression of two high molecular weight isoforms of tropomyosin similar to the change in type 2 diabetic patients. PLoS One. 2016;11:1–13.
Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, et al. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation. 2020;Dcm:387–98.
Zhang SY, Lv Y, Zhang H, Gao S, Wang T, Feng J, et al. Adrenomedullin 2 improves early obesity-induced adipose insulin resistance by inhibiting the class II MHC in adipocytes. Diabetes. 2016;65:2342–55.
Fang M, Fan Z, Tian W, Zhao Y, Li P, Xu H, et al. HDAC4 mediates IFN-γ induced disruption of energy expenditure-related gene expression by repressing SIRT1 transcription in skeletal muscle cells. Biochim Biophys Acta - Gene Regul Mech. 2016;1859:294–305.
Merid SK, Novoloaca A, Sharp GC, Küpers LK, Kho AT, Roy R, et al. Epigenome-wide meta-analysis of blood DNA methylation in newborns and children identifies numerous loci related to gestational age. Genome Med. 2020;12:1–17.
Waterland R, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–300.
Li CCY, Young PE, Maloney CA, Eaton SA, Cowley MJ, Buckland ME, et al. Maternal obesity and diabetes induces latent metabolic defects and widespread epigenetic changes in isogenic mice. Epigenetics. 2013;8:602–11.
Quilter CR, Cooper WN, Cliffe KM, Skinner BM, Prentice PM, Nelson L, et al. Impact on offspring methylation patterns of maternal gestational diabetes mellitus and intrauterine growth restraint suggest common genes and pathways linked to subsequent type 2 diabetes risk. FASEB J. 2014;28:4868–79.
Slieker RC, Bos SD, Goeman JJ, Bovée JV, Talens RP, van der Breggen R, et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 2013;6:26.
Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, et al. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet. 2009;18:4808–17.
Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–9.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13:97–109.
West NA, Kechris K, Dabelea D. Exposure to maternal diabetes in utero and DNA methylation patterns in the offspring. Immunometabolism. 2013;3:1–9.
Acknowledgements
The Danish Diabetes Association is acknowledged for originally assisting in the creation of a registry of pregnant women with T1DM. PD contributed to the establishment of the original registry. In addition, data collection in the original registry was performed by Lars Mølsted-Pedersen, Joachim Klebe, Niels Hahnemann, Margrethe Møller, Jes G. Westergaard, Hans Gjessing, Jens Kragh Mostrup, K. H. Frandsen, Edna Stage, Anders Thomsen, Thea Lousen, Kresten Rubeck Petersen, Bjarne Øvlisen, Jan Kvetny, and Hedvig Poulsen. Apart from PD, the original registry working group included Henning Beck-Nielsen, Anders Frøland, Lars Mølsted-Pedersen, Joachim Klebe, and Carl Erik Mogensen. Margaret Gellet and Pamela Celis performed bisulfite converting and DNA and RNA isolation.
Funding
European Foundation for the Study of Diabetes, Danish Ministry of Science, Innovation and Higher Education, Lundbeck Foundation, Aarhus University, Danish Diabetes Academy, Beckett Foundation, the Novo Nordisk Foundation, Danielsen Foundation, Hede Nielsen Foundation, and Central Denmark Region. None of the funding sources played any part in the study design, data collection, analysis, or interpretation of the data.
Author information
Authors and Affiliations
Contributions
SK, ZL, and BB contributed to data collection in the EPICOM cohort. SK, CH, ZL, BB, PD, KH, DMJ, KS, and CHG all contributed substantial to conception and design of the study. SK, AS, JJ, EJ, CT, SV, and CHG analyzed the data, and SK drafted the manuscript and designed the tables. All authors were involved in the interpretation of the data and critically revised the article. All authors had full access to the data in the study and take full responsibility for the integrity of the data and the accuracy of the data analysis. SK is the guarantor of the study and, as such, had full access to all data and affirms that the manuscript is honest, accurate, and transparent and no important aspects of the study have been omitted. The data has not been published previously and is not under consideration for publication elsewhere. The data has partially been presented at the 47th Annual Diabetes and Pregnancy Study Group Meeting. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All participants provided informed consent. The study was approved by The Danish Data Protection Agency and approved by The Danish National Committee on Health Research Ethics in Central Region Denmark (M-20110239). All clinical investigations were conducted according to the Declaration of Helsinki. The research has been registered at ClinicalTrials.gov (no. NCT01559181).
Consent for publication
Not applicable.
Competing interests
CHG has given talks for Novo Nordisk (NN). PD is participating in multicenter and multinational clinical studies on the use of insulin in pregnant women with pre-existing diabetes in collaboration with NN; no personal honorarium is involved. None of the study funders had any role in study design, collection, analysis, interpretation, preparation of the manuscript, or in the decision to submit the article for publication.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table T1.
List of differentially methylated positions between type 1 diabetes exposed offspring and controls. Displayed are positions with a crude p < 0.005 and a M-value ≥ 1. The mean M values and mean B values displayed are corrected for cell count and age.
Additional file 2: Table T2.
List of differentially methylated regions between type 1 diabetes exposed offspring and controls. Displayed are regions with a crude p < 0.005.
Additional file 3: Figure F1.
Boxplots illustrating differences in CPM (Count Per Million) at each differentially expressed gene with a crude p < 0.005 between type 1 diabetes exposed offspring and controls.
Additional file 4: Table T3.
List of differentially expressed non-coding RNA between type 1 diabetes exposed offspring and controls. Displayed are genes with a crude p < 0.005 and a log2FC ≥ 0.3.
Additional file 5: Figure F2.
Network plots depicting the genes that are involved in the significant GOBP terms from the Functional Enrichment Analysis of the gene expression changes observed in type 1 diabetes exposed offspring compared to matched controls.
Additional file 6: Table T4.
Gene Set Enrichment Analysis of differentially methylated positions. List of biological processes (Gen Ontology, GOBP) associated with the differentially methylated positions (p < 0.01).
Additional file 7: Table T5.
Gene Set Enrichment Analysis of differentially methylated positions. List of biological pathways (REACTOME) associated with the differentially methylated positions (p < 0.01).
Additional file 8: Figure F3.
Weighted gene correlation network analysis (WGCNA) of all expressed genes. The genes were sorted into modules, with each module appointed a color, based on co-expression patters. The eigengene of each module was correlated to the measured clinical traits. The correlation values and p-values were depicted for each correlation, which were colored based on positive (red) or negative (blue) correlation.
Additional file 9: Table T6.
Correlation between DNA methylation and RNA expression between type 1 diabetes-exposed offspring and controls. Displayed are all correlations reaching a p < 0.05 and marked with grey are correlations reaching a p < 0.10 using Spearman Ranks correlation between level of methylation and gene expression (normalized counts). P-value (correlation analysis) and FDR (correlation analysis) is the p-value/FDR for the DEG used as input for the correlation analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Knorr, S., Skakkebæk, A., Just, J. et al. Epigenetic and transcriptomic alterations in offspring born to women with type 1 diabetes (the EPICOM study). BMC Med 20, 338 (2022). https://doi.org/10.1186/s12916-022-02514-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-022-02514-x