
Abstract
Poly(ADP-ribose) polymerases (PARP) are enzymes involved in DNA-damage repair. Inhibition of PARPs is a promising strategy for targeting cancers with defective DNA-damage repair, including BRCA1 and BRCA2 mutation-associated breast and ovarian cancers. Several PARP inhibitors are currently in trials in the adjuvant, neoadjuvant, and metastatic settings for the treatment of ovarian, BRCA-mutated breast, and other cancers. We herein review the development of PARP inhibitors and the basis for the excitement surrounding these agents, their use as single agents and in combinations, as well as their toxicities, mechanisms of acquired resistance, and companion diagnostics.
Similar content being viewed by others

Background
Modern strategies for the development of novel cancer therapies include agents targeting specific molecular defects that characterize certain cancer cells in order to increase treatment efficacy and reduce toxicities. In breast cancer, targeted therapies have long been effective, as agents targeting hormone receptors in tumors expressing them and as antibodies or tyrosine kinase inhibitors targeting overexpressed or amplified HER2 molecules. Breast tumors expressing none of these are called triple-negative breast cancers (TNBC), which comprise about 15 % of breast cancers overall, about 70 % of breast cancers in individuals harboring a germline BRCA1 mutation, and 20 % in BRCA2 mutation carriers [1–4]. The discovery of the family of nuclear enzymes poly(ADP-ribose) polymerases (PARPs) and their role in DNA-damage repair pathways opened the possibility of developing a new class of antineoplastic drugs with the ability to interfere with the DNA damage repair systems of cancer cells – PARP inhibitors (PARPi). One characteristic of BRCA-mutated cancers is defective function of one of the major DNA damage repair pathways, the homologous recombination (HR) pathway. The original concept of the activity of PARP inhibitors was that they acted through synthetic lethality by targeting the base excision repair pathway (BER); in tumor cells with defects in a different DNA repair mechanism, disruption of both pathways led to cell death. The preferential sensitivity of BRCA-associated breast and ovarian cancers was therefore predicted as the tumor cells are characterized by defective homologous recombination repair. Subsequently, PARPi have shown significant activity in BRCA-associated breast, ovarian, and other cancers [5, 6]. However, the activity in sporadic ovarian cancers suggests a more complex mechanism of action described below [7].
PARPs and DNA damage repair
PARPs are a family of enzymes involved in various activities in response to DNA damage [8]. Eighteen components of this family have been discovered; PARP-1 to -3 are so far the only members defined as DNA damage-dependent PARPs [9].
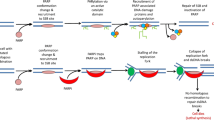
PARP activation, largely driven by DNA damage (other mechanisms may occur, as reviewed by Bürkle et al. [10]), determines post-transcriptional modification of nuclear proteins such as histones [9]. PARP-1 activation is one of the earliest responses to DNA damage in human cells [11, 12]. The ADP-ribosylation of histones and the recruitment of chromatin remodeling enzymes create a relaxed chromatin state that is appropriate for DNA repairing activities (Fig. 1a). The ADP-ribose polymer synthesized by PARP acts as a “flag” that drives the assembly of DNA-repair complex at sites of DNA damage, mainly promoting BER and single strand break repair (SSBR) pathways [9], while involvement of PARPs in double strand break repair (DSB – an error-free DNA repair system) is likely limited [13].

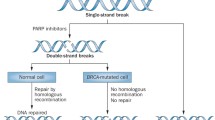
Current model for PARP role in DNA damage repair and PARP inhibition – BRCA mutation synthetic lethality. a When single-strand break (SSB) is detected, PARP recruitment and activation leads to SSB repair through poly(ADP-ribosyl)ation (PARylation) of histones and chromatin remodeling enzymes, auto-PARylation of PARP, and recruitment of PARP-dependent DNA repair proteins. Repaired DNA can undergo replication determining cell survival. b In the presence of PARP inhibitors, PARPs recruited to DNA-damage sites are no longer able to activate PARP-dependent repair systems and to dissociate from DNA (due to catalytic activity inhibition and/or direct trapping), determining replication fork (RF) stalling during DNA replication. Stalled RF eventually collapse creating double strand break (DSB). DSB can be repaired by homologous recombination (HR) and replication may restart, leading to cell survival. In BRCA-deficient cells, HR is impaired, thus DSB cannot be efficiently repaired; in this context, DSB accumulate determining cell death
Rationale for development of PARPi in breast cancer
Since cancer is a disease in which DNA replication is critical, replication errors are prominent, and deficiencies in DNA-repair pathways are common [14], the involvement of PARPs in DNA-repair pathways stimulated the development of agents capable of targeting PARP activity.
To maintain DNA integrity, HR-deficient cells rely on secondary DNA repair pathways, such as BER, SSBR, and non-homologous end joining. When PARP-dependent activation of BER/SSBR and non-homologous end joining is defective, cells rely on the HR pathway to restore DNA integrity. BRCA1 and BRCA2 proteins are key actors of the HR apparatus and deficiency of either (secondary to germline mutation in one copy and loss of heterozygosity inactivating or removing the other copy) results in inefficient activation of HR (Fig. 1b). Using BRCA1- and BRCA2-deficient cell lines and mouse xenografts, Bryant et al. [15] and Farmer et al. [16] demonstrated marked in vitro and in vivo cytotoxicity of PARPi monotherapy in tumor cells with intrinsic HR deficiency, with close to no effect on BRCA-proficient cells.
The model explaining this “synthetic lethality” effect of PARP inhibition in HR-deficient cells is comprehensively reviewed by Helleday [17]. Briefly, suppression of PARP catalytic activity blocks the formation of ADP-ribose polymers at site of SSB, hence PARP-dependent DNA-damage repair complexes cannot be efficiently recruited. Unrepaired SSB eventually lead to stalling of replication forks [17]. Stalled replication forks collapse into double strand breaks that are highly cytotoxic lesions if not repaired by HR [17], the repair mechanism inefficiently activated in BRCA-mutated cancers. Recent data suggest that another mechanism of action of PARPi, so-called “PARP trapping”, is more important in determining PARPi cytotoxicity. Murai et al. [18] showed that PARPi prevent dissociation of recruited PARPs from DNA-damage sites: these stabilized PARP/DNA complexes determine stalling of the replication fork during DNA replication, with subsequent formation of double strand breaks.
The observation that BRCA-mutated breast cancers show an impairment in HR pathways [19], and that some sporadic TNBC are phenocopies of BRCA1-mutated cancers (i.e. they display a phenotype resembling BRCA1-mutated cancers without harboring a BRCA1 mutation, a feature also defined as “BRCAness”, see below) [20, 21], led to exploration of the application of PARP inhibition to the treatment of breast cancer (BRCA-associated and TNBC).
Clinical application in breast cancer
Clinical development of PARPi started in 2003 and focused on two strategies: utilizing PARPi in combination with other drugs in a range of solid malignancies or using PARPi monotherapy in specific cancer types with features (like impairment of DNA-damage repair systems alternative to the PARP-dependent ones) that would be predicted to be highly sensitive to PARP inhibition. Testing of PARPi in combination with cytotoxic drugs showed the feasibility of this approach with overall good tolerability, but there was little evidence of activity in unselected patients [22]. In contrast, promising data emerged in the treatment of patients with breast and ovarian cancers [23, 24], the two malignancies most frequently associated with BRCA mutations.
Clinical testing of PARPi was initially slowed by negative results from a phase 3 trial of iniparib, a compound inaccurately classified as a PARPi [25]. Subsequently, it was shown that iniparib and its metabolites do not inhibit PARP in intact cells [26], and clinical development of genuine PARPi gained new vigor. Currently, five compounds with the ability to inhibit the activity of various PARPs are being investigated in clinical trials (Table 1). Below, we will present the most important findings from phase 1 and 2 clinical trials evaluating the efficacy of PARPi in the treatment of breast cancer. These data are also summarized in Tables 2 and 3.
Clinical trials in advanced disease
PARPi as single agent therapy
Following the demonstration by Bryant and Farmer [15, 16] of the cytotoxic effect of PARP inhibition in HR-deficient cells, there was interest in studying the activity of PARPi as monotherapy in solid tumors. In earlier studies, the population enrolled in these trials was not restricted to patients with known BRCA mutations, but encompassed also those whose cancer displayed a phenotype similar to BRCA-mutated cancers. Clinically, this group included triple-negative breast cancers and high-grade serous or poorly differentiated ovarian cancer. The term “BRCAness” was introduced to identify sporadic tumors that shared common phenotypic features with familial BRCA tumors [20]. Attempts to identify cancers with BRCAness included evaluation of epigenetic silencing of BRCA genes [27], measurement of levels of proteins involved in HR [28], and of foci of DNA-repair proteins like gammaH2AX [5, 29]. However, after preliminary data showing minimal efficacy of PARPi in sporadic breast cancers, some of the trials were amended to enrich the study cohorts for BRCA-associated tumors [5, 30].
Initial phase 1 testing of olaparib as monotherapy in BRCA-associated breast and ovarian cancers showed encouraging results: 47 % of patients with BRCA-associated breast, ovarian, or prostate cancers treated with olaparib achieved a partial response, and 63 % of them derived clinical benefit (tumor marker decrease or radiologic response or stable disease for 4 or more months) [5]. A phase 1 study of niraparib in patients with advanced solid tumors enriched for BRCA-associated cancers reported an overall response rate of 40 % (8 of 20) in patients with BRCA-associated ovarian cancer and 50 % (2 of 4) in patients with BRCA-associated breast cancer [31]. Talazoparib monotherapy has shown antitumor activity in patients with BRCA mutations, with an objective response rate of 65 % in ovarian and peritoneal tumors and 33 % (2 of 6 patients) in breast cancers [32]. Data presented at ASCO 2014 on single agent rucaparib showed efficacy in BRCA-associated ovarian, breast, and pancreatic cancers [33].
These data from phase 1 trials guided the development of phase 2 studies in the population of patients with BRCA-associated cancers or with cancer usually associated with “BRCAness”, namely triple-negative breast cancer and high-grade serous ovarian cancer (HGSOC).
Tutt et al. [34] reported efficacy of olaparib as monotherapy in 54 patients with advanced breast cancer and germline BRCA1/2 mutations. At the maximum tolerated olaparib dose of 400 mg bid, a 41 % objective response rate was observed, with responses in both TNBC and hormone receptor-positive HER2-negative patients. Toxicities were generally manageable, with treatment-related adverse events reported in 81 % of patients, but grade 3 or 4 events occurred in only 24 % of patients. Efficacy data from this study compare favorably with response rates in studies of single agent cytotoxics (capecitabine [35], vinorelbine [36], eribulin [37], ixabepilone [38–40]) and of new anti-HER2 targeted therapies (pertuzumab [41] and T-DM1 [42]) in advanced breast cancer treatment. Similar results from a parallel phase 2 study of olaparib monotherapy in recurrent ovarian, fallopian tube, or peritoneal cancers were reported by Audeh et al. In germline BRCA1/2 mutation-positive patients, the objective response rate was 33 % [43]. It should be noted that in both trials, for the first time, a documented germline BRCA mutation was an enrollment criterion [34, 43].
Gelmon et al. [7] assessed safety and efficacy of olaparib as a single agent in HGSOC and TNBC in an important trial that also demonstrated the feasibility of pre- and post-treatment biopsies. While sustained responses were documented in HGSOC, no confirmed objective response was shown in TNBC, regardless of BRCA-mutation status, although 50 % of BRCA-mutation carriers had a greater than 30 % reduction in the target lesion. The authors speculated that the lack of evidence of efficacy in BRCA-associated breast cancers in this trial could be due to chance because of small sample size or population characteristics (heavily pretreated patients) [7].
Kaufman et al. [6] reported data of a phase 2 study (NCT01078662) of olaparib monotherapy in 298 patients with diverse recurrent cancers (mostly ovarian, breast, pancreatic, and prostate) and confirmed BRCA1/2 mutations (a study design called “basket trial”). Breast cancer tumor response rate was 12.9 % in 62 patients, and 47 % of patients had disease stabilization for ≥8 weeks. The lower objective response rate in this study compared with previous studies [5, 34] could be due to the fact that the study population was more heavily pretreated than in other trials (mean of 4.6 prior chemotherapy regimens in the metastatic setting vs. 3 in Tutt et al. [6]).
When tested in ovarian cancer, PARPi showed efficacy regardless of BRCA status. In the previously cited Gelmon et al. [7] study, olaparib induced sustained responses in non-BRCA mutant HGSOC. Responses to olaparib were also observed in ovarian cancer patients with wild type or unknown BRCA status in a study of maintenance therapy after platinum-based chemotherapy [44] and in a study of olaparib plus cediranib [45]. Molecular studies suggested that up to 20 % of HGSOC lose BRCA1 or BRCA2 function through epigenetic events [46], thus expressing an HR-deficient phenotype with sensitivity to PARPi even in the absence of somatic/germline BRCA mutation.
Studies of veliparib monotherapy in metastatic breast cancer are currently in progress [47, 48]; data on veliparib efficacy as single agent in gynecological cancers are already available. Coleman et al. [49] reported data from a multicenter phase 2 study in BRCA-associated persistent or recurrent ovarian, fallopian tube, or primary peritoneal cancer: objective response rate to single agent veliparib was 26 % and progression-free survival at 6 months was 54 %, without significant difference between platinum-sensitive or platinum-resistant tumors.
PARPi in combination therapy
PARPi have been tested in the treatment of metastatic breast cancer in combination with multiple compounds in phase 1 and 2 studies [22]. Preclinical data showed that veliparib exerts remarkable synergic activity with other cytotoxic compounds [50]: in particular, veliparib enhanced temozolomide’s cytotoxic effect even in tumor types not typically responsive to temozolomide [51] with a good safety profile. Veliparib has been further clinically explored mainly as a part of combination therapy. In a phase 2 trial in BRCA-associated breast cancers, treatment with veliparib and temozolomide offered a response rate of 22 % and a clinical benefit rate of 50 % (defined as complete response, partial response, or stable disease) [52]. Efficacy was successively confirmed in a larger expansion cohort with patients previously treated with platinum compounds or PARPi [30].
Other combinations between PARPi and chemotherapy drugs have been proven effective in early clinical trials: the best results in terms of efficacy emerged from combination with cisplatin [23, 53] and carboplatin [54, 55], as well as topotecan [56], with response rates in BRCA-related breast cancers up to 73 % [23, 53]. Contrasting data about the safety of the combination therapy approach emerged from these studies. The combination topotecan-olaparib showed dose-limiting hematological adverse events at sub-therapeutic doses of olaparib [57]; in contrast, veliparib combinations have been better tolerated overall.
It is still not clear which is the best chemotherapeutic companion for a PARPi, and studies show that different PARPi may combine more or less efficiently with cytotoxic drugs with different mechanisms of action [58, 59]. The differences in synergistic effect between cytotoxic drugs and PARPi may be explained by PARPi mechanisms of action. Indeed, some PARPi exert their cytotoxic effect mainly suppressing PARPs’ catalytic activity (veliparib), while others more by trapping PARPs to DNA (olaparib, talazoparib, rucaparib, niraparib) [18]. It has been proposed that PARP trapping is synergistic with alkylating agents, while PARP catalytic inhibition synergizes with topoisomerase I inhibitors [58]. In preclinical models, proliferation of breast cancer cells is more potently suppressed when both mechanisms of PARP inhibition are present [18]. On the other hand, the higher toxicity of this class of PARPi may render them more toxic in combination with cytotoxic therapies.
Ongoing studies in the metastatic setting
Ongoing randomized phase 3 studies of PARPi in metastatic breast cancer are limited to patients with documented BRCA1/2 mutations (Table 4). Three parallel study designs will test oral PARPi monotherapy vs. physician’s choice single agent chemotherapy in breast cancer patients with PARPi-naive metastatic disease with germline BRCA1/2 mutations: BRAVO (niraparib, NCT01905592 [60]), EMBRACA (talazoparib, NCT01945775 [61]), and OlympiAD (olaparib, NCT02000622 [62]). Finally, study NCT02163694 [63] will test the efficacy of veliparib versus placebo in combination with carboplatin and paclitaxel in HER2-negative metastatic or locally advanced, unresectable, BRCA-associated breast cancer.
Results from these studies are eagerly awaited and, if positive, will form the basis of applications for Food and Drugs Administration approval of PARPi for the treatment of metastatic BRCA-associated breast cancer. Approval will require an acceptable safety profile (see below) in a well-characterized and defined target population that currently lacks a specific targeted therapy. In 2014, both the European Medicines Agency and the Food and Drugs Administration [64, 65] granted accelerated approval to olaparib in high-grade serous ovarian, fallopian tube, and primary peritoneal cancer based on the results of two phase 2 trials [44, 66].
Going beyond the metastatic setting
Conventionally, new antineoplastic drugs are tested as adjuvant treatments for breast cancer after solid data from phase 3 trials in the metastatic setting become available. In the case of PARPi, the remarkable activity of olaparib and veliparib in multiple phase 2 trials and their manageable toxicity profiles have led to trials of several PARPi in the adjuvant and neoadjuvant settings (Table 4). The adjuvant trial OlympiA is evaluating 1 year of the PARPi olaparib [67]. Data for the acceptability of olaparib given for extended periods of time come from a phase 2 study of single agent olaparib as maintenance therapy in platinum-sensitive ovarian cancer – median duration of treatment 206 days – but some patients stayed on the medication for years [44].
The OlympiA trial (NCT02032823 [68]) will assess the efficacy and safety of up to 12 months of olaparib versus placebo as adjuvant treatment in patients with germline BRCA1/2 mutations and high-risk hormone receptor-negative HER2-negative primary breast cancer who have completed definitive local treatment and neoadjuvant or adjuvant chemotherapy. Eligibility criteria have recently been expanded to allow for enrollment of high-risk hormone receptor-positive patients. Randomization will be stratified by prior neoadjuvant versus adjuvant chemotherapy, and according to the use of prior platinum-based chemotherapy for breast cancer. The post-neoadjuvant treatment group will comprise patients in whom pathologic complete response was not achieved following at least six cycles of neo-adjuvant chemotherapy. The primary end point will be interval disease-free survival, and the secondary end points will be overall survival, distant disease-free survival, and the development of new primary invasive cancers.
Rucaparib is being tested in a phase 2 trial as adjuvant treatment for TNBC or BRCA-mutated HER2-negative breast cancers with residual disease after preoperative chemotherapy (NCT01074970) [69]; preliminary data presented at ASCO 2014 showed no improvement in 1-year disease-free survival with rucaparib plus cisplatin versus cisplatin alone in the intent-to-treat population; rucaparib did not add substantial toxicity to the cisplatin treatment [70].
The I-SPY2 study assesses sequential novel agents in the neoadjuvant treatment of breast cancer. In the I-SPY-2 trial assessing the addition of veliparib and carboplatin to standard neoadjuvant therapy in TNBC, an estimated 52 % pathologic complete response rate was observed in the experimental arm versus 26 % in the standard treatment arm [71]. In the cooperative group neoadjuvant trials GeparSixto and Alliance 40603, the addition of carboplatin to standard neoadjuvant chemotherapy increased pathological complete response rates in TNBC from 42.7 % to 53.2 % and from 41 % to 54 %, respectively [72, 73]. In GeparSixto, this effect is most evident in patients with germline BRCA1/2 or RAD51 mutations (the pathological complete response rate with carboplatin was 66.7 % versus 43.5 % without carboplatin). Participants are currently being accrued to a randomized three arms phase 3 trial that will test the efficacy of the addition of carboplatin plus veliparib, carboplatin alone, or placebo to standard neoadjuvant chemotherapy (Brightness Study – NCT02032277) [74].
A pilot phase 2 study of neoadjuvant talazoparib monotherapy in BRCA-associated breast cancer is ongoing at MD Anderson Cancer Center in Texas [75].
An interesting possibility for the future development of PARP inhibition in BRCA-related breast cancers has been raised by To et al. [76], who demonstrated a chemopreventive effect of veliparib and olaparib in delaying mammary tumor development in BRCA1-deficient mice. Data in this field are still too limited to speculate whether these findings could be translated to humans, but the concept of a chemopreventive drug active in a population at high risk of developing breast cancer is nonetheless intriguing. The future of PARPi in prevention is not clear at this time because of some chemotherapy-like toxic effects on bone marrow, in particular [22].
Safety of PARPi
Toxicities of PARPi monotherapy appear to be similar to cytotoxic chemotherapeutic agents. Data from prominent phase 1 and 2 studies are summarized in Table 5: the most frequently reported adverse events in published studies are grade 1–2 nausea, vomiting, diarrhea, fatigue, headache, and anemia. The most common grade 3–4 toxicities were nausea, vomiting, and hematological toxicity, with anemia, lymphopenia, and thrombocytopenia being the most common dose-limiting toxicities in dose-finding studies [5, 34].
Conversely, dose-limiting toxicities observed in trials of PARPi in combination with cytotoxic agents include primarily hematologic toxicities [77, 78]. These potentiated toxicities might restrict the future development of some olaparib-cytotoxic combinations [79]. However, using an intermittent schedule of PARPi administration instead of continuous dosing has proved effective in overcoming this limitation [23].
One major concern with drugs that inhibit DNA damage repair mechanisms is the risk of development of new primary malignancies. A small number of cases of myelodysplastic syndrome and acute myeloid leukemia have been described in PARPi studies, with an incidence of <1 % [22]. It is noteworthy that most patients had already been treated with DNA-damaging classic chemotherapeutic drugs, which per se, represents a risk factor for development of new malignancies. Nonetheless, the increased concentration of gammaH2AX (a marker of DNA damage [80–82]) in tissues of patients treated with PARPi implies an accumulation of DSB in normal tissues and thus could lead to an increased risk of cancer secondary to DNA damage [22], warranting a high level of attention when developing PARPi therapy, especially in the adjuvant setting.
Resistance to PARP inhibition
As with most targeted therapies, cancers develop resistance to PARPi. All tumors that responded initially to treatment with PARPi have ultimately progressed. So far, three mechanisms of resistance to PARPi have been demonstrated, while two others have been hypothesized [83–85] (Table 6). The first of the three established mechanisms is the development of secondary mutations that restore BRCA functionality. Preclinical and clinical evidence indicates that genomic instability promoted by PARPi in HR-deficient cells may result in secondary mutations in the mutated BRCA1 or BRCA2 gene with restoration of functional protein expression and induction of PARPi resistance [86–88]. The second mechanism involves increased drug efflux with consequent reduction of intracellular PARPi concentrations. PARP1 knock-out cells show dramatic overexpression of P-glycoprotein [89]; PARP inhibition induces up-regulation of P-glycoprotein expression in an in vivo mammary tumor model [59]. The third mechanism of PARPi resistance is based on loss of p53 binding protein 1 (53BP1). In vitro and in vivo experiments showed that mutations causing loss of 53BP1 are able to restore the HR in BRCA1/2 mutated cells, at least partially [90–92]: this “DNA damage repair rewiring” ultimately leads to reduced sensitivity to PARPi [93].
Another hypothesized, but still unconfirmed, mechanism of resistance to PARPi at the time of this submission is the presence of BRCA1/2 forms with low level of expression, but that can be enhanced in the presence of opportune stimuli (such as increase in DSB due to PARP inhibition) – so called hypomorphic BRCA1/2 [84]. Furthermore, the hypomorphs may lead to the reduced formation of PARP-DNA complexes because of decreased PARP expression (for example, by epigenetic silencing of the gene or increased turnover of the protein) [85].
Some of the aforementioned mechanisms of resistance are shared between PARPi and platinum compounds [94], but the degree of overlap is not clear. For example, Audeh et al. [43] reported response to olaparib in ovarian cancer regardless of previous platinum sensitivity or resistance, while in the basket trial by Kaufman et al. [6], response rate to olaparib across breast cancer patients showed a trend in favor of patients without prior platinum exposure. However, platinum sensitivity can persist after resistance to PARPi develops [95]. It is notable that most of the ongoing studies of PARPi in advanced breast cancer exclude patients who had been previously treated with platinum compounds [61, 63, 96, 97] or who progressed on platinum-based chemotherapy regimens [60, 62].
The existence of resistance mechanisms can limit the clinical utility of PARPi; strategies to overcome acquired resistance are needed. For example, it has been shown that drugs able to block efflux pumps may revert their PARPi resistance [59]. Further, when PARPi resistance is due to restoration of BRCA-proficiency, induction of a BRCAness phenotype via CDK1 inhibition may render the tumor cells again susceptible to PARPi [98].
Predicting response to PARPi
No established biomarker of response to PARPi is currently available. A candidate biomarker is the homologous recombination deficiency (HRD) score, which combines three different DNA-based metrics of genomic instability that are highly associated with BRCA1/2 mutational status or predictive of sensitivity to platinum chemotherapy [99]; Richardson et al. [100] demonstrated that the HRD score is able to identify patients with breast tumors with underlying HR deficiency (including BRCA1/2 non-mutated tumors) that benefit from neoadjuvant platinum therapy. In PrECOG 0105, high HRD scores identified patients with a higher likelihood of achieving pathological complete response to platinum-based neoadjuvant chemotherapy [101]. However, data from the GeparSixto study showed a statistically significant increase in pathological complete response rates in patients with high HRD score; the benefit was observed irrespective of BRCA1/2 status (mutated versus intact) [102]. These results could not be replicated in the advanced setting, although the fact that the HRD assay was performed on primary tumor specimens rather than metastatic samples may have limited its ability to predict responsiveness to carboplatin in metastatic breast cancers (TNT trial) [103]. The value of HRD score in predicting response to therapy is being prospectively tested in both neoadjuvant and advanced settings using platinum compounds [104] and PARPi [97, 105], respectively. Other promising biomarkers are the assessment of PARP activity through measurement of poly(ADP-ribose) levels [93, 106], the evaluation of HR proficiency through the formation of nuclear RAD51 foci [107, 108], the presence of miRNAs involved in the regulation of BRCA proteins (such as miR-182) [109], and the evaluation of levels of 53BP1 expression [17, 93].
Strategies to expand PARPi application to BRCA-proficient breast cancers
Theoretically, PARPi activity could be expanded to breast cancers without BRCA1/2 mutations; several preclinical experiments support this possibility by focusing on the impairment of the HR pathway. PTEN [110] and ATM [111, 112] deficiencies correlate with sensitivity to PARPi both in vitro and in vivo; moreover, CDK1 inhibition [98] and histone deacetylase inhibition [113] have been shown to efficiently sensitize BRCA-proficient cells to PARPi in vitro and, in animal models, in vivo. A phase 1 study is ongoing in patients with solid tumors testing the association of veliparib, a selective CDK inhibitor (dinaciclib) and carboplatin: an expanded cohort of BRCA-proficient tumors is planned [114]. Unfortunately, no validated biomarker of HR dysfunction other than germline BRCA1/2 mutations is currently available.
Alterations in the HR pathway different from BRCA1/2 mutations may determine an HR-deficient phenotype similar to BRCA-deficient tumor (namely, BRCAness) [20]. Such alterations include BRCA1/2 suppression (for example, by promoter methylation) or mutations in genes encoding other proteins involved in HR (such as PTEN, FANCF, RAD51, ATM, and CDK1) [20, 28, 110]. In line with this hypothesis, talazoparib will be tested in BRCA1/2 wild type breast cancer with high HRD score or deleterious germline or somatic mutation implicated in the HR pathway [97].
Other options to exploit PARP inhibition in BRCA-proficient breast cancers currently under investigation (mainly in cell lines and animal models, but also in clinical trials) include PI3K inhibition [115, 116] and TGFβ activation [117]. Preliminary positive data of clinical efficacy of PARPi/PI3K inhibitors in BRCA wild type ovarian and breast cancer have been presented by Matulonis et al. [118] at the 2015 American Association for Cancer Research Annual Meeting.
Conclusions
PARP inhibition is a promising strategy for the treatment of breast cancer associated with germline BRCA1/2 mutations and papillary serous ovarian cancers. Efficacy data from phase 1 and 2 studies showed encouraging objective response rates with acceptable toxicity profiles for PARPi monotherapy. The initial data are consistent with those of other targeted therapies in identifiable subsets of tumors. There is great excitement about the ongoing phase 3 trials in the metastatic, adjuvant, and neoadjuvant settings.
However, other questions apart from clinical efficacy need to be addressed before PARPi will become part of clinical practice. For example, the long-term effects of continuous administration of this class of drugs are not yet fully characterized: will prolonged exposure to PARPi confer increased risk of hematological toxicity or development of new primary malignancies? This is a concern of particular importance in the adjuvant setting. Increasing use of platinum in early triple-negative disease may influence the way PARPi are used given the overlapping mechanisms of action and resistance.
New strategies are being examined to expand the application of PARPi in BRCA-associated cancers beyond breast and ovarian, and in some sporadic tumors. PARPi should be more fully studied in ER-positive BRCA-associated tumors as well. PARPi appear likely to assume an important role in the management of patients with BRCA-associated tumors, and possibly in other carefully defined tumor subsets as well.
Abbreviations
- 53BP1:
-
p53 binding protein 1
- BER:
-
Base excision repair pathway
- HGSOC:
-
High-grade serous ovarian cancer
- HR:
-
Homologous recombination
- HRD:
-
Homologous recombination deficiency
- PARPi:
-
PARP inhibitors
- PARPs:
-
Poly(ADP-ribose) polymerases
- SSBR:
-
Single strand break repair
- TNBC:
-
Triple-negative breast cancers
References
Gonzalez-Angulo AM, Timms KM, Liu S, Chen H, Litton JK, Potter J, et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res. 2011;17:1082–9. doi:10.1158/1078-0432.CCR-10-2560.
Mavaddat N, Barrowdale D, Andrulis IL, Domchek SM, Eccles D, Nevanlinna H, et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol Biomarkers Prev. 2012;21:134–47. doi:10.1158/1055-9965.EPI-11-0775.
Sharma P, Klemp JR, Kimler BF, Mahnken JD, Geier LJ, Khan QJ, et al. Germline BRCA mutation evaluation in a prospective triple-negative breast cancer registry: implications for hereditary breast and/or ovarian cancer syndrome testing. Breast Cancer Res Treat. 2014;145:707–14. doi:10.1007/s10549-014-2980-0.
Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 2015;33:304–11. doi:10.1200/JCO.2014.57.1414.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi:10.1056/NEJMoa0900212.
Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–50. doi:10.1200/JCO.2014.56.2728.
Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852–61. doi:10.1016/S1470-2045(11)70214-5.
Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–93. doi:10.1002/bies.20085.
De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol. 2012;84:137–46. doi:10.1016/j.bcp.2012.03.018.
Burkle A, Virag L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol Aspects Med. 2013;34:1046–65. doi:10.1016/j.mam.2012.12.010.
Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet. 2008;9:619–31. doi:10.1038/nrg2380.
Ali AA, Timinszky G, Arribas-Bosacoma R, Kozlowski M, Hassa PO, Hassler M, et al. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat Struct Mol Biol. 2012;19:685–92. doi:10.1038/nsmb.2335.
Rulten SL, Fisher AE, Robert I, Zuma MC, Rouleau M, Ju L, et al. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol Cell. 2011;41:33–45. doi:10.1016/j.molcel.2010.12.006.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi:10.1016/j.cell.2011.02.013.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi:10.1038/nature03443.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi:10.1038/nature03445.
Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5:387–93. doi:10.1016/j.molonc.2011.07.001.
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–99. doi:10.1158/0008-5472.CAN-12-2753.
Tutt AN, Lord CJ, McCabe N, Farmer H, Turner N, Martin NM, et al. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb Symp Quant Biol. 2005;70:139–48. doi:10.1101/sqb.2005.70.012.
Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–9. doi:10.1038/nrc1457.
Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–32. doi:10.1016/j.ccr.2006.01.013.
Sonnenblick A, de Azambuja E, Azim Jr HA, Piccart M. An update on PARP inhibitors-moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12:27–41. doi:10.1038/nrclinonc.2014.163.
Balmana J, Tung NM, Isakoff SJ, Grana B, Ryan PD, Saura C, et al. Phase I trial of olaparib in combination with cisplatin for the treatment of patients with advanced breast, ovarian and other solid tumors. Ann Oncol. 2014;25:1656–63. doi:10.1093/annonc/mdu187.
Del Conte G, Sessa C, von Moos R, Vigano L, Digena T, Locatelli A, et al. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumours. Br J Cancer. 2014;111:651–9. doi:10.1038/bjc.2014.345.
O’Shaughnessy J, Schwartzberg L, Danso MA, Miller KD, Rugo HS, Neubauer M, et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2014;32:3840–7. doi:10.1200/JCO.2014.55.2984.
Patel AG, De Lorenzo SB, Flatten KS, Poirier GG, Kaufmann SH. Failure of iniparib to inhibit poly(ADP-Ribose) polymerase in vitro. Clin Cancer Res. 2012;18:1655–62. doi:10.1158/1078-0432.CCR-11-2890.
Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006;8:R38. doi:10.1186/bcr1522.
McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. doi:10.1158/0008-5472.CAN-06-0140.
Lukas J, Lukas C, Bartek J. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13:1161–9. doi:10.1038/ncb2344.
Isakoff SJ, Overmoyer B, Tung NM, Gelman RS, Habin K, Qian J, et al. A phase II trial expansion cohort of the PARP inhibitor veliparib (ABT888) and temozolomide in BRCA1/2 associated metastatic breast cancer. Cancer Res. 2011;71:P3-16-05.
Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14:882–92. doi:10.1016/S1470-2045(13)70240-7.
De Bono JS, Mina LA, Gonzalez M, Curtin NJ, Wang E, Henshaw JW, et al. First-in-human trial of novel oral PARP inhibitor BMN 673 in patients with solid tumors. J Clin Oncol. 2013;31:2580.
Kristeleit RS, Burris HA, LoRusso P, Patel MR, Asghar US, El-Khouly F, et al. Phase 1/2 study of oral rucaparib: final phase 1 results. J Clin Oncol. 2014;32:2573.
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–44. doi:10.1016/S0140-6736(10)60892-6.
Ershler WB. Capecitabine monotherapy: safe and effective treatment for metastatic breast cancer. Oncologist. 2006;11:325–35. doi:10.1634/theoncologist.11-4-325.
Martin M, Ruiz A, Munoz M, Balil A, Garcia-Mata J, Calvo L, et al. Gemcitabine plus vinorelbine versus vinorelbine monotherapy in patients with metastatic breast cancer previously treated with anthracyclines and taxanes: final results of the phase III Spanish Breast Cancer Research Group (GEICAM) trial. Lancet Oncol. 2007;8:219–25. doi:10.1016/S1470-2045(07)70041-4.
Cortes J, O’Shaughnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, et al. Eribulin monotherapy versus treatment of physician's choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet. 2011;377:914–23. doi:10.1016/S0140-6736(11)60070-6.
Low JA, Wedam SB, Lee JJ, Berman AW, Brufsky A, Yang SX, et al. Phase II clinical trial of ixabepilone (BMS-247550), an epothilone B analog, in metastatic and locally advanced breast cancer. J Clin Oncol. 2005;23:2726–34. doi:10.1200/JCO.2005.10.024.
Perez EA, Lerzo G, Pivot X, Thomas E, Vahdat L, Bosserman L, et al. Efficacy and safety of ixabepilone (BMS-247550) in a phase II study of patients with advanced breast cancer resistant to an anthracycline, a taxane, and capecitabine. J Clin Oncol. 2007;25:3407–14. doi:10.1200/JCO.2006.09.3849.
Thomas E, Tabernero J, Fornier M, Conte P, Fumoleau P, Lluch A, et al. Phase II clinical trial of ixabepilone (BMS-247550), an epothilone B analog, in patients with taxane-resistant metastatic breast cancer. J Clin Oncol. 2007;25:3399–406. doi:10.1200/JCO.2006.08.9102.
Baselga J, Gelmon KA, Verma S, Wardley A, Conte P, Miles D, et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol. 2010;28:1138–44. doi:10.1200/JCO.2009.24.2024.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91. doi:10.1056/NEJMoa1209124.
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–51. doi:10.1016/S0140-6736(10)60893-8.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366:1382–92. doi:10.1056/NEJMoa1105535.
Liu JF, Tolaney SM, Birrer M, Fleming GF, Buss MK, Dahlberg SE, et al. A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor olaparib (AZD2281) in combination with the anti-angiogenic cediranib (AZD2171) in recurrent epithelial ovarian or triple-negative breast cancer. Eur J Cancer. 2013;49:2972–8. doi:10.1016/j.ejca.2013.05.020.
Press JZ, De Luca A, Boyd N, Young S, Troussard A, Ridge Y, et al. Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC Cancer. 2008;8:17. doi:10.1186/1471-2407-8-17.
National Cancer Institute. Veliparib with or without carboplatin in treating patients with stage III or stage IV breast cancer. https://www.clinicaltrials.gov/ct2/show/NCT01149083. Accessed 15 Jun 2015.
Abbvie. A study evaluating veliparib as a single agent or in combination with chemotherapy in subjects with solid tumors. https://www.clinicaltrials.gov/ct2/show/NCT02033551. Accessed 15 Jun 2015.
Coleman RL, Sill MW, Bell-McGuinn K, Aghajanian C, Gray HJ, Tewari KS, et al. A phase II evaluation of the potent, highly selective PARP inhibitor veliparib in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in patients who carry a germline BRCA1 or BRCA2 mutation - An NRG Oncology/Gynecologic Oncology Group study. Gynecol Oncol. 2015;137:386–91. doi:10.1016/j.ygyno.2015.03.042.
Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–37. doi:10.1158/1078-0432.CCR-06-3039.
Palma JP, Wang YC, Rodriguez LE, Montgomery D, Ellis PA, Bukofzer G, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15:7277–90. doi:10.1158/1078-0432.CCR-09-1245.
Isakoff SJ, Overmoyer B, Tung NM, Gelman RS, Giranda VL, Bernhard KM, et al. A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer. J Clin Oncol. 2010;28:1019.
Rodler ET, Gralow J, Kurland BF, Griffin M, Yeh R, Thompson JA, et al. Phase I: Veliparib with cisplatin (CP) and vinorelbine (VNR) in advanced triple-negative breast cancer (TNBC) and/or BRCA mutation-associated breast cancer. J Clin Oncol. 2014;32:2569.
Somlo G, Frankel PH, Luu TH, Ma C, Arun B, Garcia A, et al. Efficacy of the combination of ABT-888 (veliparib) and carboplatin in patients with BRCA-associated breast cancer. J Clin Oncol. 2013;31:1024.
Pahuja S, Beumer JH, Appleman LJ, Tawbi HA, Stoller RG, Lee JJ, et al. A phase I study of veliparib (ABT-888) in combination with weekly carboplatin and paclitaxel in advanced solid malignancies and enriched for triple-negative breast cancer (TNBC). J Clin Oncol. 2015;33:1015.
Kummar S, Chen A, Ji J, Zhang Y, Reid JM, Ames M, et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011;71:5626–34. doi:10.1158/0008-5472.CAN-11-1227.
Samol J, Ranson M, Scott E, Macpherson E, Carmichael J, Thomas A, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs. 2012;30:1493–500. doi:10.1007/s10637-011-9682-9.
Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther. 2014;349:408–16. doi:10.1124/jpet.113.210146.
Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–84. doi:10.1073/pnas.0806092105.
Tesaro, Inc. A phase III trial of niraparib versus physician’s choice in Her2 negative, germline BRCA mutation-positive breast cancer patients (BRAVO). https://clinicaltrials.gov/ct2/show/NCT01905592. Accessed 15 Jun 2015.
BioMarin Pharmaceutical. A study evaluating talazoparib (BMN 673), a PARP inhibitor, in advanced and/or metastatic breast cancer patients with BRCA mutation (EMBRACA Study). https://clinicaltrials.gov/ct2/show/NCT01945775. Accessed 15 Jun 2015.
AstraZeneca. Assessment of the efficacy and safety of olaparib monotherapy versus physicians choice chemotherapy in the treatment of metastatic breast cancer patients with germline BRCA1/2 mutations. (OlympiAD). https://clinicaltrials.gov/ct2/show/NCT02000622. Accessed 15 Jun 2015.
AbbVie. A phase 3 randomized, placebo-controlled trial of carboplatin and paclitaxel with or without veliparib (ABT-888) in HER2-negative metastatic or locally advanced unresectable BRCA-associated breast cancer. https://clinicaltrials.gov/ct2/show/NCT02163694. Accessed 15 Jun 2015.
European Medicines Agency. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/003726/smops/Positive/human_smop_000744.jsp&mid=WC0b01ac058001d127. Accessed 15 Jan 2015.
U. S. Food and Drug Administration. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm427554.htm. Accessed 15 Jan 2015.
Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16:87–97. doi:10.1016/S1470-2045(14)71135-0.
Tutt A, Kaufman B, Gelber R, McFadden E, Goessl CD, Viale G, et al. OlympiA: A randomized phase III trial of olaparib as adjuvant therapy in patients with high-risk HER2-negative breast cancer (BC) and a germline BRCA1/2 mutation (gBRCAm). J Clin Oncol. 2015;33, TPS1109.
AstraZeneca. Olaparib as adjuvant treatment in patients with germline BRCA mutated high risk HER2 negative primary breast cancer (OlympiA). https://clinicaltrials.gov/ct2/show/NCT02032823. Accessed 15 Jun 2015.
Hoosier Cancer Research Network. PARP inhibition for triple negative breast cancer (ER-/PR-/HER2-) with BRCA1/2 mutations. https://clinicaltrials.gov/ct2/show/NCT01074970. Accessed 15 Jun 2015.
Dwadasi S, Tong Y, Walsh T, Danso MA, Ma CX, Silverman P, et al. Cisplatin with or without rucaparib after preoperative chemotherapy in patients with triple-negative breast cancer (TNBC): Hoosier Oncology Group BRE09-146. J Clin Oncol. 2014;32:1019.
Rugo HS, Olopade O, DeMichele A, van’t Veer L, Buxton M, Hylton N, et al. Veliparib/carboplatin plus standard neoadjuvant therapy for high-risk breast cancer: first efficacy results from the I-SPY 2 TRIAL. Cancer Res. 2013;73:S5–02.
von Minckwitz G, Schneeweiss A, Loibl S, Salat C, Denkert C, Rezai M, et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet Oncol. 2014;15:747–56. doi:10.1016/S1470-2045(14)70160-3.
Sikov WM, Berry DA, Perou CM, Singh B, Cirrincione CT, Tolaney SM, et al. Impact of the addition of carboplatin and/or Bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (alliance). J Clin Oncol. 2015;33:13–21. doi:10.1200/JCO.2014.57.0572.
AbbVie. A study evaluating safety and efficacy of the addition of ABT-888 plus carboplatin versus the addition of carboplatin to standard chemotherapy versus standard chemotherapy in subjects with early stage triple negative breast cancer. https://clinicaltrials.gov/ct2/show/NCT02032277. Accessed 15 Jun 2015.
BioMarin Pharmaceutical. Neoadjuvant bmn673 for patients with a BRCA deleterious mutation. https://www.clinicaltrials.gov/ct2/show/NCT02282345. Accessed 15 Jun 2015.
To C, Kim EH, Royce DB, Williams CR, Collins RM, Risingsong R, et al. The PARP inhibitors, veliparib and olaparib, are effective chemopreventive agents for delaying mammary tumor development in BRCA1-deficient mice. Cancer Prev Res (Phila). 2014;7:698–707. doi:10.1158/1940-6207.CAPR-14-0047.
Khan OA, Gore M, Lorigan P, Stone J, Greystoke A, Burke W, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer. 2011;104:750–5. doi:10.1038/bjc.2011.8.
Rajan A, Carter CA, Kelly RJ, Gutierrez M, Kummar S, Szabo E, et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin Cancer Res. 2012;18:2344–51. doi:10.1158/1078-0432.CCR-11-2425.
Dean E, Middleton MR, Pwint T, Swaisland H, Carmichael J, Goodege-Kunwar P, et al. Phase I study to assess the safety and tolerability of olaparib in combination with bevacizumab in patients with advanced solid tumours. Br J Cancer. 2012;106:468–74. doi:10.1038/bjc.2011.555.
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68.
Lowndes NF, Toh GW. DNA repair: the importance of phosphorylating histone H2AX. Curr Biol. 2005;15:R99–R102. doi:10.1016/j.cub.2005.01.029.
Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi:10.1038/nrc2523.
Chiarugi A. A snapshot of chemoresistance to PARP inhibitors. Trends Pharmacol Sci. 2012;33:42–8. doi:10.1016/j.tips.2011.10.001.
Bouwman P, Jonkers J. Molecular pathways: how can BRCA-mutated tumors become resistant to PARP inhibitors? Clin Cancer Res. 2014;20:540–7. doi:10.1158/1078-0432.CCR-13-0225.
Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19:1381–8. doi:10.1038/nm.3369.
Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5. doi:10.1038/nature06548.
Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69:6381–6. doi:10.1158/0008-5472.CAN-09-1178.
Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68:2581–6. doi:10.1158/0008-5472.CAN-08-0088.
Wurzer G, Herceg Z, Wesierska-Gadek J. Increased resistance to anticancer therapy of mouse cells lacking the poly(ADP-ribose) polymerase attributable to up-regulation of the multidrug resistance gene product P-glycoprotein. Cancer Res. 2000;60:4238–44.
Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–95. doi:10.1038/nsmb.1831.
Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi:10.1016/j.cell.2010.03.012.
Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3:68–81. doi:10.1158/2159-8290.CD-12-0049.
Oplustilova L, Wolanin K, Mistrik M, Korinkova G, Simkova D, Bouchal J, et al. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle. 2012;11:3837–50. doi:10.4161/cc.22026.
Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008–15. doi:10.1200/JCO.2010.34.2980.
Ang JE, Gourley C, Powell CB, High H, Shapira-Frommer R, Castonguay V, et al. Efficacy of chemotherapy in BRCA1/2 mutation carrier ovarian cancer in the setting of PARP inhibitor resistance: a multi-institutional study. Clin Cancer Res. 2013;19:5485–93. doi:10.1158/1078-0432.CCR-13-1262.
AbbVie. The study evaluating efficacy and tolerability of veliparib in combination with temozolomide or in combination with carboplatin and paclitaxel versus placebo in subjects with BRCA1 and BRCA2 mutation and metastatic breast cancer. https://clinicaltrials.gov/ct2/show/NCT01506609. Accessed 15 Jun 2015.
PARP inhibitor BMN-673 in treating patients with BRCA1 and BRCA2 wild-type, metastatic or recurrent, triple-negative or HER2-negative breast cancer. https://clinicaltrials.gov/ct2/show/NCT02401347. Accessed 15 Jun 2015.
Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. 2011;17:875–82. doi:10.1038/nm.2377.
Timms KM, Abkevich V, Hughes E, Neff C, Reid J, Morris B, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16:475. doi:10.1186/s13058-014-0475-x.
Richardson AL, Silver DP, Szallasi Z, Birkbak NJ, Wang ZC, Iglehart JD, et al. Homologous recombination deficiency (HRD) score predicts response to cisplatin neoadjuvant chemotherapy in patients with triple negative breast cancer. San Antonio Breast Cancer Symposium Proceedings. 2014;2014:P3-06-11.
Telli ML, Timms K, Reid JE, Neff C, Abkevich V, Gutin A, et al. Combined homologous recombination deficiency (HRD) scores and response to neoadjuvant platinum-based chemotherapy in triple-negative and/or BRCA1/2 mutation-associated breast cancer. J Clin Oncol. 2015;33:1018.
von Minckwitz G, Timms K, Untch M, Elkin EP, Fasching PA, Schneeweiss A, et al. Prediction of pathological complete response (pCR) by homologous recombination deficiency (HRD) after carboplatin-containing neoadjuvant chemotherapy in patients with TNBC: results from GeparSixto. J Clin Oncol. 2015;33:1004.
Tutt A, Ellis P, Kilburn L, Gilett C, Pinder S, Abraham J, et al. TNT: a randomized phase III trial of carboplatin (C) compared with docetaxel (D) for patients with metastatic or recurrent locally advanced triple negative or BRCA1/2 breast cancer (CRUK/07/012). San Antonio Breast Cancer Symposium Proceedings. 2014;2014:S3–01.
Cisplatin vs paclitaxel for triple neg. https://clinicaltrials.gov/show/NCT01982448. Accessed 15 Jun 2015.
Phase II study of BMN 673. https://clinicaltrials.gov/ct2/show/NCT02286687. Accessed 15 Jun 2015.
Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, Djureinovic T, et al. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010;70:5389–98. doi:10.1158/0008-5472.CAN-09-4716.
Balmana J, Domchek SM, Tutt A, Garber JE. Stumbling blocks on the path to personalized medicine in breast cancer: the case of PARP inhibitors for BRCA1/2-associated cancers. Cancer Discov. 2011;1:29–34. doi:10.1158/2159-8274.CD-11-0048.
Lee JM, Ledermann JA, Kohn EC. PARP inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25:32–40. doi:10.1093/annonc/mdt384.
Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell. 2011;41:210–20. doi:10.1016/j.molcel.2010.12.005.
Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–22. doi:10.1002/emmm.200900041.
Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–87. doi:10.1182/blood-2010-01-265769.
Williamson CT, Muzik H, Turhan AG, Zamo A, O’Connor MJ, Bebb DG, et al. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol Cancer Ther. 2010;9:347–57. doi:10.1158/1535-7163.MCT-09-0872.
Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SG, et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget. 2014;5:5637–50.
Veliparib and dinaciclib with or without carboplatin in treating patients with advanced solid tumors. https://clinicaltrials.gov/ct2/show/NCT01434316. Accessed 15 Jun 2015.
De P, Sun Y, Carlson JH, Friedman LS, Leyland-Jones BR, Dey N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia. 2014;16:43–72.
Phase I study of the oral PI3kinase inhibitor BKM120 or BYL719 and the oral PARP inhibitor olaparib in patients with recurrent triple negative breast cancer or high grade serous ovarian cancer. https://clinicaltrials.gov/ct2/show/NCT01623349. Accessed 15 Jun 2015.
Liu L, Zhou W, Cheng CT, Ren X, Somlo G, Fong MY, et al. TGFbeta induces ‘BRCAness’ and sensitivity to PARP inhibition in breast cancer by regulating DNA-repair genes. Mol Cancer Res. 2014;12:1597–609. doi:10.1158/1541-7786.MCR-14-0201.
Matulonis U, Wulf G, Barry W, Birrer M, Westin S, Spagnoletti T, et al. Phase I of oral BKM120 or BLY719 and olaparib for high-grade serous ovarian cancer or triple-negative breast cancer: Final results of the BKM120 plus olaparib cohort. Proceedings of the 106th Annual Meeting of the American Association for Cancer Research. 2015 Apr 18–22:CT324.
Yamamoto N, Nokihara H, Yamada Y, Goto Y, Tanioka M, Shibata T, et al. A phase I, dose-finding and pharmacokinetic study of olaparib (AZD2281) in Japanese patients with advanced solid tumors. Cancer Sci. 2012;103:504–9. doi:10.1111/j.1349-7006.2011.02179.x.
Pahuja S, Beumer JH, Appleman LJ, Tawbi HA, Stoller RG, Lee JJ, et al. Outcome of BRCA 1/2-mutated (BRCA+) and triple-negative, BRCA wild type (BRCA-wt) breast cancer patients in a phase I study of single-agent veliparib (V). J Clin Oncol. 2014;32:135.
Drew Y, Ledermann JA, Jones A, Hall G, Jayson GC, Highley M, et al. Phase II trial of the poly(ADP-ribose) polymerase (PARP) inhibitor AG-014699 in BRCA-1 and -2 mutated, advanced ovarian and/or locally advanced or metastatic breast cancer. J Clin Oncol. 2011;29:3104.
Dent RA, Lindeman GJ, Clemons M, Wildiers H, Chan A, McCarthy NJ, et al. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013;15:R88. doi:10.1186/bcr3484.
Lee JM, Hays JL, Annunziata CM, Noonan AM, Minasian L, Zujewski JA, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst. 2014;106:dju089.
Appleman LJ, Beumer JH, Jiang Y, Puhalla S, Lin Y, Owonikoko TK, et al. A phase I study of veliparib (ABT-888) in combination with carboplatin and paclitaxel in advanced solid malignancies. J Clin Oncol. 2012;30:3049.
Tan AR, Toppmeyer D, Stein MN, Moss RA, Gounder M, Lindquist DC, et al. Phase I trial of veliparib, (ABT-888), a poly(ADP-ribose) polymerase (PARP) inhibitor, in combination with doxorubicin and cyclophosphamide in breast cancer and other solid tumors. J Clin Oncol. 2011;29:3041.
Bell-McGuinn KM, Gray HJ, Fleming GF, Cristea MC, Medina DM, Xiong H, et al. Phase I study of ABT-888 in combination with carboplatin and gemcitabine in subjects with advanced solid tumors. J Clin Oncol. 2013;31:2584.
Wesolowski R, Zhao M, Geyer SM, Lustberg MB, Mrozek E, Layman RM, et al. Phase I trial of the PARP inhibitor veliparib (V) in combination with carboplatin (C) in metastatic breast cancer (MBC). J Clin Oncol. 2014;32:1075.
Kummar S, Ji J, Morgan R, Lenz HJ, Puhalla SL, Belani CP, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res. 2012;18:1726–34. doi:10.1158/1078-0432.CCR-11-2821.
Molife LR, Roxburgh P, Wilson RH, Gupta A, Middleton MR, Evans TRJ, et al. A phase I study of oral rucaparib in combination with carboplatin. J Clin Oncol. 2013;31:2586.
Plummer R, Stephens P, Aissat-Daudigny L, Cambois A, Moachon G, Brown PD, et al. Phase 1 dose-escalation study of the PARP inhibitor CEP-9722 as monotherapy or in combination with temozolomide in patients with solid tumors. Cancer Chemother Pharmacol. 2014;74:257–65. doi:10.1007/s00280-014-2486-9.
BioMarin Pharmaceutical. A phase 2, 2-stage, 2-cohort study of talazoparib (BMN 673), in locally advanced and/or metastatic breast cancer patients with BRCA mutation (ABRAZO Study). https://clinicaltrials.gov/ct2/show/NCT02034916. Accessed 15 Jun 2015.
Cancer Research UK. Rucaparib (CO-338; formally called AG-014699 or PF-0136738) in treating patients with locally advanced or metastatic breast cancer or advanced ovarian cancer. https://clinicaltrials.gov/ct2/show/NCT00664781. Accessed 15 Jun 2015.
PARP inhibition for triple negative breast cancer (ER-/PR-/HER2-) with BRCA1/2 mutations. https://clinicaltrials.gov/ct2/show/NCT01074970. Accessed 15 Jun 2015.
Bundred N, Gardovskis J, Jaskiewicz J, Eglitis J, Paramonov V, McCormack P, et al. Evaluation of the pharmacodynamics and pharmacokinetics of the PARP inhibitor olaparib: a phase I multicentre trial in patients scheduled for elective breast cancer surgery. Invest New Drugs. 2013;31:949–58. doi:10.1007/s10637-012-9922-7.
Kummar S, Kinders R, Gutierrez ME, Rubinstein L, Parchment RE, Phillips LR, et al. Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009;27:2705–11. doi:10.1200/JCO.2008.19.7681.
Acknowledgements
The authors thank Dr. Daniel Silver (Dana-Farber Cancer Institute, Boston, MA) for advising on the creation of figures for this review.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LL and JEG drafted and revised the manuscript. Both authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Livraghi, L., Garber, J.E. PARP inhibitors in the management of breast cancer: current data and future prospects. BMC Med 13, 188 (2015). https://doi.org/10.1186/s12916-015-0425-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-015-0425-1