
Abstract
Changes in cell metabolism and metabolic adaptation are hallmark features of many cancers, including leukemia, that support biological processes involved into tumor initiation, growth, and response to therapeutics. The discovery of mutations in key metabolic enzymes has highlighted the importance of metabolism in cancer biology and how these changes might constitute an Achilles heel for cancer treatment. In this Review, we discuss the role of metabolic and mitochondrial pathways dysregulated in acute myeloid leukemia, and the potential of therapeutic intervention targeting these metabolic dependencies on the proliferation, differentiation, stem cell function and cell survival to improve patient stratification and outcomes.
Similar content being viewed by others
Acute myeloid leukemia (AML) is a heterogeneous group of hematological malignancies and represents the most frequent cause of leukemia-related deaths [1]. It arises from genetic abnormalities in hematopoietic stem or progenitor cells, inducing uncontrolled growth and an accumulation of abnormal myeloblasts, leading to bone marrow failure and often death. For the past three decades, standard intensive induction therapy involved a combination of cytarabine plus anthracycline cytotoxic chemotherapy. Despite a high rate (70–80%) of complete remission after standard front-line chemotherapy, the prognosis remains poor, especially for older patients. This mainly results from the high frequency of distant relapses caused by tumor regrowth initiated by chemoresistant leukemic clones after chemotherapy [2, 3]. Therefore, more specific and safe therapeutics are urgently needed. One area of high interest and potential is targeting metabolic and mitochondrial pathways that are important in AML biology and that may constitute an Achilles heel of AML cells. This review focuses on metabolic pathways dysregulated in AML, and especially in several cytogenetically defined patient subgroups, and how targeting these metabolic dependencies impacts proliferation and cell survival in this disease.
Major metabolic dysregulations in acute myeloid leukemia
Metabolism is altered in most, if not all, cancer cells, regardless of the tumor type [4]. A key alteration in cancer metabolism is the increase in glucose uptake required to satisfy energetic and anabolic demands. It is now well established that the metabolic reprogramming undergone by transformed cells extends far beyond glycolysis and the Warburg effect, and changes in cell metabolism have fundamental implications for tumor biology and therapy [5, 6].
Glucose metabolism
Higher aerobic glycolysis in cancer cells, reported almost one century ago by Otto Warburg and known as the Warburg effect [7, 8], has sparked debate over the role of glycolysis and oxidative phosphorylation in normal and cancer cells. Since Warburg’s discovery and especially during the past 20 years, considerable efforts have been made to better understand glucose utilization in cancer cells, in particular to determine if inhibiting glycolysis or other glucose-dependent pathways could represent promising therapeutic approaches. It has been suggested that AML patients exhibit a high glycolytic metabolism at diagnosis that is potentially associated with favorable outcomes [9], even if the number of patients in this study remains small. Another study reported that a six-metabolite signature (including pyruvate and lactate) related to the crosstalk between glycolysis and mitochondria was specifically enriched in the serum of patients at diagnosis compared to healthy controls and demonstrated prognostic value in cytogenetically normal AML (CN-AML) patients as it could predict poor survival for these patients [10]. Interestingly, deletions of the two glycolytic enzymes PKM2 and LDHA, which catalyze the production of cytosolic pyruvate and lactate, respectively, inhibit leukemia initiation in vivo in AML mice models while preserving normal hematopoietic stem cell function [11] (Fig. 1).

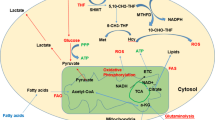
Metabolic pathways relative to deregulated reactions in myeloid leukemia. Enzymes discussed in this review are in blue. Compound abbreviations: F1P fructose-1-phosphate, G1P glucose-1-phosphate, G6P glucose-6-phosphate, F6P fructose-6-phosphate, F1,6BP fructose-1,6-biphosphate, GA3P glyceraldehyde 3-phosphate, DHAP dihydroxyacetone phosphate, 3PG 3-phosphoglycerate, P-Serine phosphoserine, 2PG 2-phosphoglycerate, PEP phosphoenolpyruvate, 6PGL 6-phosphogluconolactone, 6PG 6-phosphogluconic acid, Rib5P ribulose-5-phosphate, X5P xylulose-5-phosphate, R5P ribose-5-phosphate, Sed7P sedoheptulose-7-phosphate, E4P erythrose-4-phosphate, PRPP phosphoribosyl pyrophosphate, Carbamoyl-P carbamoyl phosphate, DHO dihydroorotate, THF tetrahydrofolate, OAA oxaloacetate, α-KG α-ketoglutarate, 2-HG 2-hydroxyglutarate, BCAA branched-chain amino acid
Glucose metabolism is also involved in other crucial metabolic pathways such as the pentose phosphate pathway (PPP) coupled to NADPH production, glutathione/redox recycling, and nucleotide biosynthesis (Fig. 1). Overexpression of glucose-6-phosphate dehydrogenase (G6PD) has been reported to correlate with an adverse prognosis in an AML cohort [12]. Moreover, in vitro and in vivo inhibition of 6-phosphogluconate dehydrogenase (6PGD) and G6PD demonstrated anti-leukemic activities and synergized with cytarabine [12,13,14,15]. Inhibition of 6PGD leads to impaired lipogenesis through reactivation of LKB1-AMPK signaling [14]. Sensitivity to G6PD inhibition is driven by mTORC1 activity as mTORC1 activation leads to glucose addiction in AML. Inhibition of mTORC1 induces a switch toward oxidative metabolism and survival of AML cells [12]. Furthermore, the anti-leukemic effects of mTOR inhibitors are enhanced when combined with anti-glycolytic agents, underscoring the strong interconnection between mTOR activity and leukemic metabolism [16]. Better characterization of mTOR-associated metabolic alterations would help in the design of new combinatory therapeutic approaches and/or help distinguish patients who could better benefit from these treatments. This will be even more important since no clear evidence of clinical efficacy has been found by several clinical trials of agents targeting mTOR kinase in myeloid leukemia [17,18,19,20,21,22] (Table 1). This modest efficacy is due to multifactorial aspects of mTOR biology and AML heterogeneity. The anti-leukemic effect of mTOR inhibition depends on the level of constitutive PI3K/Akt/mTOR pathway activation, leukemia-microenvironment crosstalk, and the release of mediators by both AML and stromal cells [71].
Amino acid metabolism
Of note, Willems et al. have shown that glutamine availability is a limiting step for mTORC1 activation and that the anti-tumor effect of L-asparaginase is mainly due to its glutaminase activity in AML [72], highlighting a major role for amino acids in leukemia biology. Indeed, intracellular glutamine concentration controls the uptake of leucine as leucine is imported into the cell in exchange for glutamine by the SLC7A5/3A2 transporter and leucine is required for Rheb-mediated mTOR activation at the lysosomal surface [73, 74]. Glutamine is a non-essential amino acid and one of the major carbon sources used by cancer cells for proliferation in vitro [75, 76]. It is also an important nitrogen donor for amino acids and nucleotides and a major substrate for TCA cycle intermediates as well as glutamate and aspartate [77,78,79] (Fig. 1). Dependence of leukemic cells on glutamine for tumor growth has been reported, and knockdown of the glutamine transporter SLC1A5 abrogates tumor development in mice [72].
An approach to extend therapeutic opportunities beyond glycolysis and glutaminolysis may be found in the identification of auxotrophic amino acids required by AML cells. It has been reported that most AML patients are deficient in arginosuccinate synthetase-1 (ASS1), an enzyme that allows the conversion of citrulline and aspartate into the arginine precursor argininosuccinate [29] (Fig. 1). The loss of ASS1 has been reported in other tumor types where it is required to support cell proliferation and nucleotide synthesis by sustaining the intracellular aspartate level [80]. A decrease in ASS1 can also lead to a dependence on arginine, which has been explored as a potential vulnerability in different cancer types, including AML [29].
Lipid and sterol metabolism
De novo lipid biosynthesis is another metabolic pathway highly reprogrammed in cancer and leukemic cells, in particular to increase biomass. Numerous studies support targeting lipid synthesis for therapeutic benefit [81, 82]. Inhibition of key lipogenic enzymes, fatty acid synthase (FASN) [83] and stearoyl CoA desaturase 1 (SCD1) [68], have been shown to disrupt lipid synthesis and induce apoptosis in AML (Fig. 1). SCD1 inhibition was obtained through treatment with BaP, a combination of lipid-regulating bezafibrate and the sex hormone medroxyprogesterone acetate [68] (Table 1). BaP disrupts prostaglandin metabolism, leading to AML growth arrest and differentiation [68,69,70]. Interestingly, it was reported that BaP treatment caused redirection of pyruvate utilization leading to conversion of α-ketoglutarate (α-KG) to succinate and of oxaloacetate into malonate to cope with oxidative stress [68, 84,85,86]. This pyruvate reprogramming by BaP includes preferential activation of pyruvate carboxylase (PC) over pyruvate dehydrogenase (PDH) to produce malonate, a competitive inhibitor of the succinate dehydrogenase [87,88,89] (Fig. 1). PC has been shown to play a key role in different solid tumors, in particular through in vivo reprogramming of glucose utilization to support anapleurosis [90,91,92,93,94,95]. Further investigations of PC activity in leukemia, especially in vivo, would be highly valuable and provide a better understanding of pyruvate metabolism and channeling between glycolysis, TCA cycle, and amino acid pathways.
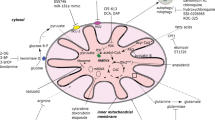
Various studies have focused on the mevalonate pathway and the inhibition of the rate-limiting enzyme 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) with statins in AML [63, 96] (Fig. 1). The end-products of the mevalonate pathway include cholesterol, a major constituent of cell membranes, but also ubiquinone, which is involved in electron transfer between the Electron transfer chain (ETC) complexes I to III (see below; Fig. 2), geranylgeranyl and farnesyl pyrophosphate, which are necessary for post-translational modification of oncogenic proteins, and tyrosine kinase (TK) receptors [97].

Pharmacological inhibitors used to disrupt mitochondrial activities in myeloid leukemia
Oxidative phosphorylation and mitochondrial metabolism
Originally, observations by Otto Warburg that cancer cells exhibited higher glycolytic activity than normal cells even in the presence of oxygen led to the assumption that cancer cell mitochondrial respiration may be impaired. Since then, major studies have clearly demonstrated that cancer cells are able to use oxygen via oxidative phosphorylation (OxPHOS) [98,99,100,101] and mitochondria are essential for cancer cell survival. In myeloid leukemia, Ṧkrtić et al. observed that AML cells had higher mitochondrial mass and an increased oxygen consumption rate compared to normal hematopoietic progenitors [32]. Of note, bulk cell populations had higher mitochondrial mass than an immature CD34+CD38− cell population, suggesting unique mitochondrial characteristics of leukemic stem cells (LSCs). However, the increased mitochondrial mass in AML did not translate into an increase in ETC complex I, III, IV, and V activities, resulting in a lower capability of AML compared to normal cells to enhance their maximal respiration with higher electron flux, known as the spare reserve capacity, suggesting a decreased ability to cope with oxidative stress [102]. In addition, different studies have reported an amplification of mitochondrial DNA (mtDNA) levels in AML [34, 103] that correlates with enhanced cytoplasmic nucleoside kinase expression [34, 104]. Almost 20 years ago, Beuneu et al. reported that dihydro-orotate dehydrogenase (DHODH), a mitochondrial enzyme of de novo pyrimidine biosynthesis that catalyzes the ubiquinone-mediated conversion of dihydro-orotate (DHO) to orotate, could provide electrons to the ETC via ubiquinone in AML cells [105]. Therefore, inhibition of DHODH could represent another promising approach to tackle mitochondria in cancer.
Fatty acids can be a major source for TCA cycle precursors and mitochondrial respiration, especially during and following metabolic challenges or limitations of other oxidizable substrates [82, 106] (Fig. 2). Increased fatty acid oxidation (FAO) and high carnitine palmitoyltransferase 1 (CPT1a) expression have been associated with a poor prognosis in normal karyotype AML patients [107, 108]. German et al. [109] observed a key role of prolyl-hydroxylase 3 (PHD3) in FAO regulation in AML. They reported that, in the setting of high nutrient abundance, PHD3 activates acetyl-CoA carboxylase 2 (ACC2) via hydroxylation, causing inhibition of CPT1a and FAO. Accordingly, when nutrients are scarce and energetic stress is induced, AMPK phosphorylates and inhibits ACC2 to activate FAO [110, 111]. Reduced expression of PHD3 could therefore represent a marker of good responders to FAO inhibitors in AML.
Targeting metabolic vulnerabilities in acute myeloid leukemia
As metabolic alterations are part of oncogenesis and tumor progression, cancer cell metabolism offers promising targets for therapeutic intervention. Hereafter, we discuss several key metabolic pathways that might be therapeutically targetable for AML treatment.
Tackling aerobic glycolysis
Treatment with 2-deoxyglucose (2-DG) to inhibit aerobic glycolysis and related glycosylation of oncogenic proteins exerts an anti-proliferative effect in different AML cell lines and patients and synergizes with conventional cytarabine chemotherapy [10, 23]. However, targeting aerobic glycolysis has not shown great success in clinical settings as 2-DG treatment necessitates high dosing that might induce hypoglycemia and cardiac and red blood cell toxicities due to PPP alteration. Moreover, LDH inhibitors have never progressed into clinical trials (Table 1). Another way to approach high glycolytic metabolism in myeloid leukemia could be through direct targeting of the glucose storage pathway or inhibition of other glycolytic sources such as glycogen and fructose (Fig. 1). It is notable that mRNA levels of glycogen biosynthetic enzymes GYS1/2 and GBE1 were associated with poor survival in AML and that invalidation of GYS1 delayed tumor growth in vivo [112]. AML cells may additionally rely on fructose under low glucose conditions through upregulation of the GLUT5 transporter to maintain glycolytic flux and overcome glucose restriction. Expression of SLC25A5, which encodes GLUT5, is associated with poor AML patient outcome and pharmacological inhibition of GLUT5 eliminates leukemic phenotypes and potentiates the effect of cytarabine in vivo [113].
Glutaminolysis inhibition and amino acid depletion
Targeting glutaminolysis has been investigated as a promising therapeutic target in myeloid leukemia [26, 114, 115]. Of particular interest, inhibition of glutaminase with CB-839 reduces mitochondrial activities and TCA cycle intermediate levels, suggesting that glutamine exerts control on mitochondrial oxidative metabolism in AML [26, 116] (Fig. 2). Clinical trials are currently in progress to assess the benefit of the allosteric glutaminase inhibitor CB-839 (Table 1) with mixed evidence of clinical efficacy. Willems et al. have shown that the ability of L-asparaginase (kidrolase and erwinase) to transform extracellular glutamine into glutamate leads to inhibition of mTORC1 and protein translation in AML cells and that L-asparaginase exhibits anti-leukemic activities [72]. L-asparaginase, which mainly catalyzes the hydrolysis of L-asparagine to L-aspartic acid, is one of the standard drugs for treatment of acute lymphoblastic leukemia (ALL). These patients lack L-asparagine synthetase (ASNS), the enzyme that catalyzes the biosynthesis of L-asparagine, leading to a higher dependency on this amino acid [117]. However, AML patients harbor variable expression of ASNS that could explain their reduced sensitivity to L-asparaginase. Another recent study proposed another explanation linked to the bone marrow microenvironment [118]. The study by Michelozzi et al. suggests that while AML cells are sensitive to L-asparaginase, mesenchymal stromal cells (MSCs) and monocytes/macrophages produce lysosomal cysteine protease cathepsin B able to inactivate L-asparaginase. This contribution of the bone marrow microenvironment to asparaginase resistance was also described in ALL through release of asparagine and glutamine by adipocytes [119].
Depletion of arginine using a mycoplasma-derived enzyme of arginine deiminase formulated with polyethylene glycol (ADI-PEG20) that degrades arginine to citrulline reduces tumor burden in AML and synergizes with cytarabine in vitro and in vivo. Normal human hematopoietic stem-progenitor cells express higher ASS1 than AML cells, supporting the idea of selective targeting of leukemia cells and highlighting a potential therapeutic window for ADI-PEG20 [29, 30], currently under phase 2 clinical evaluation (Table 1).
Inhibition of the mevalonate pathway
The anti-leukemic effects of statins, HMG-CoA inhibitors, have been studied [96, 120] and found to be additive with conventional chemotherapies such as cytarabine and daunorubicin in primary AML samples compared to healthy donors [63,64,65]. Phase I and then phase II clinical trials combining pravastatin with idarubicin and cytarabine for relapse cases of AML have shown an encouraging response rate of 75% [66, 67] (Table 1). However, a subsequent investigation of this regimen has not confirmed these encouraging results in patients with newly diagnosed AML or MDS [121]. These differences between response in newly diagnosed AML or patients at relapse could be due to rewiring of intracellular cholesterol metabolism and sterol membrane transport following chemotherapy and suggest that statins could play a role in overcoming chemoresistance rather than synergizing with frontline therapies. The focus of this review does not include deciphering all the adaptive mechanisms induced by chemotherapeutic agents or new drugs in AML, although this is important for understanding the clinical relevance of these metabolic inhibitors.
Hitting at OxPHOS, BCL2, and mitochondrial dependencies
Mitochondria are dynamic organelles that play a crucial role in several fundamental signaling and metabolic processes such as reactive oxygen species (ROS) regulation, energy production, calcium signaling, TCA cycle, and pyrimidine or heme biosynthesis. Mitochondrial metabolism represents a targetable vulnerability due to the enhanced dependency on mitochondrial energetics of AML cells. Various strategies to disable mitochondrial function have been investigated in myeloid leukemia, including inhibition of mitochondrial translation with tigecycline [32], inhibition of the mitochondrial protease ClpP, thereby decreasing ETC complex II activity [33], and inhibition of mtDNA polymerase using 2’,3’-dideoxycytidine, a drug already used in the treatment of AIDS [34] (Fig. 2; Table 1). Each of these treatments had anti-leukemic properties in vitro and in vivo. Moreover, we and other investigators have shown that metformin, a common biguanide used to treat type 2 diabetes, exhibits anti-leukemic activities in AML [35, 36] (Table 1). However, metformin pharmacokinetics and its maximum efficient dose do not allow its use as an anti-AML agent alone in a clinical setting. Nevertheless, metformin (or other biguanides) might be promising in combination with chemotherapies or other targeted therapies, as recently shown in diffuse large B cell lymphoma refractory to all anti-CD20-based therapies using L-asparaginase, mTOR inhibitor, and metformin (called KTM therapy) [122]. Whereas metformin inhibits ETC complex I activity and thus mitochondrial oxygen consumption, high basal glucose consumption and Akt levels can also affect metformin sensitivity, suggesting combinatory therapies with AKT inhibitors may be effective [35]. More recently, the new ETC complex I inhibitor IACS-010759, which inhibits OxPHOS and nucleotide biosynthesis by decreasing aspartate levels [37], is in a phase I clinical trial for AML and solid tumors [37] (Fig. 2; Table 1).
FAO is a key catabolic pathway involved in the generation of NADH and FADH2, which are the electron donors of complex I and complex II of the ETC, respectively, and leading to the production of acetyl-CoA. This latter plays a crucial role in energy generation, biosynthesis, and epigenetic control through post-translational protein modifications. Inhibiting FAO has been investigated in myeloid leukemia [42, 44, 46, 114, 123]. Inhibition of CPT1a, which catalyzes the transfer of the acyl group from fatty acyl CoA to carnitine and constitutes the rate limiting step of FAO, with the aminocarnitine derivative ST1326 [45, 46], lipid Avocatin B [44], or etomoxir [42, 43] has shown anti-leukemic properties (Table 1).
As an inner mitochondrial membrane protein associated with the ETC, DHODH links de novo pyrimidine biosynthesis to mitochondrial bioenergetics. In this context, Sykes et al. [38] found that its inhibition with brequinar sodium (BRQ) abrogates the myeloid differentiation blockade and leads to anti-leukemic activities in a diverse range of AML subtypes. This can be rescued by addition of extracellular uridine. Very recently, two other newly developed DHODH inhibitors for AML and one for chronic myeloid leukemia (CML) have been described [39,40,41] (Fig. 2; Table 1). Although BRQ has not shown benefits in early phase clinical trials with solid cancers, it has not yet been studied in hematological malignancies [124,125,126,127,128]. Because BRQ has been shown to lead to a potent induction of myeloid differentiation and decrease leukemic burden, the role of DHODH in AML metabolism merits further study.
Another very exciting approach to trigger mitochondrial priming of cell death is through treatment with anti-apoptotic BCL2 inhibitors [52, 129] (Fig. 2). Lagadinou et al. demonstrated that LSCs are characterized by low levels of ROS. These ROS-low LSCs are dependent on OxPHOS via amino acid uptake for respiration rather than glycolysis and overexpress BCL2 anti-apoptotic proteins [47, 48]. Thus, pharmacological inhibition of BCL2 with the drug ABT-199 (venetoclax) impairs mitochondrial respiration and selectively targets ROS-low LSCs unable to switch to glycolysis/glucose or FAO to maintain energy production [47]. Clinical trials with venetoclax monotherapy in relapsed/refractory AML have shown a very low response rate due to a lack of apoptosis induction while mitochondrial priming is activated by this treatment to induce cell death. However, results from a phase 1b study in elderly patients with previously untreated AML on venetoclax treatment in combination with hypomethylating agents (azacitidine and decitabine) reported a 61% overall response [50] (Table 1). Treatment with venetoclax plus azacitidine inhibited amino acid uptake and induced disruption of the TCA cycle, inhibition of ETC complex II, and impairment of OxPHOS in ROS-low LSCs [48, 49]. Pharmacological inhibition of amino acid metabolism also decreased OxPHOS and induced cell death in AML [48] (Table 1). Previous work has suggested that FAO could be involved in BCL2 regulation and BAX- and BAK-dependent mitochondrial permeability transition pore formation through interactions between CPT1 and the pro-apoptotic BH3-only protein Bid [130] or BCL2 [131], highlighting a dual interest in FAO inhibition and synergy with BH3 mimetics in AML. Because many of the reported manipulations of metabolic pathways have been shown to modulate BCL2 expression or dependence, combinations of metabolic inhibitors and BCL2 inhibitors are of special interest. For example, statins also enhanced ABT-199 efficacy in AML through the inhibition of protein geranyl-geranylation, which leads to BCL2 modulation and upregulation of pro-apoptotic BH3 only proteins PUMA [132] and etomoxir, increasing the therapeutic efficacy of ABT-737 in vivo [43]. Very recently, a study has elegantly mapped metabolic pathways that are specifically implicated in ABT-199-induced apoptotic cell death, and demonstrated that the heme biosynthetic pathway is the major regulator of mitochondrial priming of apoptosis through ETC and OxPHOS in AML [133]. Altogether, these studies strengthen the scientific rationale for clinical development of new combinations of venetoclax and OxPHOS (or FAO) inhibitors (Table 1).
Metabolic stratification to decipher specific vulnerabilities and develop more efficient therapies in patient genetic subgroups
For diagnosis and management of AML, a prognostic stratification has been proposed based on criteria for progressive disease and for the genomic landscape of the disease [134]. However, metabolic features have not been taken into consideration yet. As more and more studies are highlighting metabolic specificities driven by mutations in AML and as specific inhibitors of some of these mutations are displaying very promising results in clinical trials, investigating the link between genetic stratification, metabolic dependencies, and response to these specific inhibitors is particularly important. This may be crucial in order to propose better combinations of these new drugs, understand mechanisms of resistance to them, and potentially identify early markers of response.
Isocitrate dehydrogenase mutations
In 2009, recurrent mutations in genes of two crucial metabolic enzymes, cytosolic isocitrate dehydrogenase (IDH)1 and mitochondrial IDH2, were observed in about 20% of AML patients [135,136,137,138], reinforcing the importance of furthering metabolic investigations in AML. While wild-type IDH (IDH WT) catalyzes the conversion of isocitrate to α-KG and generates NADPH, mutant IDH catalyzes a neomorphic enzyme activity that oxidizes NADPH and produces the oncometabolite 2-hydroxyglutarate (2-HG) from α-KG [139, 140]. The impact of monoallelic IDH mutation and the related accumulation of 2-HG have been well documented, in particular its effect on α-KG-dependent dioxygenase activity and subsequent effects on numerous cellular functions in these cancers, such as alteration of DNA and histone methylation and biased myeloid/erythroid differentiation [141,142,143,144,145,146,147,148,149,150,151,152].
Beyond epigenetic modifications and chromatin remodeling, 2-HG has multi-faceted roles in AML biology and leukemic transformation by competitively inhibiting multiple classes of αKG-dependent dioxygenases involved in metabolic reprogramming, BCL2-dependent cell survival, and cellular defense against oxidative stress. As IDH mutations are early events in oncogenesis and are systematically conserved at relapse [153, 154], IDH1/2 mutated enzymes represent attractive therapeutic targets [53, 144, 155,156,157] and small molecules selectively inhibiting the mutated forms of these enzymes have been developed and very recently approved for clinical studies [54, 56, 58,59,60,61,62] (Table 1). Both the IDH2m- and IDH1m-specific inhibitors promote differentiation and reduce methylation levels as well as significantly decreasing 2-HG levels [53, 54, 57, 157, 158]. However, while clinical trials are highly encouraging (up to 40% overall response rate in monotherapy in phase I/II for relapsed or refractory AML patients), resistance is routinely observed [54,55,56,57, 159].
Moreover, suppression of serum 2-HG levels alone did not predict response in AML patients, as non-responders also displayed a significant decrease in the amount of 2-HG [54, 55, 57, 160, 161]. Thus, targeting IDH mutant activity alone is not sufficient to achieve a durable clinical response in relapsed AML and new combinatory approaches need to be designed. Given the crucial roles of wild type IDH1/2 in cell metabolism (e.g. Krebs cycle, OxPHOS, cytosolic and mitochondrial redox, anabolism including lipid biosynthesis), a better understanding of the contribution of oncogenic IDH mutations to AML cell intermediary metabolism and α-KG homeostasis is expected to lead to new therapeutic strategies.
Because α-KG is the direct precursor of 2-HG, various studies have investigated the glutaminolysis pathway in IDH mutant cells and reported that glutamine was indeed the main source of 2-HG production [139, 162]. Therefore, inhibition of glutaminolysis with different glutaminase inhibitors (BPTES, CB-839) has shown higher in vitro anti-leukemic activities in IDH mutant cells than in IDH wild-type cells [28, 114], in line with the results obtained in gliomas [163]. However, although CB-839 clinical efficiency is currently being assessed in a phase 1 study in patients with AML (NCT02071927), in vivo preclinical studies have not been highly encouraging [27].
Interestingly, in IDH1 mutant glioma, 2-HG has been shown to inhibit branched-chain amino acid transaminases BCAT1 and BCAT2, which catalyze the degradation of BCAA into glutamate, increasing the dependency on glutamine to sustain glutamate and glutathione production and leading to synergy between glutaminase inhibition with CB-839 and radiation therapy [164, 165]. It would be particularly relevant to investigate BCAA in IDH mutant cells as Raffel et al. have already shown that BCAT1 mediates α-KG homeostasis in IDH WT AML and could represent a good therapeutic opportunity [166]. As demonstrated in gliomas, investigating the consequences of decreasing the BCAA pathway in IDH mutant AML and/or following treatments with IDH mutant inhibitors could pave the way toward a more efficient combinatory approach in myeloid leukemia. Furthermore, IDH mutation leads to higher mitochondrial activities in various solid cancers [162, 167,168,169], and the decreased NADPH levels associated with reduced wild-type activity in brain tumors and colorectal carcinomas [170,171,172] was partly restored by enhanced PPP activity in mutant astrocytes [173]. However, no detailed investigations of redox homeostasis in IDH mutant cells in AML have been reported to date, though Ward et al. suggested an increase in the activity of IDH wild-type enzyme may make a significant contribution to maintaining cellular and subcellular NADPH levels [140].
Key metabolic differences such as sensitivity to OxPHOS inhibitors seem to emerge in regard to cell lineage or cell types. Indeed, it has recently been reported that IDH1 mutant glioma cells were more resistant to rotenone (ETC complex I inhibitor) due to enhanced activity of pyrroline 5-carboxylate reductase 1 (PYCR1), which can oxidize NADH and produce proline as a ‘metabolic bypass’ of ETC complex I [174] (Fig. 1), while breast and colon cancer IDH1 mutant cells have been reported to be more sensitive to ETC complex I inhibition by metformin [167]. Of particular interest, overall response to a combination of venetoclax with azacitidine increased to 33% in IDH mutant subgroups of AML patients [15]. Chan et al. observed that (R)-2-HG inhibited cytochrome c oxidase activity (ETC complex IV), increasing the dependence on BCL2, and this led to higher sensitivity to ABT-199 in AML primary cells with an IDH mutation [175]. Notably, they observed a partial rescue of ABT-199 sensitivity with addition of specific IDH mutant inhibitors, which lower 2-HG levels [175].
FMS-like tyrosine kinase 3 mutations
FMS-like tyrosine kinase 3 (FLT3) mutations, predominantly including internal tandem duplication defect (FLT3-ITD), are found in 30% of AML patients and confer a poor prognosis with enhanced relapse rate [176,177,178,179]. Clinical success of tyrosine kinase inhibitors (TKIs) against the oncogenic kinase BCR-ABL for CML treatment raised great expectations for FLT3 inhibitors in AML. However, although the initial response to monotherapy was promising (44% response in FLT3-ITD patients with relapsed/refractory AML treated with AC220, quizartinib [180, 181]), this did not result in prolonged disease-free survival [182]. The necessity to find new combinations has thus become apparent, underscoring the importance of better understanding FLT3-ITD specificities and linking this with inhibitor resistance (Table 1). Ju et al. first compared murine BaF3 cells with BaF3 cells overexpressing FLT3-ITD and observed enhanced glycolytic activity in FLT3-ITD cells, which was associated with higher phosphorylation of HK2 localized preferentially to mitochondria, favoring ATP transfer from OxPHOS to promote glycolysis. This also provides mitochondrial protection against mitochondrial death pathways by preventing opening of the mitochondrial permeability transition pore. Thus, a combination of glycolytic inhibitors with FLT3-ITD inhibitors produced encouraging results in vivo [24, 25], corroborating previous observations about 2-DG antileukemic activity in AML with FLT3-ITD or KIT mutations through glycosylation of oncogenic proteins [23].
Gregory et al. performed a synthetic lethality screen in AML cell line MOLM13 harboring a FLT3-ITD mutation and found that a number of the genes able to sensitize AML FLT3-ITD cells to FLT3 inhibitors were involved in metabolic processes [183], in particular the ataxia telangiectasia mutated (ATM) gene shown to activate G6PD to maintain redox homeostasis [184]. Furthermore, while AC220 treatment largely reverses the glycolytic phenotype, it also induces decreased glutathione metabolism, accumulation of mitochondrial ROS, and higher mitochondrial membrane potential, leading to an increased dependency on glutamine uptake to compensate. Thus, while not conveying benefit alone, AC220 efficacy in FLT3-ITD AML in vivo was increased by the addition of OxPHOS inhibitors or glutaminase inhibitors [51, 183, 185,186,187].
While described in many cancer types as a key deregulated metabolic pathway and promising therapeutic target [188,189,190,191,192], one-carbon metabolism in myeloid leukemia remains mostly unexplored. One carbon metabolism plays a crucial role in nucleotide synthesis, methylation processes, and redox homeostasis. Serine availability resulting from both increased uptake and de novo synthesis also appears to be a key player in tumorigenesis for various cancers [188, 193,194,195] but, to date, has not been reported in myeloid leukemia. However, Pikman et al. demonstrated that inhibition of methylenetetrahydrofolate dehydrogenase-cyclohydrolase 2 (MTHFD2) decreased AML growth, in particular in the FLT3-ITD subgroup [196]. MTHFD2 catalyzes the mitochondrial conversion of methylene-THF to formyl-THF using either NAD+ or NADP+ and is thus involved in purine biosynthesis, OxPHOS, redox homeostasis, and lipogenesis (Fig. 1).
Interestingly, an increasing number of studies focus on using current preclinical and clinical trials of these new drugs to better define their mechanisms of action and propose combinations with already FDA-approved treatments. In this context, the relevance of combining IDH mutant inhibitors with inhibition of oncogenic kinase signaling using TKIs has been demonstrated in two studies in AML [197, 198]. In one hand, Shih et al. have shown that combination of AC220 with the IDH2 mutant inhibitor AG-221 promotes better recovery of normal hematopoiesis and a reduction in mutant allele burden, targeting the mutant clone in vivo in Idh2R140QFlt3ITD AML mice [197]. On the other hand, Chen et al. recently pinpointed that both FLT3 WT and FLT3-ITD mutation increased the activity of IDH1 mutant AMLs through the activation of JAK2 by phosphorylation, providing a clinical rationale to combine FLT3 inhibitor and IDH1 mutant inhibitor regardless of FLT3 mutational status [198].
Other AML patient mutational and cytogenetic subgroups
Surprisingly, metabolic dysregulation and/or specific biochemical characteristics are almost completely unknown in other karyotype and mutational patient subgroups with adverse risks, such as patients with p53, RAS, or CEBPα mutations, or monosomic complex karyotypes in AML. Notably, Fenouille et al. have shown that mitochondrial function was specifically driven by the creatine kinase pathway in the EVI1 subgroup of patients associated with poor prognosis [31]. EVI1 represses the myeloid differentiation regulator RUNX1, thus promoting expression of creatine kinase mitochondrial 1 (CKMT1). CKMT1 contributes to the conversion of arginine into creatinine. Pharmacological inactivation or genetic invalidation of CKMT1 abrogates ATP production and mitochondrial respiration, decreases viability of EVI1 AML, and prolongs the survival of the mice engrafted with high EVI1-expressing AML cells compared to xenograft with low EVI1-expressing AML cells. These observations highlight the therapeutic potential of targeting metabolic dependency specific to this EVI1 patient subgroup and show the necessity of identifying specific liabilities to achieve the best clinical outcome (Table 1).
Current limitations in cancer metabolism studies and metabolism-based therapeutic strategies
Over the last 10 years, a number of increasing concerns emerged in cancer (metabolism) research about 1) reproducibility of published data [199,200,201], 2) differences of efficacy between in vitro and in vivo studies [93, 94, 202, 203] and 3) high attrition rates for cancer drugs [200, 204]. The models to use, the culture conditions, and the experimental design are undoubtedly at the heart of these discussions.
The example of striking discrepancies in anticancer efficacy of glutaminase inhibitor CB839 observed in vitro and in vivo highlights the crucial importance of tumor cell environment. Indeed, human non-small cell lung cancer (NSCLC) cells exhibit high sensitivity to CB839 treatment and displayed enhanced glutamine catabolism in vitro, while resistance to this inhibitor was observed in vivo [94]. Isotopic profiling experiments using 13C-glucose and/or 13C-glutamine performed in vivo in mouse KRAS-driven NSCLC and directly in patients using intraoperative 13C-glucose infusions revealed NSCLC tumors rely much more on glucose than on glutamine for TCA cycle anaplerosis in vivo [93, 94, 202]. Interestingly, Muir et al. cultured NSCLC cells in adult bovine serum, a medium in which component concentrations are much closer to in vivo models. In this culture medium, they observed that glutamine contribution to TCA was significantly lower compared to the classic in vitro conditions using fetal bovine serum, and thus comparable to in vivo data on glutamine metabolism and response to CB839 [203]. They went further, demonstrating that these differences relied on the level of a single nutrient, cystine (the oxidized dimer of the amino acid cysteine), present in classic in vitro conditions in concentrations 100-fold higher than in in vivo conditions. As the cystine level regulates glutamate export through the cystine/glutamate antiporter xCT, high levels of cystine in vitro lead to an increased export of intracellular glutamate and therefore a higher dependence on glutaminase activity to maintain glutamate level, and thus ultimately to enhanced sensitivity to CB839. Accordingly, these in vitro observations were not translatable to mouse and patient models [203]. These crucial studies highlighted the importance of taking into account how nutrient conditions can impact cell metabolism and response to therapies.
In this same vein, various efforts have been made to develop media with nutrient levels closer to those found in human serum such as human plasma-like medium (HPLM) [205] and Plasmax [206]. Cultures with these two media revealed that nutrient compositions of routinely used culture media can induce metabolic dependencies and rewiring that are not observed in vivo. One example of this is that growth of cancer and AML cells in HPLM containing human plasma levels of uric acid led to the inhibition of de novo pyrimidine synthesis. Indeed, uric acid is tenfold higher in human blood than in culture media and mice serum and can inhibit uridine monophosphate synthase (UMPS), and consequently reduces the sensitivity of cancer cells to the chemotherapeutic agent 5-fluorouracil [205]. Finally, large-scale RNAi and CRISPR screens are powerful tools to identify metabolic genes essential for cancer/AML cell proliferation and response to therapies. However, metabolic gene essentiality depends on cell culture medium, which is the major confounding factor affecting the reproducibility of such approaches [207]. This should especially be accounted for when investigating metabolic abnormalities in the context of tumor metabolic heterogeneity and to develop more effective metabolism-focused treatment strategies.
The above-mentioned studies indicate the importance of addressing metabolic reprogramming in the context of the microenvironment and developing combinatory therapeutic strategies. Directly linked to nutrient amounts and substrate availability in the niche, the notion of crosstalk between cancer cells and their neighbors should be taken into account. As we briefly mentioned in the previous section, MSCs and adipocytes have been shown to participate in and modulate the response to several therapies in AML, in particular through nutrient and metabolite releases or transfers. Co-cultures of AML cells with MSCs or with bone marrow adipocytes significantly reduced the sensitivity to CPT1a inhibitors [43, 208], reinforcing the major role of the microenvironment in sustaining energetic and anabolic demands. Notably, Tabe et al. reported that inhibition of CPT1a in AML increases free fatty acids and glucose uptake only in bone marrow adipocyte co-cultures, allowing blasts to preserve their viability [208].
A consideration of tumor metabolic systems biology is also allowing a better understanding of metabolic regulation, substrate utilization, and energy balance in whole organisms and will ultimately lead to better therapeutic strategies. Interestingly, AML cells were recently shown to hijack systemic glucose metabolism, inducing an insulin resistance with aberrant homeostasis in adipose tissues, pancreas, gut, and microbiota to desensitize normal tissues to glucose and support their own growth [209]. This study strongly suggests that 1) AML cells have a parasitic behavior in systemic host metabolism and that 2) organismal metabolic status is a key component of cancer/AML progression. Accordingly, recent studies have shown that nutrient availability from the environment/host, dietary regimens, and hormonal status can affect host insulin homeostasis and cancer cell metabolism to enhance drug efficacy [210, 211]. Moreover, non-cell autonomous autophagy, also called secretory autophagy (i.e., autophagy of cells in the tumor microenvironment), has recently been implicated in cancer metabolism by providing nutrients required to support anabolic cell growth and to satisfy cell demands in vivo for proliferation [212,213,214].
Finally, chemoresistance is the main cause of poor prognosis in AML patients and assessing the metabolic reprogramming of resistant LSCs after conventional chemotherapy or new treatments is an area of intensive research. A crucial point is that cells at relapse have been shown to be dramatically different in terms of phenotype or metabolism [48, 108, 215, 216]. Strikingly, Jones et al. have reported fundamental differences between therapy-naïve LSCs and LSCs at relapse [48]. As mentioned previously, they demonstrated that naïve LSCs are more dependent on amino acid uptake for OxPHOS maintenance and cannot up-regulate FA metabolism to preserve TCA cycle fueling in the absence of amino acids. However, they indicated that LSCs from relapse patients after conventional chemotherapy exhibit a new ability to compensate amino acid loss by enhancing FAO [48]. This could explain the decreased overall response to a combination of venetoclax with azacitidine in clinical trials for relapsed patients [217] compared to previously untreated patients [50]. Therefore, if de novo AML LSCs seem to be metabolically inflexible, at least regarding OxPHOS dependency, the ones resistant to chemotherapy and contributing to relapse are AML cells able to acquire metabolic flexibility and adapt [48, 218].
Altogether, these studies highlight the importance of better defining, better characterizing, and better designing our in vitro and preclinical studies as cell culture medium composition can significantly affect the response to metabolic pathway inhibition. Interestingly, comparisons between classic in vitro and more physiological medium also led to understanding some tumor metabolic specificities and dependencies and to propose new combinations of standard chemotherapeutic treatment or newly FDA-approved targeted therapies with metabolism-based drugs. Such studies should be conducted in AML and could, at least in part, explain the unsuccessful clinical translation of glutaminase or metabolic inhibitors, even though they displayed promising results in vitro and even in some studies in mice. These also show two major points in studying metabolic reprogramming to identify efficient clinical targets: 1) understanding metabolic cooperation, competition and symbiosis in the tumor microenvironment/niche is fundamental to tackling flexibility; and 2) primary tumor cell culture conditions impose critical experimental limitations to the study of cancer.
Conclusion and perspectives
In the past decade, tremendous research efforts have uncovered key metabolic specificities and Achilles heels of cancer cells, including AML cells. These studies strongly suggest that myeloid leukemias are metabolic disorders and should be regarded in this light for metabolic-based personalized medicine treatments as well as for monitoring clinical responses to treatment. Several studies have further shown that AML cells, like other normal and cancer cells, are able to undergo compensatory metabolic and energetic adaptations in response to the inhibition of metabolic pathways, indicating that AML cells display complex metabolic capacities and flexibility that limit sustained drug efficacy, especially when challenged by chemotherapeutic drugs. However, targeting metabolic flexibility per se is not a feasible approach. By contrast, non-exclusive therapeutic strategies, which impede this metabolic flexibility by targeting its consequence(s), such as mitochondrial dependency, blocking the utilization of nutrients from the microenvironment, and/or targeting metabolic checkpoints, are emerging. Most of the metabolic pathways described in this review also occur in normal cells, although they are frequently less active, making the determination of the right therapeutic window difficult. Thus, if we are able to distinguish particular requirements of cancer cells to take up and utilize or eliminate certain metabolites, specifically targeting these exchanges may provide more effective treatment strategies. Finally, as already described in several solid tumors, an in vitro examination of metabolic flux networks does not reflect what occurs in situ, in vivo, and in patients due mainly to the enormous plasticity and heterogeneity of their metabolism [219, 220, 202]. AML, in common with many tumors, is highly genetically heterogeneous and its metabolism should be directly studied in patients in situ.
Availability of data and materials
Not applicable.
Abbreviations
- 2-DG:
-
2-Deoxyglucose
- 2-HG:
-
2-Hydroxyglutarate
- 6PGD:
-
6-Phosphogluconate dehydrogenase
- α-KG:
-
α-Ketoglutarate
- ACC2:
-
Acetyl-CoA carboxylase 2
- Akt:
-
Protein kinase B
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- AMPK:
-
5' Adenosine monophosphate-activated protein kinase
- ASNS:
-
L-asparagine synthetase
- ASS1:
-
Arginosuccinate synthetase-1
- ATM:
-
Ataxia telangiectasia mutated
- ATP:
-
Adenosine triphosphate
- BAK:
-
Bcl-2 homologous antagonist/killer
- BAX:
-
Bcl-2-associated X protein
- BCAA:
-
Branched-chain amino acid
- BCAT1/2:
-
Branched-chain amino acid transaminases
- BCL2:
-
B-cell lymphoma 2
- BH3:
-
Bcl-2 homology domain 3
- BRQ:
-
Brequinar sodium
- CKMT1:
-
Creatine kinase mitochondrial 1
- CML:
-
Chronic myeloid leukemia
- CN-AML:
-
Cytogenetically normal AML
- CPT1:
-
Carnitine palmitoyltransferase 1
- DHODH:
-
Dihydro-orotate dehydrogenase
- DHO:
-
Dihydro-orotate
- ETC:
-
Electron transfer chain
- EVI1:
-
Ecotropic virus integration site 1 protein homolog
- FADH2 :
-
Flavin adenine dinucleotide (hydroquinone form)
- FAO:
-
Fatty acid oxidation
- FASN:
-
Fatty acid synthase
- FDA:
-
Food and Drug Administration
- FLT3:
-
FMS-like tyrosine kinase 3
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- GBE1:
-
Glycogen branching enzyme
- GYS1/2:
-
Glycogen synthase 1/2
- HMG-CoA:
-
3-Hydroxy-3-methylglutaryl-coenzyme A
- HPLM:
-
Human plasma-like medium
- IDH:
-
Isocitrate dehydrogenase
- JAK2:
-
Janus kinase 2 non-receptor tyrosine kinase
- LDHA:
-
Lactate dehydrogenase A
- LKB1:
-
Liver kinase B1 protein
- LSCs:
-
Leukemic stem cell
- MDS:
-
Myelodysplastic syndrome
- MSC:
-
Mesenchymal stromal cell
- mtDNA:
-
Mitochondrial DNA
- mTORC1:
-
Mammalian target of rapamycin complex 1 protein
- MTHF2:
-
Methylenetetrahydrofolate dehydrogenase-cyclohydrolase 2
- NADH:
-
Nicotinamide adenine dinucleotide
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NSCLC:
-
Non-small cell lung cancer
- OxPHOS:
-
Oxidative phosphorylation
- PC:
-
Pyruvate carboxylase
- PDH:
-
Pyruvate dehydrogenase
- PHD3:
-
Prolyl-hydroxylase 3
- PI3K:
-
Phosphoinositide 3-kinase
- PKM2:
-
Pyruvate kinase PKM
- PPP:
-
Pentose phosphate pathway
- PUMA:
-
p53 upregulated modulator of apoptosis
- PYCR1:
-
Pyrroline 5-carboxylate reductase 1
- ROS:
-
Reactive oxygen species
- SCD1:
-
Stearoyl CoA desaturase 1
- TCA:
-
Tricarboxylic acid cycle or Krebs cycle
- TK:
-
Tyrosine kinase
- TKI:
-
Tyrosine kinase inhibitor
- UMPS:
-
Uridine monophosphate synthase
- WT:
-
Wild type
References
Montalban-Bravo G, Garcia-Manero G. Novel drugs for older patients with acute myeloid leukemia. Leukemia. 2015;29:760–9. https://doi.org/10.1038/leu.2014.244.
Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2016;127:53–61. https://doi.org/10.1182/blood-2015-08-604520.
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–52. https://doi.org/10.1056/NEJMra1406184.
Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Sullivan LB, Gui DY, Vander HMG. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer. 2016;16:680–93. https://doi.org/10.1038/nrc.2016.85.
Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. https://doi.org/10.1016/j.cmet.2015.12.006.
Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–30. https://doi.org/10.1085/JGP.8.6.519.
Warburg O. On the origin of cancer cells. Science. 1956;123:309–14 https://doi.org/10.1126/science.123.3191.309.
Herst PM, Howman RA, Neeson PJ, Berridge MV, Ritchie DS. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. J Leukoc Biol. 2011;89:51–5. https://doi.org/10.1189/jlb.0710417.
Chen W-L, Wang J-H, Zhao A-H, Xu X, Wang Y-H, Chen T-L, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood. 2014;124:1645–54. https://doi.org/10.1182/blood-2014-02-554204.
Wang Y-H, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158:1309–23. https://doi.org/10.1016/j.cell.2014.07.048.
Poulain L, Sujobert P, Zylbersztejn F, Barreau S, Stuani L, Lambert M, et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia. 2017;31:2326–35. https://doi.org/10.1038/leu.2017.81.
Bhanot H, Weisberg EL, Reddy MM, Nonami A, Neuberg D, Stone RM, et al. Acute myeloid leukemia cells require 6-phosphogluconate dehydrogenase for cell growth and NADPH-dependent metabolic reprogramming. Oncotarget. 2017;8:67639–50. https://doi.org/10.18632/oncotarget.18797.
Lin R, Elf S, Shan C, Kang H-B, Ji Q, Zhou L, et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1–AMPK signalling. Nat Cell Biol. 2015;17:1484–96. https://doi.org/10.1038/ncb3255.
Elf S, Lin R, Xia S, Pan Y, Shan C, Wu S, et al. Targeting 6-phosphogluconate dehydrogenase in the oxidative PPP sensitizes leukemia cells to antimalarial agent dihydroartemisinin. Oncogene. 2017;36:254–62. https://doi.org/10.1038/onc.2016.196.
Akers LJ, Fang W, Levy AG, Franklin AR, Huang P, Zweidler-McKay PA. Targeting glycolysis in leukemia: a novel inhibitor 3-BrOP in combination with rapamycin. Leuk Res. 2011;35:814–20. https://doi.org/10.1016/j.leukres.2010.12.028.
Tabe Y, Tafuri A, Sekihara K, Yang H, Konopleva M. Inhibition of mTOR kinase as a therapeutic target for acute myeloid leukemia. Expert Opin Ther Targets. 2017;21:705–14. https://doi.org/10.1080/14728222.2017.1333600.
Carneiro BA, Kaplan JB, Altman JK, Giles FJ, Platanias LC. Targeting mTOR signaling pathways and related negative feedback loops for the treatment of acute myeloid leukemia. Cancer Biol Ther. 2015;16:648–56. https://doi.org/10.1080/15384047.2015.1026510.
Recher C, Beyne-Rauzy O, Demur C, Chicanne G, Dos Santos C, VM-D M, et al. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood. 2005;105:2527–34. https://doi.org/10.1182/blood-2004-06-2494.
Willems L, Chapuis N, Puissant A, Maciel TT, Green AS, Jacque N, et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia. 2012;26:1195–202. https://doi.org/10.1038/leu.2011.339.
Tamburini J, Green AS, Bardet V, Chapuis N, Park S, Willems L, et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood. 2009;114:1618–27. https://doi.org/10.1182/blood-2008-10-184515.
Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL, et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene. 2011;30:1855–67. https://doi.org/10.1038/onc.2010.561.
Larrue C, Saland E, Vergez F, Serhan N, Delabesse E, Mansat-De Mas V, et al. Antileukemic activity of 2-deoxy-D-glucose through inhibition of N-linked glycosylation in acute myeloid leukemia with FLT3-ITD or c-KIT mutations. Mol Cancer Ther. 2015;14:2364–73. https://doi.org/10.1158/1535-7163.MCT-15-0163.
Ju H-Q, Zhan G, Huang A, Sun Y, Wen S, Yang J, et al. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia. 2017;31:2143–50. https://doi.org/10.1038/leu.2017.45.
Huang A, Ju H-Q, Liu K, Zhan G, Liu D, Wen S, et al. Metabolic alterations and drug sensitivity of tyrosine kinase inhibitor resistant leukemia cells with a FLT3/ITD mutation. Cancer Lett. 2016;377:149–57. https://doi.org/10.1016/J.CANLET.2016.04.040.
Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. 2015;126:1346–56. https://doi.org/10.1182/blood-2015-01-621870.
Wang ES, Frankfurt O, Orford KW, Bennett M, Flinn IW, Maris M, et al. Phase 1 study of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase in patients with relapsed/refractory leukemia. Blood. 2015;126 http://www.bloodjournal.org/content/126/23/2566.
Emadi A, Jun SA, Tsukamoto T, Fathi AT, Minden MD, Dang CV. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp Hematol. 2014;42:247–51. https://doi.org/10.1016/j.exphem.2013.12.001.
Miraki-Moud F, Ghazaly E, Ariza-McNaughton L, Hodby KA, Clear A, Anjos-Afonso F, et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood. 2015;125:4060–8. https://doi.org/10.1182/blood-2014-10-608133.
Tsai H-J, Jiang SS, Hung W-C, Borthakur G, Lin S-F, Pemmaraju N, et al. A phase II study of arginine deiminase (ADI-PEG20) in relapsed/refractory or poor-risk acute myeloid leukemia patients. Sci Rep. 2017;7:11253. https://doi.org/10.1038/s41598-017-10542-4.
Fenouille N, Bassil CF, Ben-Sahra I, Benajiba L, Alexe G, Ramos A, et al. The creatine kinase pathway is a metabolic vulnerability in EVI1-positive acute myeloid leukemia. Nat Med. 2017;23:301–13. https://doi.org/10.1038/nm.4283.
Škrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–88. https://doi.org/10.1016/j.ccr.2011.10.015.
Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, et al. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2015;27:864–76. https://doi.org/10.1016/j.ccell.2015.05.004.
Liyanage SU, Hurren R, Voisin V, Bridon G, Wang X, Xu C, et al. Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML. Blood. 2017;129:2657–66. https://doi.org/10.1182/blood-2016-10-741207.
Scotland S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia. 2013;27:2129–38. https://doi.org/10.1038/leu.2013.107.
Green AS, Chapuis N, Maciel TT, Willems L, Lambert M, Arnoult C, et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood. 2010;116:4262–73. https://doi.org/10.1182/blood-2010-02-269837.
Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24:1036–46. https://doi.org/10.1038/s41591-018-0052-4.
Sykes DB, Kfoury YS, Mercier FE, Wawer MJ, Law JM, Haynes MK, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167:171–186.e15. https://doi.org/10.1016/j.cell.2016.08.057.
Ladds MJGW, van Leeuwen IMM, Drummond CJ, Chu S, Healy AR, Popova G, et al. A DHODH inhibitor increases p53 synthesis and enhances tumor cell killing by p53 degradation blockage. Nat Commun. 2018;9:1107. https://doi.org/10.1038/s41467-018-03441-3.
Wu D, Wang W, Chen W, Lian F, Lang L, Huang Y, et al. Pharmacological inhibition of dihydroorotate dehydrogenase induces apoptosis and differentiation in acute myeloid leukemia cells. Haematologica. 2018;103:1472–83. https://doi.org/10.3324/haematol.2018.188185.
Cao L, Weetall M, Trotta C, Cintron K, Ma J, Kim MJ, et al. Targeting of hematologic malignancies with PTC299, a novel potent inhibitor of dihydroorotate dehydrogenase with favorable pharmaceutical properties. Mol Cancer Ther. 2019;18:3–16. https://doi.org/10.1158/1535-7163.MCT-18-0863.
Estañ MC, Calviño E, Calvo S, Guillén-Guío B, Boyano-Adánez Mdel C, de Blas E, et al. Apoptotic efficacy of etomoxir in human acute myeloid leukemia cells. Cooperation with arsenic trioxide and glycolytic inhibitors, and regulation by oxidative stress and protein kinase activities. PLoS One. 2014;9:e115250. https://doi.org/10.1371/journal.pone.0115250.
Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–56. https://doi.org/10.1172/JCI38942.
Lee EA, Angka L, Rota S-G, Hanlon T, Mitchell A, Hurren R, et al. Targeting mitochondria with avocatin B induces selective leukemia cell death. Cancer Res. 2015;75:2478–88. https://doi.org/10.1158/0008-5472.CAN-14-2676.
Samudio I, Konopleva M. Targeting leukemia’s “fatty tooth.”. Blood. 2015;126:1874–5. https://doi.org/10.1182/BLOOD-2015-08-665125.
Ricciardi MR, Mirabilii S, Allegretti M, Licchetta R, Calarco A, Torrisi MR, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015;126:1925–9. https://doi.org/10.1182/BLOOD-2014-12-617498.
Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–41. https://doi.org/10.1016/j.stem.2012.12.013.
Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell. 2018;34:724–740.e4. https://doi.org/10.1016/j.ccell.2018.10.005.
Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24:1859–66. https://doi.org/10.1038/s41591-018-0233-1.
DiNardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19:216–28. https://doi.org/10.1016/S1470-2045(18)30010-X.
Mali RS, Lasater EA, Doyle K, Malla R, Boghaert E, Souers A, et al. FLT3-ITD activation mediates resistance to the BCL-2 selective antagonist, venetoclax, in FLT3-ITD mutant AML models. Blood. 2017;130:1348.
Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6:1106–17. https://doi.org/10.1158/2159-8290.CD-16-0313.
Yen K, Travins J, Wang F, David MD, Artin E, Straley K, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7:478–93. https://doi.org/10.1158/2159-8290.CD-16-1034.
Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–31. https://doi.org/10.1182/blood-2017-04-779405.
Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130:732–41. https://doi.org/10.1182/blood-2017-04-779447.
DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386–98. https://doi.org/10.1056/NEJMoa1716984.
DiNardo C, de Botton S, Pollyea DA, Stein EM, Fathi AT, Roboz GJ, et al. Molecular profiling and relationship with clinical response in patients with IDH1 mutation-positive hematologic malignancies receiving AG-120, a first-in-class potent inhibitor of mutant idh1, in addition to data from the completed Dose Escalation Portion of the Phase 1 Study. Blood. 2015;126:1306 http://www.bloodjournal.org/content/126/23/1306.
Chaturvedi A, Herbst L, Pusch S, Klett L, Goparaju R, Stichel D, et al. Pan-mutant-IDH1 inhibitor BAY1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Leukemia. 2017;31:2020–8. https://doi.org/10.1038/leu.2017.46.
Heuser M, Herbst L, Pusch S, Klett L, Goparaju R, Stichel D, et al. Pan-mutant-IDH1 inhibitor Bay-1436032 is highly effective against human IDH1 mutant acute myeloid leukemia in vivo. Blood. 2016;128:2020–8 http://www.bloodjournal.org/content/128/22/745.
Chaturvedi A, Gupta C, Goparaju R, Gabdoulline R, Kaulfuss S, Görlich K, et al. Synergistic activity of IDH1 inhibitor Bay-1436032 with azacitidine in IDH1 mutant acute myeloid leukemia. Blood. 2017;130(Suppl 1) http://www.bloodjournal.org/content/130/Suppl_1/1352.
DiNardo CD, Schimmer AD, Yee KWL, Hochhaus A, Kraemer A, Carvajal RD, et al. A phase I study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations. Blood. 2016;128 http://www.bloodjournal.org/content/128/22/1073?sso-checked=true.
Yen K, Konteatis Z, Sui Z, Artin E, Dang L, Straley K, et al. Abstract B126: AG-881, a brain penetrant, potent, pan-mutant IDH (mIDH) inhibitor for use in mIDH solid and hematologic malignancies. In: Therapeutic agents: Other topics: American Association for Cancer Research; 2018. p. B126–6. https://doi.org/10.1158/1535-7163.TARG-17-B126.
Stirewalt DL, Appelbaum FR, Willman CL, Zager RA, Banker DE. Mevastatin can increase toxicity in primary AMLs exposed to standard therapeutic agents, but statin efficacy is not simply associated with ras hotspot mutations or overexpression. Leuk Res. 2003;27:133–45. https://doi.org/10.1016/S0145-2126(02)00085-1.
Lishner M, Bar-Sef A, Elis A, Fabian I. Effect of simvastatin alone and in combination with cytosine arabinoside on the proliferation of myeloid leukemia cell lines. J Investig Med. 2001;49:319–24. https://doi.org/10.2310/6650.2001.33896.
Holstein SA, Hohl RJ. Interaction of cytosine arabinoside and lovastatin in human leukemia cells. Leuk Res. 2001;25:651–60. https://doi.org/10.1016/S0145-2126(00)00162-4.
Kornblau SM, Banker DE, Stirewalt D, Shen D, Lemker E, Verstovsek S, et al. Blockade of adaptive defensive changes in cholesterol uptake and synthesis in AML by the addition of pravastatin to idarubicin + high-dose Ara-C: a phase 1 study. Blood. 2007;109:2999–3006. https://doi.org/10.1182/blood-2006-08-044446.
Advani AS, Li H, Michaelis LC, Medeiros BC, Liedtke M, List AF, et al. Report of the relapsed/refractory cohort of SWOG S0919: A phase 2 study of idarubicin and cytarabine in combination with pravastatin for acute myelogenous leukemia (AML). Leuk Res. 2018;67:17–20. https://doi.org/10.1016/J.LEUKRES.2018.01.021.
Southam AD, Khanim FL, Hayden RE, Constantinou JK, Koczula KM, Michell RH, et al. Drug redeployment to kill leukemia and lymphoma cells by disrupting SCD1-mediated synthesis of monounsaturated fatty acids. Cancer Res. 2015;75:2530–40. https://doi.org/10.1158/0008-5472.CAN-15-0202.
Khanim FL, Hayden RE, Birtwistle J, Lodi A, Tiziani S, Davies NJ, et al. Combined bezafibrate and medroxyprogesterone acetate: Potential novel therapy for acute myeloid leukaemia. PLoS One. 2009;4:e8147. https://doi.org/10.1371/journal.pone.0008147.
Murray JA, Khanim FL, Hayden RE, Craddock CF, Holyoake TL, Jackson N, et al. Combined bezafibrate and medroxyprogesterone acetate have efficacy without haematological toxicity in elderly and relapsed acute myeloid leukaemia (AML). Br J Haematol. 2010;149:65–9. https://doi.org/10.1111/j.1365-2141.2009.08055.x.
Mirabilii S, Ricciardi M, Piedimonte M, Gianfelici V, Bianchi M, Tafuri A, et al. Biological aspects of mTOR in leukemia. Int J Mol Sci. 2018;19:2396. https://doi.org/10.3390/ijms19082396.
Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood. 2013;122:3521–32. https://doi.org/10.1182/blood-2013-03-493163.
Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan K-L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–45. https://doi.org/10.1038/ncb1753.
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–34. https://doi.org/10.1016/J.CELL.2008.11.044.
Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678–84. https://doi.org/10.1172/JCI69600.
Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–34. https://doi.org/10.1038/nrc.2016.71.
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–50. https://doi.org/10.1073/pnas.0709747104.
Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. 2017;168:657–69. https://doi.org/10.1016/j.cell.2016.12.039.
Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17:351–9. https://doi.org/10.1038/ncb3124.
Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature. 2015;527:379–83. https://doi.org/10.1038/nature15529.
Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol. 2017;24:1161–80. https://doi.org/10.1016/j.chembiol.2017.08.028.
Currie E, Schulze A, Zechner R, Walther TC, Farese RV. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–61. https://doi.org/10.1016/j.cmet.2013.05.017.
Pizer ES, Wood FD, Pasternack GR, Kuhajda FP. Fatty acid synthase (FAS): a target for cytotoxic antimetabolites in HL60 promyelocytic leukemia cells. Cancer Res. 1996;56:745–51 http://www.ncbi.nlm.nih.gov/pubmed/8631008.
Reed MAC, Ludwig C, Bunce CM, Khanim FL, Günther UL. Malonate as a ROS product is associated with pyruvate carboxylase activity in acute myeloid leukaemia cells. Cancer Metab. 2016;4:15. https://doi.org/10.1186/s40170-016-0155-7.
Fedotcheva NI, Sokolov AP, Kondrashova MN. Nonezymatic formation of succinate in mitochondria under oxidative stress. Free Radic Biol Med. 2006;41:56–64. https://doi.org/10.1016/J.FREERADBIOMED.2006.02.012.
Tiziani S, Lodi A, Khanim FL, Viant MR, Bunce CM, Günther UL. Metabolomic profiling of drug responses in acute myeloid leukaemia cell lines. PLoS One. 2009;4:e4251. https://doi.org/10.1371/journal.pone.0004251.
Kimura T, Hauber J, Singer TP. Studies on succinate dehydrogenase. 13. Reversible activation of the mammalian enzyme. J Biol Chem. 1967;242:4987–93 http://www.ncbi.nlm.nih.gov/pubmed/6058941.
Thorn MB. Inhibition by malonate of succinic dehydrogenase in heart-muscle preparations. Biochem J. 1953;54:540–7 http://www.ncbi.nlm.nih.gov/pubmed/13058948.
Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–5. https://doi.org/10.1038/nature13909.
Phannasil P, Thuwajit C, Warnnissorn M, Wallace JC, MacDonald MJ, Jitrapakdee S. Pyruvate carboxylase is up-regulated in breast cancer and essential to support growth and invasion of MDA-MB-231 cells. PLoS One. 2015;10:e0129848. https://doi.org/10.1371/journal.pone.0129848.
Phannasil P, Ansari IH, El Azzouny M, Longacre MJ, Rattanapornsompong K, Burant CF, et al. Mass spectrometry analysis shows the biosynthetic pathways supported by pyruvate carboxylase in highly invasive breast cancer cells. Biochim Biophys Acta Mol Basis Dis. 2017;1863:537–51. https://doi.org/10.1016/J.BBADIS.2016.11.021.
Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Matés JM, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108:8674–9. https://doi.org/10.1073/pnas.1016627108.
Sellers K, Fox MP, Bousamra M, Slone SP, Higashi RM, Miller DM, et al. Pyruvate carboxylase is critical for non–small-cell lung cancer proliferation. J Clin Invest. 2015;125:687–98. https://doi.org/10.1172/JCI72873.
Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, et al. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 2016;23:517–28. https://doi.org/10.1016/j.cmet.2016.01.007.
Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, et al. Breast cancer-derived lung metastases show increased pyruvate carboxylase-dependent anaplerosis. Cell Rep. 2016;17:837–48. https://doi.org/10.1016/j.celrep.2016.09.042.
Clutterbuck RD, Millar BC, Powles RL, Newman A, Catovsky D, Jarman M, et al. Inhibitory effect of simvastatin on the proliferation of human myeloid leukaemia cells in severe combined immunodeficient (SCID) mice. Br J Haematol. 1998;102:522–7. https://doi.org/10.1046/j.1365-2141.1998.00783.x.
Cerqueira NMFSA, Oliveira EF, Gesto DS, Santos-Martins D, Moreira C, Moorthy HN, et al. Cholesterol biosynthesis: A mechanistic overview. Biochemistry. 2016;55:5483–506. https://doi.org/10.1021/acs.biochem.6b00342.
Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–37. https://doi.org/10.1038/nrc3038.
Weinhouse S, Warburg O, Burk D, Schade A. On respiratory impairment in cancer cells. Science. 1956;124:267–9. https://doi.org/10.1126/SCIENCE.124.3215.267.
Weinhouse S. Studies on the fate of isotopically labeled metabolites in the oxidative metabolism of tumors. Cancer Res. 1951;11:585–91 http://www.ncbi.nlm.nih.gov/pubmed/14859221.
Weinhouse S. Oxidative metabolism of neoplastic tissues. Adv Cancer Res. 1955;3:269–325. https://doi.org/10.1016/S0065-230X(08)60922-7.
Sriskanthadevan S, Jeyaraju DV, Chung TE, Prabha S, Xu W, Skrtic M, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood. 2015;125:2120–30. https://doi.org/10.1182/blood-2014-08-594408.
Boultwood J, Fidler C, Mills KI, Frodsham PM, Kusec R, Gaiger A, et al. Amplification of mitochondrial DNA in acute myeloid leukaemia. Br J Haematol. 1996;95:426–31. https://doi.org/10.1046/j.1365-2141.1996.d01-1922.x.
Pinton P. A mitochondrial drug to treat AML. Blood. 2017;129:2597–9. https://doi.org/10.1182/BLOOD-2017-03-773200.
Beuneu C, Auger R, Löffler M, Guissani A, Lemaire G, Lepoivre M. Indirect inhibition of mitochondrial dihydroorotate dehydrogenase activity by nitric oxide. Free Radic Biol Med. 2000;28:1206–13. https://doi.org/10.1016/S0891-5849(00)00239-2.
Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227–32. https://doi.org/10.1038/nrc3483.
Shi J, Fu H, Jia Z, He K, Fu L, Wang W. High expression of CPT1A predicts adverse outcomes: A potential therapeutic target for acute myeloid leukemia. EBioMedicine. 2016;14:55–64. https://doi.org/10.1016/j.ebiom.2016.11.025.
Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7:716–35. https://doi.org/10.1158/2159-8290.CD-16-0441.
German NJ, Yoon H, Yusuf RZ, Murphy JP, Finley LWS, Laurent G, et al. PHD3 Loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of ACC2. Mol Cell. 2016;63:1006–20. https://doi.org/10.1016/j.molcel.2016.08.014.
Cho YS, Il LJ, Shin D, Kim HT, Jung HY, Lee TG, et al. Molecular mechanism for the regulation of human ACC2 through phosphorylation by AMPK. Biochem Biophys Res Commun. 2010;391:187–92. https://doi.org/10.1016/J.BBRC.2009.11.029.
Hardie DG, Pan DA. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans. 2002;30(Pt 6):1064–70. http://www.biochemsoctrans.org/content/30/6/1064.long.
Bhanot H, Reddy MM, Nonami A, Weisberg EL, Bonal D, Kirschmeier PT, et al. Pathological glycogenesis through glycogen synthase 1 and suppression of excessive AMP kinase activity in myeloid leukemia cells. Leukemia. 2015;29:1555–63. https://doi.org/10.1038/leu.2015.46.
Chen W-L, Wang Y-Y, Zhao A, Xia L, Xie G, Su M, et al. Enhanced fructose utilization mediated by SLC2A5 is a unique metabolic feature of acute myeloid leukemia with therapeutic potential. Cancer Cell. 2016;30:779–91. https://doi.org/10.1016/j.ccell.2016.09.006.
Matre P, Velez J, Jacamo R, Qi Y, Su X, Cai T, et al. Inhibiting glutaminase in acute myeloid leukemia: metabolic dependency of selected AML subtypes. Oncotarget. 2016;7:79722–35. https://doi.org/10.18632/oncotarget.12944.
Cai T, Lorenzi PL, Rakheja D, Pontikos MA, Lodi A, Han L, et al. Gls inhibitor CB-839 modulates cellular metabolism in AML and potently suppresses AML cell growth when combined with 5-azacitidine. Blood. 2016;128:4064.
Emadi A. Exploiting AML vulnerability: glutamine dependency. Blood. 2015;126:1269–70. https://doi.org/10.1182/blood-2015-07-659508.
Akagi T, Yin D, Kawamata N, Bartram CR, Hofmann W-K, Wolf I, et al. Methylation analysis of asparagine synthetase gene in acute lymphoblastic leukemia cells. Leukemia. 2006;20:1303–6. https://doi.org/10.1038/sj.leu.2404216.
Michelozzi IM, Granata V, De Ponti G, Alberti G, Tomasoni C, Antolini L, et al. Acute myeloid leukaemia niche regulates response to L-asparaginase. Br J Haematol. 2019. https://doi.org/10.1111/bjh.15920.
Ehsanipour EA, Sheng X, Behan JW, Wang X, Butturini A, Avramis VI, et al. Adipocytes cause leukemia cell resistance to L-asparaginase via release of glutamine. Cancer Res. 2013;73:2998–3006. https://doi.org/10.1158/0008-5472.CAN-12-4402.
Dimitroulakos J, Nohynek D, Backway KL, Hedley DW, Yeger H, Freedman MH, et al. Increased sensitivity of acute myeloid leukemias to lovastatin-induced apoptosis: A potential therapeutic approach. Blood. 1999;93:1308–18.
Shadman M, Mawad R, Dean C, Chen TL, Shannon-Dorcy K, Sandhu V, et al. Idarubicin, cytarabine, and pravastatin as induction therapy for untreated acute myeloid leukemia and high-risk myelodysplastic syndrome. Am J Hematol. 2015;90:483–6. https://doi.org/10.1002/ajh.23981.
Chiche J, Reverso-Meinietti J, Mouchotte A, Rubio-Patiño C, Mhaidly R, Villa E, et al. GAPDH expression predicts the response to R-CHOP, the tumor metabolic status, and the response of DLBCL patients to metabolic inhibitors. Cell Metab. 2019. https://doi.org/10.1016/j.cmet.2019.02.002.
Wu Y, Hurren R, MacLean N, Gronda M, Jitkova Y, Sukhai MA, et al. Carnitine transporter CT2 (SLC22A16) is over-expressed in acute myeloid leukemia (AML) and target knockdown reduces growth and viability of AML cells. Apoptosis. 2015;20:1099–108. https://doi.org/10.1007/s10495-015-1137-x.
Noe DA, Rowinsky EK, Shen HS, Clarke BV, Grochow LB, McGuire WB, et al. Phase I and pharmacokinetic study of brequinar sodium (NSC 368390). Cancer Res. 1990;50:4595–9 http://www.ncbi.nlm.nih.gov/pubmed/2369734.
Burris HA, Raymond E, Awada A, Kuhn JG, O’Rourke TJ, Brentzel J, et al. Pharmacokinetic and phase I studies of brequinar (DUP 785; NSC 368390) in combination with cisplatin in patients with advanced malignancies. Invest New Drugs. 1998;16:19–27 http://www.ncbi.nlm.nih.gov/pubmed/9740540.
Arteaga CL, Brown TD, Kuhn JG, Shen HS, O’Rourke TJ, Beougher K, et al. Phase I clinical and pharmacokinetic trial of Brequinar sodium (DuP 785; NSC 368390). Cancer Res. 1989;49:4648–53 http://www.ncbi.nlm.nih.gov/pubmed/2743343.
Schwartsmann G, Dodion P, Vermorken JB, ten Bokkel Huinink WW, Joggi J, Winograd B, et al. Phase I study of Brequinar sodium (NSC 368390) in patients with solid malignancies. Cancer Chemother Pharmacol. 1990;25:345–51 http://www.ncbi.nlm.nih.gov/pubmed/2306795.
Peters GJ. Re-evaluation of Brequinar sodium, a dihydroorotate dehydrogenase inhibitor. Nucleosides Nucleotides Nucleic Acids. 2018;37:1–13. https://doi.org/10.1080/15257770.2018.1508692.
Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–32. https://doi.org/10.1038/nrc2297.
Giordano A, Calvani M, Petillo O, Grippo P, Tuccillo F, Melone MAB, et al. tBid induces alterations of mitochondrial fatty acid oxidation flux by malonyl-CoA-independent inhibition of carnitine palmitoyltransferase-1. Cell Death Differ. 2005;12:603–13. https://doi.org/10.1038/sj.cdd.4401636.
Paumen MB, Ishida Y, Han H, Muramatsu M, Eguchi Y, Tsujimoto Y, et al. Direct interaction of the mitochondrial membrane protein carnitine palmitoyltransferase I with Bcl-2. Biochem Biophys Res Commun. 1997;231:523–5. https://doi.org/10.1006/BBRC.1997.6089.
Lee JS, Roberts A, Juarez D, Vo T-TT, Bhatt S, Herzog L-O, et al. Statins enhance efficacy of venetoclax in blood cancers. Sci Transl Med. 2018;10:eaaq1240. https://doi.org/10.1126/scitranslmed.aaq1240.
Lin KH, Xie A, Rutter JC, Ahn Y, Lloyd-Cowden JM, Nichols AG, et al. Systematic dissection of the metabolic-apoptotic interface in AML reveals heme biosynthesis to be a regulator of drug sensitivity. Cell Metab. 2019. https://doi.org/10.1016/J.CMET.2019.01.011.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47. https://doi.org/10.1182/blood-2016-08-733196.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66. https://doi.org/10.1056/NEJMoa0903840.
Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116:2122–6. https://doi.org/10.1182/blood-2009-11-250878.
Marcucci G, Maharry K, Wu Y-Z, Radmacher MD, Mrózek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28:2348–55. https://doi.org/10.1200/JCO.2009.27.3730.
Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. 2010;28:3636–43. https://doi.org/10.1200/JCO.2010.28.3762.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–44. https://doi.org/10.1038/nature08617.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34. https://doi.org/10.1016/j.ccr.2010.01.020.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67. https://doi.org/10.1016/j.ccr.2010.11.015.
Turcan S, Makarov V, Taranda J, Wang Y, Fabius AWM, Wu W, et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat Genet. 2018;50:62–72. https://doi.org/10.1038/s41588-017-0001-z.
Sasaki M, Knobbe CB, Itsumi M, Elia AJ, Harris IS, Chio IIC, et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012;26:2038–49. https://doi.org/10.1101/gad.198200.112.
Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–6. https://doi.org/10.1126/science.1234769.
Chou AP, Chowdhury R, Li S, Chen W, Kim AJ, Piccioni DE, et al. Identification of retinol binding protein 1 promoter hypermethylation in isocitrate dehydrogenase 1 and 2 mutant gliomas. JNCI J Natl Cancer Inst. 2012;104:1458–69. https://doi.org/10.1093/jnci/djs357.
Guilhamon P, Eskandarpour M, Halai D, Wilson GA, Feber A, Teschendorff AE, et al. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun. 2013;4:2166. https://doi.org/10.1038/ncomms3166.
Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110–4. https://doi.org/10.1038/nature16490.
Mazor T, Chesnelong C, Pankov A, Jalbert LE, Hong C, Hayes J, et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc Natl Acad Sci U S A. 2017;114:10743–8. https://doi.org/10.1073/pnas.1708914114.
Jiang B, Zhang J, Xia J, Zhao W, Wu Y, Shi M, et al. IDH1 Mutation promotes tumorigenesis by inhibiting JNK activation and apoptosis induced by serum starvation. Cell Rep. 2017;19:389–400. https://doi.org/10.1016/j.celrep.2017.03.053.
Boutzen H, Saland E, Larrue C, de Toni F, Gales L, Castelli FA, et al. Isocitrate dehydrogenase 1 mutations prime the all-trans retinoic acid myeloid differentiation pathway in acute myeloid leukemia. J Exp Med. 2016;213:483–97. https://doi.org/10.1084/jem.20150736.
Kats LM, Reschke M, Taulli R, Pozdnyakova O, Burgess K, Bhargava P, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. 2014;14:329–41. https://doi.org/10.1016/j.stem.2013.12.016.
Losman J-A, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–5. https://doi.org/10.1126/science.1231677.
Corces-Zimmerman MR, Majeti R. Pre-leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia. 2014;28:2276–82. https://doi.org/10.1038/leu.2014.211.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–33. https://doi.org/10.1038/nature13038.
Okoye-Okafor UC, Bartholdy B, Cartier J, Gao EN, Pietrak B, Rendina AR, et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol. 2015;11:878–86. https://doi.org/10.1038/nchembio.1930.
Popovici-Muller J, Saunders JO, Salituro FG, Travins JM, Yan S, Zhao F, et al. Discovery of the first potent inhibitors of mutant IDH1 that lower tumor 2-HG in vivo. ACS Med Chem Lett. 2012;3:850–5. https://doi.org/10.1021/ml300225h.
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–30. https://doi.org/10.1126/science.1236062.
De Botton S, Pollyea DA, Stein EM. Clinical safety and activity of AG-120, a first-in-class, potent inhibitor of the IDH1 mutant protein, in a phase 1 study of patients with advanced IDH1-mutant hematologic malignancies [abstract]. Haematologica. 2015;100:214–5.
Intlekofer AM, Shih AH, Wang B, Nazir A, Rustenburg AS, Albanese SK, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature. 2018;559:125–9. https://doi.org/10.1038/s41586-018-0251-7.
Thomas D, Majeti R. Optimizing next-generation AML therapy: Activity of mutant IDH2 inhibitor AG-221 in preclinical models. Cancer Discov. 2017;7:459–61. https://doi.org/10.1158/2159-8290.CD-17-0270.
Waitkus MS, Diplas BH, Yan H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell. 2018. https://doi.org/10.1016/J.CCELL.2018.04.011.
Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014;74:3317–31. https://doi.org/10.1158/0008-5472.CAN-14-0772-T.
Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–7. https://doi.org/10.1158/0008-5472.CAN-10-1666.
McBrayer SK, Mayers JR, DiNatale GJ, Shi DD, Khanal J, Chakraborty AA, et al. Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell. 2018;175:101–16 e25. https://doi.org/10.1016/j.cell.2018.08.038
Tönjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med. 2013;19:901–8. https://doi.org/10.1038/nm.3217.
Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature. 2017;551:384–8. https://doi.org/10.1038/nature24294.
Cuyàs E, Fernández-Arroyo S, Corominas-Faja B, Rodríguez-Gallego E, Bosch-Barrera J, Martin-Castillo B, et al. Oncometabolic mutation IDH1 R132H confers a metformin-hypersensitive phenotype. Oncotarget. 2015;6:12279–96. https://doi.org/10.18632/oncotarget.3733.
Farshidfar F, Zheng S, Gingras M-C, Newton Y, Shih J, Robertson AG, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep. 2017;19:2878–80. https://doi.org/10.1016/j.celrep.2017.06.008.
Khurshed M, Molenaar RJ, Lenting K, Leenders WP, van Noorden CJF. In silico gene expression analysis reveals glycolysis and acetate anaplerosis in IDH1 wild-type glioma and lactate and glutamate anaplerosis in IDH1-mutated glioma. Oncotarget. 2017;8:49165–77. https://doi.org/10.18632/oncotarget.17106.
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–5. https://doi.org/10.1126/science.1170944.
Bleeker FE, Atai NA, Lamba S, Jonker A, Rijkeboer D, Bosch KS, et al. The prognostic IDH1 R132 mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010;119:487–94. https://doi.org/10.1007/s00401-010-0645-6.
Molenaar RJ, Botman D, Smits MA, Hira VV, van Lith SA, Stap J, et al. Radioprotection of IDH1-mutated cancer cells by the IDH1-mutant inhibitor AGI-5198. Cancer Res. 2015;75:4790–802. https://doi.org/10.1158/0008-5472.CAN-14-3603.
Gelman SJ, Naser F, Mahieu NG, McKenzie LD, Dunn GP, Chheda MG, et al. Consumption of NADPH for 2-HG synthesis increases pentose phosphate pathway flux and sensitizes cells to oxidative stress. Cell Rep. 2018;22:512–22. https://doi.org/10.1016/j.celrep.2017.12.050.
Hollinshead KER, Munford H, Eales KL, Bardella C, Li C, Escribano-Gonzalez C, et al. Oncogenic IDH1 mutations promote enhanced proline synthesis through PYCR1 to support the maintenance of mitochondrial redox homeostasis. Cell Rep. 2018;22:3107–14. https://doi.org/10.1016/j.celrep.2018.02.084.
Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong W-J, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21:178–84. https://doi.org/10.1038/nm.3788.
Estey EH. Acute myeloid leukemia: 2019 update on risk-stratification and management. Am J Hematol. 2018;93:1267–91. https://doi.org/10.1002/ajh.25214.
Moarii M, Papaemmanuil E. Classification and risk assessment in AML: integrating cytogenetics and molecular profiling. Hematol Am Soc Hematol Educ Progr. 2017;2017:37–44. https://doi.org/10.1182/asheducation-2017.1.37.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21. https://doi.org/10.1056/NEJMoa1516192.
Network TCGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74. https://doi.org/10.1056/NEJMoa1301689.
Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31:3681–7. https://doi.org/10.1200/JCO.2013.48.8783.
Levis MJ, Perl AE, Dombret H, Döhner H, Steffen B, Rousselot P, et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukemia after second-line chemotherapy or hematopoietic stem cell transpplantation. Blood. 2012;120:673 http://www.bloodjournal.org/content/120/21/673.
Pratz KW, Luger SM. Will FLT3 inhibitors fulfill their promise in acute meyloid leukemia? Curr Opin Hematol. 2014;21:72–8. https://doi.org/10.1097/MOH.0000000000000022.
Gregory MA, D’Alessandro A, Alvarez-Calderon F, Kim J, Nemkov T, Adane B, et al. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc Natl Acad Sci U S A. 2016;113:E6669–78. https://doi.org/10.1073/pnas.1603876113.
Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011;30:546–55. https://doi.org/10.1038/emboj.2010.330.
Gregory MA, Nemkov T, Reisz JA, Zaberezhnyy V, Hansen KC, D’Alessandro A, et al. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp Hematol. 2018;58:52–8. https://doi.org/10.1016/j.exphem.2017.09.007.
Alvarez-Calderon F, Gregory MA, Pham-Danis C, DeRyckere D, Stevens BM, Zaberezhnyy V, et al. Tyrosine kinase inhibition in leukemia induces an altered metabolic state sensitive to mitochondrial perturbations. Clin Cancer Res. 2015;21:1360–72. https://doi.org/10.1158/1078-0432.CCR-14-2146.
Gallipoli P, Giotopoulos G, Tzelepis K, Costa ASH, Vohra S, Medina-Perez P, et al. Glutaminolysis is a metabolic dependency in FLT3ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood. 2018;131:1639–53. https://doi.org/10.1182/blood-2017-12-820035.
Labuschagne CF, van den Broek NJF, Mackay GM, Vousden KH, Maddocks ODK. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014;7:1248–58. https://doi.org/10.1016/j.celrep.2014.04.045.
Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–83. https://doi.org/10.1038/nrc3557.
Newman AC, Maddocks ODK. One-carbon metabolism in cancer. Br J Cancer. 2017;116:1499–504. https://doi.org/10.1038/bjc.2017.118.
Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16:650–62. https://doi.org/10.1038/nrc.2016.81.
Ducker GS, Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab. 2017;25:27–42. https://doi.org/10.1016/j.cmet.2016.08.009.
Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. J Cell Biol. 2016;214:249–57. https://doi.org/10.1083/jcb.201604085.
Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature. 2017;544:372–6. https://doi.org/10.1038/nature22056.
Amelio I, Cutruzzolá F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39:191–8. https://doi.org/10.1016/J.TIBS.2014.02.004.
Pikman Y, Puissant A, Alexe G, Furman A, Chen LM, Frumm SM, et al. Targeting MTHFD2 in acute myeloid leukemia. J Exp Med. 2016;213:1285–306. https://doi.org/10.1084/jem.20151574.
Shih AH, Meydan C, Shank K, Garrett-Bakelman FE, Ward PS, Intlekofer AM, et al. Combination targeted therapy to disrupt aberrant oncogenic signaling and reverse epigenetic dysfunction in IDH2- and TET2-mutant acute myeloid leukemia. Cancer Discov. 2017;7:494–505. https://doi.org/10.1158/2159-8290.CD-16-1049.
Chen D, Xia S, Wang M, Lin R, Li Y, Mao H, et al. Mutant and wild-type isocitrate dehydrogenase 1 share enhancing mechanisms involving distinct tyrosine kinase cascades in cancer. Cancer Discov. 2019. https://doi.org/10.1158/2159-8290.CD-18-1040.
Kaiser J. Rigorous replication effort succeeds for just two of five cancer papers. Science. 2017. https://doi.org/10.1126/science.aal0628.
Begley CG, Ellis LM. Raise standards for preclinical cancer research. Nature. 2012;483:531–3. https://doi.org/10.1038/483531a.
Baker M, Dolgin E. Cancer reproducibility project releases first results. Nature. 2017;541:269–70. https://doi.org/10.1038/541269a.
Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, et al. Metabolic heterogeneity in human lung tumors. Cell. 2016;164:681–94. https://doi.org/10.1016/j.cell.2015.12.034.
Muir A, Danai LV, Gui DY, Waingarten CY, Lewis CA. Vander Heiden MG. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife. 2017;6. https://doi.org/10.7554/eLife.27713.
Hutchinson L, Kirk R. High drug attrition rates—where are we going wrong? Nat Rev Clin Oncol. 2011;8:189–90. https://doi.org/10.1038/nrclinonc.2011.34.
Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A, et al. Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell. 2017;169:258–272.e17. https://doi.org/10.1016/j.cell.2017.03.023.
Vande Voorde J, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci Adv. 2019;5:eaau7314. https://doi.org/10.1126/sciadv.aau7314.
Lagziel S, Lee WD, Shlomi T. Inferring cancer dependencies on metabolic genes from large-scale genetic screens. BMC Biol. 2019;17:37. https://doi.org/10.1186/s12915-019-0654-4.
Tabe Y, Saitoh K, Yang H, Sekihara K, Yamatani K, Ruvolo V, et al. Inhibition of FAO in AML co-cultured with BM adipocytes: mechanisms of survival and chemosensitization to cytarabine. Sci Rep. 2018;8:16837. https://doi.org/10.1038/s41598-018-35198-6.
Ye H, Adane B, Khan N, Alexeev E, Nusbacher N, Minhajuddin M, et al. Subversion of systemic glucose metabolism as a mechanism to support the growth of leukemia cells. Cancer Cell. 2018;34:659–673.e6. https://doi.org/10.1016/j.ccell.2018.08.016.
Muir A, Vander Heiden MG. The nutrient environment affects therapy. Science. 2018;360:962–3. https://doi.org/10.1126/science.aar5986.
Pollak M. Diet boosts the effectiveness of a cancer drug. Nature. 2018;560:439–40. https://doi.org/10.1038/d41586-018-05871-x.
Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479–83. https://doi.org/10.1038/nature19084.
Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Rahman MM, et al. Microenvironmental autophagy promotes tumour growth. Nature. 2017;541:417–20. https://doi.org/10.1038/nature20815.
Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, et al. Autophagy maintains tumour growth through circulating arginine. Nature. 2018;563:569–73. https://doi.org/10.1038/s41586-018-0697-7.
Ho T-C, LaMere M, Stevens BM, Ashton JM, Myers JR, O’Dwyer KM, et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood. 2016;128:1671–8. https://doi.org/10.1182/blood-2016-02-695312.
Boyd AL, Aslostovar L, Reid J, Ye W, Tanasijevic B, Porras DP, et al. Identification of chemotherapy-induced leukemic-regenerating cells reveals a transient vulnerability of human AML recurrence. Cancer Cell. 2018;34:483–498.e5. https://doi.org/10.1016/J.CCELL.2018.08.007.
DiNardo CD, Rausch CR, Benton C, Kadia T, Jain N, Pemmaraju N, et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol. 2018;93:401–7. https://doi.org/10.1002/ajh.25000.
Nachmias B, Schimmer AD. Metabolic flexibility in leukemia-adapt or die. Cancer Cell. 2018;34:695–6. https://doi.org/10.1016/j.ccell.2018.10.012.
Courtney KD, Bezwada D, Mashimo T, Pichumani K, Vemireddy V, Funk AM, et al. Isotope tracing of human clear cell renal cell carcinomas demonstrates suppressed glucose oxidation in vivo. Cell Metab. 2018;28:793–800.e2. https://doi.org/10.1016/j.cmet.2018.07.020.
Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate metabolism in human lung tumors. Cell. 2017;171:358–371.e9. https://doi.org/10.1016/j.cell.2017.09.019.
Acknowledgements
The authors thank Dr. M.A. Selak for critical reading of the manuscript.
Funding
The authors thank the Région Midi-Pyrénées, Plan Cancer 2014-BioSys (FLEXAML), and the Institut national de la santé et de la recherche médicale (Inserm) for their support.
Author information
Authors and Affiliations
Contributions
LS, MS, and J-ES wrote the manuscript and read and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Stuani, L., Sabatier, M. & Sarry, JE. Exploiting metabolic vulnerabilities for personalized therapy in acute myeloid leukemia. BMC Biol 17, 57 (2019). https://doi.org/10.1186/s12915-019-0670-4
Published:
DOI: https://doi.org/10.1186/s12915-019-0670-4