
Abstract
Signaling pathways direct organogenesis, often through concentration-dependent effects on cells. The hedgehog pathway enables cells to sense and respond to hedgehog ligands, of which the best studied is sonic hedgehog. Hedgehog signaling is essential for development, proliferation, and stem cell maintenance, and it is a driver of certain cancers. Lipid metabolism has a profound influence on both hedgehog signal transduction and the properties of the ligands themselves, leading to changes in the strength of hedgehog signaling and cellular functions. Here we review the evolving understanding of the relationship between lipids and hedgehog signaling.
Similar content being viewed by others

Functional interactions between hedgehog signaling and lipid metabolism
Hedgehog proteins are secreted ligands that enable long-range communication between cells of developing and adult tissues [1, 2]. The core molecular components of the pathway are evolutionarily conserved and were first identified in the fruit fly Drosophila melanogaster nearly a century ago, first through mutant analysis and later by systematic genetic screens [3, 4]. These studies elucidated the signaling mechanism by which cells sense the concentration of hedgehog in their vicinity [5], which in certain contexts can be integrated with the duration of hedgehog exposure [6]. These signal transduction events converge onto downstream gene-regulatory networks to regulate processes including cell proliferation, stem cell maintenance, survival, and fate specification [7] (Fig. 1). Many of the genes that encode hedgehog pathway components have subsequently been associated with a range of inherited human developmental disorders and other pathologies [6, 8]. The phenotype of congenital hedgehog deficiency is similar to that seen with genetic mutations causing defective cholesterol metabolism [9]. At the molecular level, three observations link hedgehog signal transduction with cholesterol biosynthesis: hedgehog ligands are covalently modified by cholesterol; the hedgehog receptor patched (PTCH) contains a sterol-sensing domain (SSD), which is found in proteins involved in cholesterol synthesis and transport; and cholesterol, its precursors, and derivatives activate or inhibit smoothened (SMO), the membrane transducer of hedgehog signaling (Fig. 1). These and other observations implicate lipids as key regulators of hedgehog signaling, which could potentially couple cellular metabolism to cell proliferation and cell fate determination. Cholesterol and phospholipids constitute the major classes of lipid and are an integral component of cellular membranes. In this review, we explore the relevance of sterols and lipids to hedgehog signaling.

Overview of hedgehog signaling. The N- and C-termini of hedgehog proteins are covalently modified with palmitate and cholesterol, respectively. Lipid-modified hedgehog is transported by exocytic vesicles from the endoplasmic reticulum to the plasma membrane and secreted into the extracellular space. In the receiving cell, in the absence of bound hedgehog, the sterol-sensing domain (SSD)-containing receptor patched (PTCH) inhibits transport of smoothened (SMO) to the primary cilium by limiting the binding of cholesterol to the cysteine-rich domain (CRD) of SMO. In the absence of cilia-localized active SMO, GLI proteins are partially proteolyzed in the cytoplasm and translocate to the nucleus, where they act as transcriptional repressors (GLIR). Binding of hedgehog to PTCH1 leads to trafficking of PTCH1 away from the primary cilium, relieving repression of SMO ciliary accumulation and activation by cholesterol binding to the CRD. Active SMO inhibits the partial proteolysis of full-length GLI (GLIFL), which translocates to the nucleus and activates transcription. The transcriptional activity of GLI proteins drive progenitor cells along distinct differentiation trajectories. Hedgehog signaling also drives biological processes, including stem cell maintenance and progenitor proliferation. Aberrant hedgehog signaling induces aberrant proliferation and cellular differentiation associated with cancer
Overview of lipid synthesis
Cholesterol is a tetracyclic aromatic lipid that is a major constituent of the lipid bilayers of cellular membranes. The biochemical reactions that lead to cholesterol synthesis begin with the conversion of acetyl-CoA to 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA), which is a precursor for mevalonate (Fig. 2). Subsequently, mevalonate is metabolized to squalene via a series of isoprenoid intermediates [10,11,12]. Lanosterol, which is derived from squalene, is used as a substrate for the production of the cholesterol precursor 7-dehydrocholesterol (7DHC), from which cholesterol is derived by the terminal enzyme 7-dehydrocholesterol reductase (DHCR7) [13]. The other major class of lipids that constitute the membranes of the cell are phospholipids, which are generated by the enzymatic derivation of glycerol-3-phosphate with two long-chain fatty acid “tails”. Like sterols, fatty acids are derived from acetyl-CoA, which becomes elongated by recurrent esterification onto a growing aliphatic chain [14]. Fatty acid chains are subsequently modified, yielding diverse combinations of saturated and unsaturated carbon–carbon (C–C) bond arrangements [15]. The ratio of cholesterol to phospholipid, combined with the degree of saturation of phospholipid tails, determines the biophysical properties of lipid bilayers [16]. Further diversity in phospholipid classes depends on the identity of the head group linked via a phosphodiester bond to the glycerol backbone of the molecule [17]. An additional class of lipids synthesized similarly to phospholipids are the triglycerides, which are formed by the derivation of glycerol-3-phosphate with a third fatty-acid chain in place of the phosphodiester-linked head group. Unlike cholesterol and phospholipids, triglycerides are not constituents of lipid bilayer membranes and have an energy-storage function [14].

Lipid synthesis pathways. Cholesterol (brown), phospholipids (blue), and fatty acids (green) are synthesized from acetyl-CoA via a series of intermediate metabolites. Oxysterols are enzymatically produced from 7-dehydrocholesterol (7DHC) and cholesterol by Cytochrome P450 (CYT P450) enzyme family members, and are also generated non-enzymatically (not shown). Enzymes described in the text are shown in red. Both 7DHC and 7-dehydrodesmosterol are substrates for 7-dehydrocholesterol reductase (DHCR7). Fatty acids contribute to the synthesis of phospholipids from glycerol-3-phosphate. Multiple enzyme reactions not shown are represented by double arrows. TG triclyceride, CDP-DAG cytidine diphosphate di-acyl glycerol, PI phosphatidylinositol, HMG-CoA 3-hydroxy-3-methylglutaryl coenzyme A, HMGCR HMG-CoA reductase, PIP phosphatidylinositol phosphate, PC phosphatidyl-choline, PE phosphatidyl-ethanolamine, PS phosphatidyl-serine
Cholesterol and its precursor 7DHC undergo sterol side chain oxidation, which generates a diverse class of bioactive sterols termed oxysterols. Depending on their specific chemical identity, these sterols regulate cellular physiology via interactions with signaling pathways, intracellular trafficking, and metabolism [18]. The signaling activities of oxysterols regulate cellular differentiation [19] and inflammation [20, 21], and they have been associated with pathologies such as atheroma [22] and macular degeneration [23]. Sterol hydroxylases, most of which are members of the cytochrome P450 family, catalyze the formation of specific oxysterol species [24]. Sterol hydroxylase-deficient mouse models have proved valuable in dissecting the specific physiological activities of several oxysterols [20, 25]. However, a major route to oxysterol formation is driven by non-enzymatic free-radical and lipid peroxide “auto-oxidation” [24, 26], confounding systematic genetic analysis of the physiological activities associated with the oxysterol metabolome. Furthermore, the low abundance of oxysterols within biological tissues compared with their precursors, coupled with the propensity for precursor auto-oxidation during sample preparation, represents a further challenge to the accurate characterization of oxysterol metabolomes [27].
Cholesterol trafficking and homeostasis
Sterol homeostasis is maintained by feedback control at transcriptional and post-transcriptional levels across a network of diverse cellular processes. As major components of cellular membranes, sterols are transported between organelles by two analogous intracellular trafficking processes [28]. Endocytosis redistributes lipids resident within the plasma membrane (PM) via endocytic vesicles that form by PM budding and internalization. Conversely, exocytosis redistributes the lipids resident within the membranes of the endoplasmic reticulum (ER) and Golgi as constituents of exocytic vesicles that move to the periphery of the cell and fuse with the PM. Endocytic and exocytic vesicles are actively transported along microtubules by the motor proteins dynein and kinesin [29]. The direction and destination of a vesicle depends on the motor protein with which it is associated, and their selection is determined by Rab proteins localized to the endosomal membrane. Rab proteins act as molecular switches to regulate vesicular transport [29, 30]. Cholesterol modifies these interactions, influencing the flux of membranes and thus its own redistribution within the cell [31, 32]. Non-vesicular mechanisms of cholesterol transport can also shift cholesterol to various target membranes, including the PM, in a process that involves lipid-binding proteins, including caveolin 1, oxysterol binding protein-related proteins (ORPs), and proteins that contain START domains [33].
Cholesterol and its oxysterol derivatives regulate lipid and vesicular transport processes via oxysterol binding proteins (OSBPs) localized at the Golgi–ER interface [18]. OSBPs act as a tether between the Golgi and ER membranes and transfer cholesterol and phosphatidylinositol-4-phosphate (PI(4)P) between them in a process that is sensitive to the cholesterol content of these membranes [34]. Like cholesterol, PI(4)P and the related lipid PI(3)P regulate intracellular transport by mediating the interaction of vesicles with microtubule-associated motor proteins [35]. The cholesterol-dependent regulation of PI(4)P localization by OSBPs highlights an additional interaction between vesicular transport and cholesterol abundance. As this transporter activity of OSBPs is negatively regulated by oxysterols, such as 25-hydroxycholesterol (25-OHC) [34], it is evident that derivatives of cholesterol exert feedback control on cholesterol-regulated cellular processes.
Cholesterol localization is further controlled by the cholesterol transporters Niemann-Pick C1 (NPC1) and NPC2, which mobilize cholesterol from endosomal membranes [36, 37]. Mutation of the genes encoding either of the NPC proteins results in Niemann-Pick disease, which is characterized by cholesterol accumulation within the endosomal system [38]. The NPC1 cholesterol transporter belongs to a family of proteins that contain an evolutionarily conserved cholesterol-binding SSD. The SSD is a membrane-spanning motif composed of five transmembrane segments that regulates protein distribution, conformation, and activity in response to local sterol concentration [39]. Although the transmembrane segments are exposed to the lipid bilayer, permitting potential interactions with embedded sterols, physical interaction between the SSD and cholesterol has not been directly demonstrated. Rather, cholesterol and the oxysterol 25-OHC interact with an N-terminal domain common to NPC1 and NPC2 that is not membrane associated [40].
The enzymes that metabolize sterols are regulatory proteins that sense sterol levels and balance their rate of production and utilization. The activity of HMG-CoA reductase (HMGCR), which catalyzes the synthesis of mevalonate within the ER and is rate-limiting, is negatively regulated by products of the mevalonate biosynthetic pathway [41]. Rising concentrations of these metabolites promote the interaction between HMGCR and the ER membrane-associated insulin-induced gene 1 protein (INSIG1) and INSIG2 proteins via a process mediated by the SSD of HMGCR [42, 43]. This interaction increases the rate of HMGCR ubiquitination and subsequent proteolysis, thereby reducing the level of the rate-limiting enzyme in response to rising sterol levels [44, 45].
Sterols also negatively regulate the transcription of key lipid biosynthetic enzymes via the cholesterol sensor sterol regulatory element-binding protein cleavage-activating protein (SCAP). Like HMGCR, SCAP contains an SSD and is localized to the ER membrane, where it regulates the activity of the ER membrane-anchored sterol regulatory element-binding protein (SREBP) family of transcriptional regulators [46, 47]. In a similar manner to HMGCR, the SCAP–SREBP complex interaction with INSIG proteins is also stabilized by both cholesterol and its oxysterol derivatives [48]. Falling cholesterol levels lead to destabilization of the interaction between INSIG and SCAP–SREBP and to vesicular transport of the SCAP–SREBP complex to the Golgi where proteolytic cleavage of the SREBP membrane anchor occurs, releasing it to translocate to the nucleus and activate target gene expression [49]. SREBP proteins regulate the expression of enzymes participating in the biosynthesis of distinct lipid classes. Whereas SREBP1a and SREBP1c regulate enzymes involved in fatty acid and triglyceride synthesis, sterol synthesis is controlled by SREBP2 via its induction of multiple sterol enzyme-encoding genes including HMG-CoA synthase, HMGCR, squalene synthase, lanosterol synthase, and DHCR7 [50, 51]. Therefore, diverse sterol products negatively regulate the rate of biosynthesis of both steroid and non-steroid lipids via feedback inhibition as their levels rise.
In response to rising intracellular sterol levels, cells not only reduce cholesterol synthesis but also convert cholesterol into its storage form as cytoplasmic lipid droplets, through esterification by the enzyme acyl-coenzyme A:cholesterol acyltransferase (ACAT) within the ER [28]. ACAT undergoes positive allosteric regulation specifically by cholesterol, which it esterifies preferentially over oxysterol substrates [52,53,54]. In short, rising free cholesterol levels increase the rate of cholesterol sequestration by ACAT (and to a lesser extent oxysterol sequestration), whereas oxysterol levels do not affect the rate of sequestration of either sterol species. However, rising levels of certain oxysterols provide negative feedback on the levels of cellular sterols via the activation of the liver-X receptors (LXRs) [55]. LXR-α and LXR-β are orphan nuclear receptors that form heterodimers with retinoid receptors [56] and regulate the expression of the cholesterol transporters ATP binding cassette subfamily A member 1 (ABCA1) and ATP binding cassette subfamily G member 1 (ABCG1), and the cholesterol carrier apolipoprotein E (ApoE) [57], which together promote the efflux of cholesterol from the cell and its sequestration as low-density or high-density lipoproteins in the circulatory system. Oxysterol-activated OSBP promotes the ubiquitination and degradation of ABCA1 [58], reducing the rate of cholesterol efflux and providing a further example of a negative feedback process ensuring cellular sterol homeostasis. Taken together, these findings highlight that, in addition to both transcriptional and post-translation regulation of sterol biosynthetic enzymes, cholesterol and oxysterols also exert feedback regulation of enzymes and transport proteins controlling their distribution within, and redistribution from, the cell (Fig. 3).

Cholesterol homeostasis is maintained by a highly interconnected network of cellular processes. The transcription factor sterol regulatory element-binding protein 2 (SREBP2) positively regulates the expression of cholesterol biosynthetic enzymes. Rising cholesterol levels reduce the rate of cholesterol biosynthesis by modulating the activities of insulin-induced gene (INSIG) proteins. When activated, INSIG both promotes the ubiquitination and consequent destabilization of key metabolic enzymes and inhibits the transcriptional activity of SREBP2 by retaining it in complex with sterol regulatory element-binding protein cleavage-activating protein (SCAP) in the endoplasmic reticulum (ER). Rising cholesterol levels also allosterically activate acyl-coenzyme A:cholesterol acyltransferase (ACAT), which esterifies cholesterol leading to its sequestration in cytosolic lipid droplets. Oxysterol products of cholesterol activate liver-X receptor (LXR) transcription factors, which positively regulate the transcription of proteins that drive cholesterol efflux from the cell (ABCA1 and ABCG1), and sequester it in lipoprotein particles in the circulatory system (ApoE). Activation of oxysterol binding proteins (OSBP) by oxysterols negatively regulates cholesterol efflux by promoting ABCA1 ubiquitination and degradation. Lipoprotein-derived cholesterol is internalized in endosomes that contain Niemann-Pick C1 (NPC1) and NPC2, which act together to redistribute cholesterol to the ER. NPC1 and NPC2 are also critical for the egress of endogenously synthesized cholesterol from endosomes
Inborn errors of cholesterol metabolism
Diseases of cholesterol synthesis manifest at birth or during infancy and belong to a class of genetically determined disorders termed inborn errors of metabolism. Three autosomal recessive disorders of the post-squalene pathway—Smith-Lemli-Opitz syndrome (SLOS), lathosterolosis, and desmosterolosis—are unusual in comparison to most other inborn errors of metabolism because of their association with characteristic morphological abnormalities that affect proper formation of tissues in the midline of the head [9]. The spectrum of anatomical defects observed are termed holoprosencephaly (HPE). Collectively, HPE abnormalities are the commonest abnormality of the head and face in humans, with a frequency of 1 in 10,000 births and 1 in 250 conceptions [59]. HPE does not affect closure of the neural tube, which is the commonest congenital malformation. In the most severe form of HPE (alobar HPE) the brain is small, the cerebral hemispheres are fused around a single ventricle, and the eye field fails to separate, resulting in a single, cyclopic eye. A trunk-like structure, or proboscis, is found above the single eye. In milder forms (semilobar and lobar HPE) the brain is larger and the hemispheres are partially separated. Facial abnormalities in milder cases take a variety of forms and include reduced separation of the eyes (hypotelorism), a single central maxillary incisor, and cleft lip/palate.
All three disorders of the post-squalene pathway are characterized by the accumulation of sterol intermediates, with or without a deficiency of cholesterol [9, 60, 61]. In SLOS, mutations in DHCR7 result in a deficiency of cholesterol and accumulation of the precursor sterol, 7DHC. Lathosterolosis is caused by a mutation in the sterol C5-desaturase-like gene, SC5DL. Defects in 3β-hydroxysterol-24-reductase, due to mutation in the desmosterol reductase gene (DHCR24), result in desmosterolosis; biochemically, there is a failure to convert desmosterol to cholesterol. SLOS is by far the commonest disorder of cholesterogenesis. The rarity of lathosterolosis and desmosterolosis has meant that the phenotype of these diseases is incompletely delineated, but in broad terms all three disorders result in morphological defects on the HPE spectrum. The range and severity of phenotypic abnormalities in SLOS are highly variable. Numerous mutations have been identified in several hundred affected individuals, and although some genotype–phenotype correlations have been reported, exceptions are often identified [62].
Hedgehog signaling
Loss-of-function mutations in the hedgehog pathway also produce HPE morphological abnormalities, suggesting a regulatory relationship between sterol metabolism and the hedgehog signaling pathway during development [63]. In familial forms of HPE, dominant loss-of-function mutations in the gene encoding the human orthologue of sonic hedgehog (SHH) are the most frequent genetic finding [64].
Ligands
Higher vertebrates have three hedgehog pathway ligands—desert hedgehog (DHH), indian hedgehog (IHH), and SHH—of which SHH is the best studied [2, 65]. Hedgehog proteins are synthesized as inactive pro-peptides, which subsequently undergo cleavage leading to covalent attachment of a cholesterol molecule to the C-terminal amino acid of the active peptide [66]. Cholesterol modification reduces the solubility and diffusion of SHH, allowing incorporation into cellular membranes [67]. The N-terminal amino acid of the SHH protein is also covalently attached to the lipid palmitate by the enzyme hedgehog acyltransferase (HHAT) (Skinny Hedgehog in Drosophila), which is required for full activity of the ligand and, like cholesterol, alters its diffusion properties [68,69,70,71]. Lipid-modified hedgehog proteins are actively transported across the PM for release extracellularly. The translocation of SHH requires the activity of the transmembrane receptor dispatched (DISP) [72,73,74]. DISP contains an SSD in common with the sterol sensor SCAP and other proteins involved in cholesterol homeostasis, and it is homologous to the cholesterol transporter NPC1 [39]. Extracellular release and subsequent spreading of cholesterol-modified SHH is enhanced by its interaction with the secreted protein SCUBE2, which was first implicated in hedgehog signaling in zebrafish [75,76,77]. Both DISP and SCUBE2 directly interact with distinct structural aspects of the cholesterol moiety of SHH, which probably increases its solubility [78] in a manner similar to the transfer of cholesterol between NPC1 and NPC2 in endosomal membranes [79]. By comparison, in Drosophila the spread of hedgehog is influenced by the association of cholesterol-modified hedgehog with lipophorin particles in the hemolymph, which are analogous to the circulating lipoproteins in mammals [80].
Cholesterol-modified SHH is also shed from the surface of producing cells as a component of exovesicles or “exosomes” derived from the budding of cellular membranes [81,82,83]. In Drosophila, endocytosis and subsequent recycling of PM-associated hedgehog is required for its long-range activity and depends on the cholesterol moiety [84]. Exosomal transport of hedgehog can occur via filopodial PM protrusions termed cytonemes [82], which might also directly associate with SHH to enable long-range signaling within developing tissues [85, 86]. Finally, the formation of large multimeric complexes of SHH depends on the addition of the cholesterol moiety. As is the case for the SHH–SCUBE2 complex, formation of these multimers increases the solubility and range of spread of hedgehog within tissues, likely owing to the self-association and sequestration of the hydrophobic cholesterol moiety within the core of the complex [87,88,89]. Therefore, although the cholesterol adduct potentially limits the diffusibility of SHH by anchoring it to membranes [67], its critical role in mediating interactions with other molecules and multimer formation has the opposite effect, extending the range and activity of hedgehog within tissues.
SHH signal transduction
Genetic analysis in mice and chicks demonstrated that the reception and transduction of the SHH signal in the cytoplasmic compartment of receiving cells is localized to the primary cilium [3, 90]. This organelle is an antennae-like projection of the PM surrounding a microtubule core, which is anchored at the basal body—a structure that is derived from the mother centriole. Owing to its intimate association with the centrosome, the primary cilium is dynamically assembled and disassembled over the course of the cell cycle, via processes mediated by Rab proteins [91, 92].
The mammalian SHH receptor PTCH1 is a transmembrane protein which is localized to the primary cilium in the absence of SHH [93]. Similar to SCAP, DISP, and the NPC1 cholesterol transporter, PTCH1 contains an SSD [39]. Similarities with DISP suggest that the SSD might mediate the interaction with the cholesterol moiety of SHH, but this is unclear. In vertebrates, the transmembrane proteins cell adhesion molecule-related downregulated by oncogenes (CDO), brother of CDO (BOC), and the GPI-anchored protein, growth arrest specific protein 1 (GAS1) also act as SHH receptors and form a complex with PTCH1 [94, 95]. These SHH receptors have overlapping activities that promote signaling, potentially by presenting SHH to PTCH1 [96]. Hedgehog-interacting protein (HHIP) also binds vertebrate hedgehog proteins but inhibits rather than promotes signaling, and does not physically interact with PTCH1 [97]. Homologues of CDO and BOC termed Ihog and Brother of Ihog (Boi), respectively, had earlier been identified in Drosophila [94]. The lipid modification of Hedgehog proteins could facilitate their simultaneous interaction with this set of cognate binding partners that modulate ligand potency [87].
In mammals, PTCH1 prevents SMO, a membrane localized GPCR-like SHH signal transducer, from entering the primary cilium. When PTCH1 binds to SHH, repression of SMO is relieved, and SMO enters the cilium where a second activating step initiates downstream signaling [93, 98, 99]. In this two-step model of mammalian SMO activation, the translocation of SMO is regarded as a prerequisite for signal transduction [100]. By contrast, Drosophila cells lack primary cilia, and PTCH instead regulates the accumulation of SMO at the PM, where it activates signaling [101, 102]. A non-cell autonomous model of SMO repression by PTCH has also been proposed, which could be mediated by the cholesterol precursor 7DHC [103, 104].
Instructive and permissive effects of sterols on SHH signaling
Inhibition of SMO by PTCH1 can be overcome by cholesterol- and 7DHC-derived oxysterols [105,106,107,108]. Endogenous and synthetic oxysterols allosterically activate SMO through their binding to the extracellular cysteine-rich domain (CRD) [106,109,, 108–110]. By contrast, the plant-derived sterol cyclopamine inhibits SMO upon binding to the transmembrane domain (TMD) at a site remote from the CRD [111]. The synthetic SMO agonist SAG competes with cyclopamine for binding at the TMD and drives SMO cilia localization and activation independently of SHH [111]. However, a B-ring oxysterol derivative of 7DHC, 3β,5α-dihydroxycholest-7-en-6-one (DHCEO), was reported to inhibit SMO by binding to a site distinct from both the CRD and the cyclopamine/SAG pocket [112]. Therefore, SMO has multiple sterol-interacting interfaces that positively or negatively affect its activity.
Cholesterol is sufficient to stimulate SHH signaling independently of oxysterols and can induce neural cell types in vitro that require moderate to high levels of SHH signaling for their differentiation [113]. Cholesterol and oxysterols compete for the same binding site in the SMO CRD [114, 115]. Furthermore, a modified version of cholesterol that cannot be metabolized to oxysterols nevertheless rescued SHH signaling in sterol-depleted cells, suggesting that cholesterol is an endogenous activator of SMO [113, 115]. As with SHH, SMO is covalently bound to cholesterol via an aspartic acid residue (Asp95) in the CRD, and its mutation results in loss of SMO cilia activation in vitro and in vivo [116].
In animal models of SLOS, the response to hedgehog signaling is reduced in receiving cells, which is consistent with a requirement for cholesterol in signal transduction [117, 118]; these models also show HPE dysmorphology characteristic of SHH deficiency. However, distinguishing between precursor accumulation versus cholesterol deficiency as the reason for attenuated hedgehog signaling has proved difficult to resolve because of the complex feedback mechanisms in the cholesterol synthesis pathway described above [119, 120]. Reduced cholesterol levels have been proposed to disrupt SMO indirectly, through an undefined mechanism involving SCAP–SREBP2-mediated transcriptional upregulation of DHCR7 [121, 122]. However, we did not find evidence to support this model in our study [118]. 7DHC and DHCEO accumulate in the brain tissue of SLOS-model animals [119, 120], but we found that 7DHC levels did not affect SHH signaling [118]. Furthermore, in SLOS-mutant fibroblasts that accumulate 7DHC and presumably DHCEO, cholesterol supplementation was sufficient to rescue SHH signaling [118]. These findings warrant further analysis of the relative contribution of 7DHC, DHCEO, and cholesterol to reduced SHH signaling in SLOS.
The reduced SHH signaling response in SLOS might be due to an additional permissive role for cholesterol, which does not depend on the oxysterol-binding CRD or on residues in the SMO TMD that are important for the binding of cyclopamine and synthetic agonists [108,124,125,, 113, 117, 118, 123–126]. Such a putative mechanism could stem from the effects of cholesterol on SMO vesicular trafficking. SMO translocation is also dependent on vesicular transport, which is influenced by cholesterol. Following its synthesis in the ER and maturation through the Golgi, SMO is trafficked to the PM. The PM pool of SMO is then internalized into the endocytic system, and SMO entry into the primary cilium has been shown to occur both by direct lateral transport from the PM and from the endocytic system [98, 99]. While SMO stimulation is generally associated with its cilia localization, these events can be dissociated by SMO inhibitors and point to distinct regulatory steps in SMO activation as described earlier [100]. Perturbations in the synthesis or subcellular localization of cholesterol alters the activities of many Rab proteins and consequently disrupts endosome motility [31,128,, 127–129]. Altered cholesterol levels within specific cellular compartments resulting from inborn errors of sterol metabolism might therefore impair SMO cilia localization and activation owing to abnormal vesicular trafficking (Fig. 4). In support of this conjecture, Rab8 and Rab23 have been shown to modulate the rates of active SMO entry into and recycling from the cilium, respectively [130]. While the function of Rab23 with respect to intracellular trafficking is relatively uncharacterized, Rab23 is a known negative regulator of SHH signaling and is localized to both the PM and endosomal system [131, 132]. Furthermore, Rab8 is a mediator of both cilium biogenesis and cholesterol transport to the PM [133, 134]. Therefore, both Rab proteins represent plausible links between SMO trafficking to the cilium and intracellular cholesterol levels.

The regulation of smoothened trafficking by cholesterol. a The levels of transmembrane receptors such as smoothened (SMO) at the plasma membrane are determined by the balance between the rates of supply by exocytosis and internalization by endocytosis. Following endocytosis, receptors are either recycled back to the membrane or degraded [29]. In the absence of sonic hedgehog (SHH), patched1 (PTCH1) reduces the concentration of cholesterol in the membranes of endosomal vesicles, biasing plasma membrane SMO toward internalization and degradation [101, 139]. b SHH binding redistributes PTCH1 from the primary cilium and SMO-containing endosomal vesicles [93, 141]. In the absence of PTCH1, the concentration of cholesterol within endosomal membranes is elevated [139]. Elevated cholesterol levels within endosomal membranes allow SMO to accumulate in the plasma membrane by reducing its rate of internalization and degradation [101, 139]. Whether PTCH1 activity also modulates cholesterol levels in the membranes around the base or within the shaft of the primary cilium remains to be determined. c In Smith-Lemli-Opitz syndrome (SLOS), the associated reduced cellular cholesterol levels decrease the accumulation of SMO in the cilium in response to SHH [118]. This may be due to a requirement for direct molecular interaction between cholesterol and SMO for SMO cilia entry. Alternatively, reduced cholesterol levels may indirectly decrease SMO levels in the cilium by disrupting the kinetics of endocytic, recycling, or exocytic vesicle trafficking to and from the plasma membrane
Compared with manipulations whereby bulk sterol is depleted, reduced SMO activation owing to DHCR7 loss of function is associated with a modest reduction in total cellular cholesterol levels [117, 118]. As DHCR7 synthesis of cholesterol is localized to the ER, cholesterol levels might be more acutely reduced in the ER and cilium-associated Golgi compartments, and thus potentially impair endosomal transport and SMO trafficking to the cilium more severely than would be predicted from bulk sterol measurements. Consistent with this hypothesis, we found that in embryonic fibroblasts from SLOS mutant mice, there was a marked reduction in SMO translocation to the cilium in response to SHH, which could be rescued by cholesterol supplementation [118]. Nevertheless, cholesterol can induce significant SMO activity that is comparable to the effect of SAG stimulation, without producing appreciable cilia localization [113]. This implies the current two-step model of SMO activation requires refinement [100].
Lipid involvement in PTCH regulation of SMO
PTCH1 belongs to the resistance-nodulation-division (RND) family of small-molecule pumps [135]. Repression of SMO by PTCH1 occurs indirectly and acts non-stoichiometrically, implying a catalytic mechanism [136]. The homology of PTCH1 with sterol sensors involved in cholesterol homeostasis and its ability to bind and transport cholesterol have led to the proposal that PTCH1 may directly mediate the transport of cholesterol between cellular membranes [123, 136]. Whereas the residues of the SSD homologous between PTCH1 and the sterol sensor of SCAP are not essential for vertebrate PTCH1 activity, they are required for the function of Drosophila PTCH, limiting structure–function comparisons between the two receptors [136,137,138]. Furthermore, local reduction of cholesterol levels in early endosomes by PTCH has been demonstrated in Drosophila cells [139]. In an analogous manner, vertebrate PTCH1 might restrict access of cholesterol to SMO at the base of the cilium, which is relieved upon SHH binding to PTCH1 [113]. It is noteworthy, however, that engineered forms of SMO lacking the CRD remain partially sensitive to PTCH1 repression, suggesting an additional CRD-independent mode of SMO regulation by PTCH1 [108, 109, 136]. In addition, mutations in the SMO TMD that prevent binding of cyclopamine or its synthetic analog GDC-0449 do not affect inhibition of SMO by PTCH1 [108, 140], further indicating that PTCH1 does not repress SMO via the TMD.
Experiments in which hedgehog signaling proteins are overexpressed in mammalian cells have demonstrated the localization of both PTCH1 and SMO in endosomes, from which SMO is recycled back to the PM for entry into the cilium and activation [98, 99, 141]. In Drosophila, the interaction between PTCH and SMO has also been shown to occur within endosomes and depends upon a lipid molecule associated with lipoprotein particles [139]. It has been proposed that, in Drosophila, PTCH regulates SMO activation by controlling the distribution of the phospholipid PI(4)P and its synthesis by phosphatidylinositol 4-kinase III alpha (PI4III kinase α) [142, 143]. In this model, PI(4)P binding to the SMO intracellular domain (ICD) is critical for SMO activation [143]. Whether mammalian SMO is regulated by a similar mechanism [143] needs be tested by in vivo deletion of PI4III kinase α. Taken together, these studies raise the possibility of co-regulation of distinct SMO domains by different lipid species, cholesterol and phospholipids, and might explain why residual repression of SMO by PTCH1 occurs in the absence of the SMO CRD [108, 109, 136].
Phosphoinositides also regulate the entry of SHH pathway negative regulators intraflagellar transport-A (IFT-A), GPR161, and Tubby-like protein 3 (TULP3) into the cilium [144,145,146,147]. The enzymatic activity of ciliary phosphoinositide 5-phosphatase regulates the ratio of PI(4)P and PI(4,5)P2 within the cilium shaft, maintaining a lipid composition that is responsive to modulation by PTCH1 [148, 149]. As these negative regulators are themselves excluded from the cilium following SHH binding of PTCH1, it is evident that multiple layers of SHH pathway repression are coordinated by PTCH1 via phosphoinositides. Together these data highlight the essential and diverse modes of SHH pathway regulation by lipids and lipid-modifying enzymes (summarized in Fig. 5a).

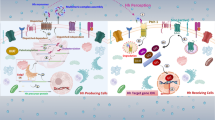
The relationship between lipids and hedgehog signaling. a Regulation of canonical hedgehog signaling by lipids. Smoothened (SMO) is the nexus of hedgehog pathway regulation by lipids. Cholesterol, oxysterols, and phosphatidylinositol-4-phosphate (PI(4)P) are SMO activators, whereas the plant sterol cyclopamine and a distinct class of B-ring oxysterols, DHCEO (7DHC, 3β,5α-dihydroxycholest-7-en-6-one) inhibit SMO. PTCH prevents activation of hedgehog signaling by restricting the access of cholesterol and PI(4)P to SMO while also inhibiting the synthesis of the latter lipid molecule. PI(4)P also prevents the entry of negative regulators of hedgehog signaling, TULP3 and GPR161, to the primary cilium. In hepatocytes, downstream of SMO full-length activated GLI was reported to repress the lipogenic transcriptional program (dashed inhibitory arrow) but the mechanism is unclear. GLIR GLI repressor, GLIFL full-length GLI. b Non-canonical signaling via AMPK in muscle and brown fat. AMPK is activated by SMO via liver kinase complex B1 (LKB1) and calcium/calmodulin-dependent kinase kinase 2 (CAMKK2). In turn, adenosine monophosphate kinase (AMPK) represses cholesterol production directly by inhibition of HMG-CoA reductase (HMGCR) and sterol regulatory element-binding protein 2 (SREBP2) and indirectly through mTORC1 inhibition. In this context cyclopamine was reported to act as a partial agonist of SMO, but whether cholesterol activates SMO has not been tested (question mark above arrow). Lipid/sterol molecules and lipogenic transcription factors are shown in red. Arrows indicate activation (arrowhead) or inhibition (orthogonal bars), except for the arrow from PI to PI(4)P, which indicates an enzymatic step
Canonical signaling
Evidence is accumulating that signaling diverges downstream of SMO activation along canonical and non-canonical pathways. The canonical pathway is the best studied and is mediated by GLI transcription factors, which regulate the developmental patterning function of sonic hedgehog by either activating gene expression, via GLI1 and GLI2, or repressing it through GLI3 repressor (GLI3R) formation [150]. Lipid metabolic homeostasis has emerged as a critical function of hedgehog signaling in the liver, which is mediated by GLI proteins. Conditional deletion of SMO in mouse hepatocytes revealed that GLI1 and GLI3 coordinately repress lipid synthesis at the transcriptional level, presumably through an indirect mechanism that is not well-defined [151] (Fig. 5a). Furthermore, mutant livers displayed a metabolic shift of glucose utilization into the fatty acid synthesis pathway. Whether the regulation of lipid metabolism by canonical hedgehog signaling has functional significance during the formation of tissues dependent on hedgehog for their identity and structure has not been addressed.
Non-canonical signaling
Evidence has begun to emerge that SMO activity reciprocally regulates lipid metabolism via a GLI-independent non-canonical pathway that is centered on adenosine monophosphate kinase (AMPK) [152]. AMPK regulates energy homeostasis within cells by switching off anabolic processes that consume adenosine triphosphate (ATP), including lipid synthesis, whereas it activates alternative catabolic pathways that generate ATP [153]. AMPK functions as an energy sensor through its binding of AMP in energy-deficient conditions, which promotes its activation by the upstream liver kinase complex B1 (LKB1) and calcium/calmodulin-dependent kinase kinase 2 (CAMKK2). In brown adipocytes, SMO activators including oxysterols stimulate rapid glucose uptake and aerobic glycolysis via AMPK that does not require GLI transcriptional activity [152]. These short-term metabolic changes are reinforced by a longer term GLI-mediated transcriptional response, resulting in extensive modulation of the cellular metabolic profile including lipid synthesis [152].
AMPK represses fatty acid, triglyceride, and cholesterol synthesis directly in several ways. Phosphorylation of acetyl-CoA carboxylase (ACC), a direct target of AMPK, inhibits the formation of malonyl CoA, the precursor for fatty acid synthesis [154]. In addition, AMPK directly represses the proteolytic processing, nuclear translocation, and transcriptional activity of SREBP1 [155]. Inhibition of sterol synthesis occurs through direct binding and phosphorylation of HMGCR [156] and SREBP2 [155], which in the latter case has broad-ranging effects similar to SREBP1 inhibition. The direct regulation of lipid metabolism is complemented by indirect inhibitory effects mediated by the mechanistic target of rapamycin complex 1 (mTORC1)-S6K kinase pathway, which in an opposite manner to AMPK is activated by nutrient availability and promotes anabolic processes, including lipid synthesis [157]. AMPK represses the activity of this complex by direct phosphorylation of mTOR and tuberous sclerosis complex (TSC) [158, 159], leading to reduced lipid synthesis (Fig. 5b). These data raise the possibility that a feedback loop involving cholesterol, SMO, and AMPK could modulate the output of non-canonical signaling to effect metabolic changes over short time scales. However, the functional significance of the reciprocal regulatory relationship between the hedgehog pathway and lipids remains unclear.
Future directions in understanding the effects of metabolism on hedgehog signaling
The similarities in the phenotypes of inborn errors of sterol metabolism and SHH deficiency prompted investigation into how lipid metabolism and hedgehog signaling intersect. Despite intensive efforts, the involvement of lipids in hedgehog signaling arguably remains the most puzzling aspect of hedgehog signal transduction. A model of how lipids are involved in PTCH1 regulation of SMO is beginning to emerge, but lipid involvement in hedgehog signaling is complex and multifaceted. Nevertheless, findings to date, which are limited by the lack of in vivo analysis in higher vertebrates, suggest there is evolutionary conservation of the core mechanism. Cholesterol seems to be the main physiological agonist of SMO in higher organisms. The differences that have emerged between Drosophila and humans in the selectivity of the CRD for sterol binding partners and the evidence of alternative binding sites in SMO [112, 160] raises the question of what the physiologically relevant lipid binding interfaces of SMO are. Whether cholesterol and phospholipids mediate the effects of PTCH on SMO through distinct SMO domains will be important to explore further. Whereas cholesterol appears to act as an allosteric regulator of SMO, further studies are needed to determine whether PI(4)P has a similar function. A broader question relates to the dependence of endogenous SMO activation on diverse lipid molecules with positive and/or negative effects on signaling and whether these lipids act directly on SMO, or by modulating vesicular trafficking. Oxysterols appear to be of lesser importance under normal physiological conditions but in certain cancers associated with aberrant hedgehog signaling and dysregulated sterol metabolism, such as medulloblastoma, a role for oxysterols in promoting tumorigenesis via hedgehog signaling might have greater significance [107]. Finally, the relevance of the reciprocal regulation of lipid metabolism by canonical and non-canonical hedgehog signaling pathways is poorly understood and could prove to be important in tumor cells. Indeed, a transcriptional analysis of the response to SHH stimulation in cultured cells revealed alterations in metabolic pathways, including lipid metabolism, associated with invasive cancer [152], further highlighting the interaction between lipid metabolism and hedgehog signaling as fertile ground for future investigation.
Abbreviations
- 7DHC:
-
7-dehydrocholesterol
- ABCA1:
-
ATP binding cassette subfamily A member 1
- ABCG1:
-
ATP binding cassette subfamily G member 1
- ACAT:
-
Acyl-coenzyme A:cholesterol acyltransferase
- ACC:
-
acetyl-CoA carboxylase
- AMPK:
-
Adenosine monophosphate kinase
- ApoE:
-
Apolipoprotein E
- ATP:
-
Adenosine triphosphate
- BOC:
-
Brother of CDO
- CAMKK:
-
Calmodulin-dependent kinase kinase
- CDO:
-
Cell adhesion molecule-related downregulated by oncogenes
- CRD:
-
Cysteine-rich domain
- DHCEO:
-
3β,5α-dihydroxycholest-7-en-6-one
- DHCR24:
-
Desmosterol reductase gene
- DHCR7:
-
7-dehydrocholesterol reductase
- DHH:
-
Desert hedgehog
- DISP:
-
Dispatched
- ER:
-
Endoplasmic reticulum
- GAS1:
-
Growth arrest specific protein 1
- GPCR:
-
G protein-coupled receptor
- HHAT:
-
Hedgehog acyltransferase
- HMGCR:
-
HMG-CoA reductase
- HPE:
-
Holoprosencephaly
- ICD:
-
Intracellular domain
- IFT:
-
Intraflagellar transport
- IHH:
-
Indian Hedgehog
- INSIG:
-
Insulin-induced gene protein
- LKB1:
-
Liver kinase complex B1
- LXR:
-
Liver-X receptor
- mTORC1:
-
Mechanistic target of rapamycin complex 1
- NPC:
-
Niemann-Pick C
- OHC:
-
Hydroxycholesterol
- ORP:
-
Oxysterol binding protein-related protein
- OSBP:
-
Oxysterol binding protein
- PIP:
-
Phosphatidylinositol-phosphate
- PM:
-
Plasma membrane
- PTCH:
-
Patched
- RND:
-
Resistance-nodulation-division
- SAG:
-
Smoothened agonist
- SC5DL:
-
Sterol C5-desaturase-like gene
- SCAP:
-
Sterol regulatory element-binding protein cleavage-activating protein
- SCUBE:
-
Signal peptide, CUB domain and EGF like domain containing
- SHH:
-
Sonic hedgehog
- SLOS:
-
Smith-Lemli-Opitz syndrome
- SMO:
-
Smoothened
- SREBF:
-
Sterol regulatory element binding transcription factor gene
- SREBP:
-
Sterol regulatory element-binding protein
- SSD:
-
Sterol-sensing domain
- TMD:
-
Transmembrane domain
- TSC:
-
Tuberous sclerosis complex
- TULP3:
-
Tubby-like protein 3
References
Lee JJ, von Kessler DP, Parks S, Beachy PA. Secretion and localized transcription suggest a role in positional signaling for products of the segmentation gene hedgehog. Cell. 1992;71(1):33–50.
Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15(23):3059–87.
Ingham PW. Drosophila segment polarity mutants and the rediscovery of the hedgehog pathway genes. Curr Top Dev Biol. 2016;116:477–88.
Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287(5785):795–801.
Ingham PW. Transducing Hedgehog: the story so far. EMBO J. 1998;17(13):3505–11.
Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14(7):416–29.
Cohen M, Briscoe J, Blassberg R. Morphogen interpretation: the transcriptional logic of neural tube patterning. Curr Opin Genet Dev. 2013;23(4):423–8.
Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–37.
Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2011;52(1):6–34.
Bucher NL, Overath P, Lynen F. beta-Hydroxy-beta-methyl-glutaryl coenzyme A reductase, cleavage and condensing enzymes in relation to cholesterol formation in rat liver. Biochim Biophys Acta. 1960;40:491–501.
Miziorko HM. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch Biochem Biophys. 2011;505(2):131–43.
Cerqueira NM, Oliveira EF, Gesto DS, Santos-Martins D, Moreira C, Moorthy HN, Ramos MJ, Fernandes PA. Cholesterol biosynthesis: a mechanistic overview. Biochemistry. 2016;55(39):5483–506.
Wilton DC, Munday KA, Skinner SJ, Akhtar M. The biological conversion of 7-dehydrocholesterol into cholesterol and comments on the reduction of double bonds. Biochem J. 1968;106(4):803–10.
Yamashita A, Hayashi Y, Nemoto-Sasaki Y, Ito M, Oka S, Tanikawa T, Waku K, Sugiura T. Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog Lipid Res. 2014;53:18–81.
Yamashita A, Sugiura T, Waku K. Acyltransferases and transacylases involved in fatty acid remodeling of phospholipids and metabolism of bioactive lipids in mammalian cells. J Biochem. 1997;122(1):1–16.
Ohvo-Rekila H, Ramstedt B, Leppimaki P, Slotte JP. Cholesterol interactions with phospholipids in membranes. Prog Lipid Res. 2002;41(1):66–97.
Dowhan W. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu Rev Biochem. 1997;66:199–232.
Olkkonen VM, Beaslas O, Nissila E. Oxysterols and their cellular effectors. Biomolecules. 2012;2(1):76–103.
Johnson JS, Meliton V, Kim WK, Lee KB, Wang JC, Nguyen K, Yoo D, Jung ME, Atti E, Tetradis S, et al. Novel oxysterols have pro-osteogenic and anti-adipogenic effects in vitro and induce spinal fusion in vivo. J Cell Biochem. 2011;112(6):1673–84.
Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science. 2014;345(6197):679–84.
Gold ES, Diercks AH, Podolsky I, Podyminogin RL, Askovich PS, Treuting PM, Aderem A. 25-Hydroxycholesterol acts as an amplifier of inflammatory signaling. Proc Natl Acad Sci U S A. 2014;111(29):10666–71.
Umetani M, Ghosh P, Ishikawa T, Umetani J, Ahmed M, Mineo C, Shaul PW. The cholesterol metabolite 27-hydroxycholesterol promotes atherosclerosis via proinflammatory processes mediated by estrogen receptor alpha. Cell Metab. 2014;20(1):172–82.
Rodriguez IR, Larrayoz IM. Cholesterol oxidation in the retina: implications of 7KCh formation in chronic inflammation and age-related macular degeneration. J Lipid Res. 2010;51(10):2847–62.
Schroepfer Jr GJ. Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev. 2000;80(1):361–554.
Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem. 2003;278(25):22980–8.
Murphy RC, Johnson KM. Cholesterol, reactive oxygen species, and the formation of biologically active mediators. J Biol Chem. 2008;283(23):15521–5.
Griffiths WJ, Wang Y. Analysis of oxysterol metabolomes. Biochim Biophys Acta. 2011;1811(11):784–99.
Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–57.
Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30(17):3481–500.
Scott CC, Vacca F, Gruenberg J. Endosome maturation, transport and functions. Semin Cell Dev Biol. 2014;31:2–10.
Lebrand C, Corti M, Goodson H, Cosson P, Cavalli V, Mayran N, Faure J, Gruenberg J. Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J. 2002;21(6):1289–300.
Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, Zwart W, Neefjes J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J Cell Biol. 2009;185(7):1209–25.
Prinz WA. Non-vesicular sterol transport in cells. Prog Lipid Res. 2007;46(6):297–314.
Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013;155(4):830–43.
Santiago-Tirado FH, Bretscher A. Membrane-trafficking sorting hubs: cooperation between PI4P and small GTPases at the trans-Golgi network. Trends Cell Biol. 2011;21(9):515–25.
Sleat DE, Wiseman JA, El-Banna M, Price SM, Verot L, Shen MM, Tint GS, Vanier MT, Walkley SU, Lobel P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci U S A. 2004;101(16):5886–91.
Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci U S A. 2008;105(40):15287–92.
Mukherjee S, Maxfield FR. Lipid and cholesterol trafficking in NPC. Biochim Biophys Acta. 2004;1685(1-3):28–37.
Kuwabara PE, Labouesse M. The sterol-sensing domain: multiple families, a unique role? Trends Genet. 2002;18(4):193–201.
Gong X, Qian H, Zhou X, Wu J, Wan T, Cao P, Huang W, Zhao X, Wang X, Wang P, et al. Structural insights into the Niemann-Pick C1 (NPC1)-mediated cholesterol transfer and Ebola infection. Cell. 2016;165(6):1467–78.
Jo Y, Debose-Boyd RA. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit Rev Biochem Mol Biol. 2010;45(3):185–98.
Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem. 2003;278(52):52479–90.
Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA. Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell. 2003;11(1):25–33.
Song BL, Javitt NB, DeBose-Boyd RA. Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 2005;1(3):179–89.
Song BL, Sever N, DeBose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005;19(6):829–40.
Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–40.
Sato R. Sterol metabolism and SREBP activation. Arch Biochem Biophys. 2010;501(2):177–81.
Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci U S A. 2007;104(16):6511–8.
Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124(1):35–46.
Horton JD, Goldstein JL, Brown MS. SREBPs: transcriptional mediators of lipid homeostasis. Cold Spring Harb Symp Quant Biol. 2002;67:491–8.
Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–31.
Chang CC, Lee CY, Chang ET, Cruz JC, Levesque MC, Chang TY. Recombinant acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) purified to essential homogeneity utilizes cholesterol in mixed micelles or in vesicles in a highly cooperative manner. J Biol Chem. 1998;273(52):35132–41.
Zhang Y, Yu C, Liu J, Spencer TA, Chang CC, Chang TY. Cholesterol is superior to 7-ketocholesterol or 7 alpha-hydroxycholesterol as an allosteric activator for acyl-coenzyme A:cholesterol acyltransferase 1. J Biol Chem. 2003;278(13):11642–7.
Liu J, Chang CC, Westover EJ, Covey DF, Chang TY. Investigating the allosterism of acyl-CoA:cholesterol acyltransferase (ACAT) by using various sterols: in vitro and intact cell studies. Biochem J. 2005;391(Pt 2):389–97.
Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383(6602):728–31.
Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000;16:459–81.
Beyea MM, Heslop CL, Sawyez CG, Edwards JY, Markle JG, Hegele RA, Huff MW. Selective up-regulation of LXR-regulated genes ABCA1, ABCG1, and APOE in macrophages through increased endogenous synthesis of 24(S),25-epoxycholesterol. J Biol Chem. 2007;282(8):5207–16.
Bowden K, Ridgway ND. OSBP negatively regulates ABCA1 protein stability. J Biol Chem. 2008;283(26):18210–7.
Muenke M, Beachy PA. Genetics of ventral forebrain development and holoprosencephaly. Curr Opin Genet Dev. 2000;10(3):262–9.
Porter FD. Malformation syndromes due to inborn errors of cholesterol synthesis. J Clin Invest. 2002;110(6):715–24.
Porter FD. Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2008;16(5):535–41.
Herman GE. Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. Hum Mol Genet. 2003;12(Spec No 1):R75–88.
Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383(6599):407–13.
Murdoch JN, Copp AJ. The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res A Clin Mol Teratol. 2010;88(8):633–52.
Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75(7):1417–30.
Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274(5285):255–9.
Peters C, Wolf A, Wagner M, Kuhlmann J, Waldmann H. The cholesterol membrane anchor of the Hedgehog protein confers stable membrane association to lipid-modified proteins. Proc Natl Acad Sci U S A. 2004;101(23):8531–6.
Taylor FR, Wen D, Garber EA, Carmillo AN, Baker DP, Arduini RM, Williams KP, Weinreb PH, Rayhorn P, Hronowski X, et al. Enhanced potency of human Sonic hedgehog by hydrophobic modification. Biochemistry. 2001;40(14):4359–71.
Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, Bixler SA, Ambrose CM, Garber EA, Miatkowski K, et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem. 1998;273(22):14037–45.
Chamoun Z, Mann RK, Nellen D, von Kessler DP, Bellotto M, Beachy PA, Basler K. Skinny hedgehog, an acyltransferase required for palmitoylation and activity of the hedgehog signal. Science. 2001;293(5537):2080–4.
Buglino JA, Resh MD. Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. J Biol Chem. 2008;283(32):22076–88.
Burke R, Nellen D, Bellotto M, Hafen E, Senti KA, Dickson BJ, Basler K. Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell. 1999;99(7):803–15.
Ma Y, Erkner A, Gong R, Yao S, Taipale J, Basler K, Beachy PA. Hedgehog-mediated patterning of the mammalian embryo requires transporter-like function of dispatched. Cell. 2002;111(1):63–75.
Tian H, Jeong J, Harfe BD, Tabin CJ, McMahon AP. Mouse Disp1 is required in sonic hedgehog-expressing cells for paracrine activity of the cholesterol-modified ligand. Development. 2005;132(1):133–42.
Creanga A, Glenn TD, Mann RK, Saunders AM, Talbot WS, Beachy PA. Scube/You activity mediates release of dually lipid-modified Hedgehog signal in soluble form. Genes Dev. 2012;26(12):1312–25.
van Eeden FJ, Granato M, Schach U, Brand M, Furutani-Seiki M, Haffter P, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, et al. Mutations affecting somite formation and patterning in the zebrafish, Danio rerio. Development. 1996;123:153–64.
Ingham PW. Zebrafish genetics gets the Scube on Hedgehog secretion. Genes Dev. 2012;26(22):2468–70.
Tukachinsky H, Kuzmickas RP, Jao CY, Liu J, Salic A. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep. 2012;2(2):308–20.
Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, Infante RE. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137(7):1213–24.
Panakova D, Sprong H, Marois E, Thiele C, Eaton S. Lipoprotein particles are required for Hedgehog and Wingless signalling. Nature. 2005;435(7038):58–65.
Liegeois S, Benedetto A, Garnier JM, Schwab Y, Labouesse M. The V0-ATPase mediates apical secretion of exosomes containing Hedgehog-related proteins in Caenorhabditis elegans. J Cell Biol. 2006;173(6):949–61.
Gradilla AC, Gonzalez E, Seijo I, Andres G, Bischoff M, Gonzalez-Mendez L, Sanchez V, Callejo A, Ibanez C, Guerra M, et al. Exosomes as Hedgehog carriers in cytoneme-mediated transport and secretion. Nat Commun. 2014;5:5649.
Vyas N, Walvekar A, Tate D, Lakshmanan V, Bansal D, Lo Cicero A, Raposo G, Palakodeti D, Dhawan J. Vertebrate Hedgehog is secreted on two types of extracellular vesicles with different signaling properties. Sci Rep. 2014;4:7357.
D’Angelo G, Matusek T, Pizette S, Therond PP. Endocytosis of Hedgehog through dispatched regulates long-range signaling. Dev Cell. 2015;32(3):290–303.
Bischoff M, Gradilla AC, Seijo I, Andres G, Rodriguez-Navas C, Gonzalez-Mendez L, Guerrero I. Cytonemes are required for the establishment of a normal Hedgehog morphogen gradient in Drosophila epithelia. Nat Cell Biol. 2013;15(11):1269–81.
Rojas-Rios P, Guerrero I, Gonzalez-Reyes A. Cytoneme-mediated delivery of hedgehog regulates the expression of bone morphogenetic proteins to maintain germline stem cells in Drosophila. PLoS Biol. 2012;10(4):e1001298.
Chen MH, Li YJ, Kawakami T, Xu SM, Chuang PT. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 2004;18(6):641–59.
Gallet A, Ruel L, Staccini-Lavenant L, Therond PP. Cholesterol modification is necessary for controlled planar long-range activity of Hedgehog in Drosophila epithelia. Development. 2006;133(3):407–18.
Zeng X, Goetz JA, Suber LM, Scott Jr WJ, Schreiner CM, Robbins DJ. A freely diffusible form of Sonic hedgehog mediates long-range signalling. Nature. 2001;411(6838):716–20.
Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11(5):331–44.
Pedersen LB, Veland IR, Schroder JM, Christensen ST. Assembly of primary cilia. Dev Dyn. 2008;237(8):1993–2006.
Das A, Guo W. Rabs and the exocyst in ciliogenesis, tubulogenesis and beyond. Trends Cell Biol. 2011;21(7):383–6.
Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–6.
Beachy PA, Hymowitz SG, Lazarus RA, Leahy DJ, Siebold C. Interactions between Hedgehog proteins and their binding partners come into view. Genes Dev. 2010;24(18):2001–12.
Izzi L, Levesque M, Morin S, Laniel D, Wilkes BC, Mille F, Krauss RS, McMahon AP, Allen BL, Charron F. Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev Cell. 2011;20(6):788–801.
Allen BL, Song JY, Izzi L, Althaus IW, Kang JS, Charron F, Krauss RS, McMahon AP. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev Cell. 2011;20(6):775–87.
Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999;397(6720):617–21.
Wang Y, Zhou Z, Walsh CT, McMahon AP. Selective translocation of intracellular Smoothened to the primary cilium in response to Hedgehog pathway modulation. Proc Natl Acad Sci U S A. 2009;106(8):2623–8.
Milenkovic L, Scott MP, Rohatgi R. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J Cell Biol. 2009;187(3):365–74.
Rohatgi R, Milenkovic L, Corcoran RB, Scott MP. Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc Natl Acad Sci U S A. 2009;106(9):3196–201.
Denef N, Neubuser D, Perez L, Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell. 2000;102(4):521–31.
Li S, Chen Y, Shi Q, Yue T, Wang B, Jiang J. Hedgehog-regulated ubiquitination controls smoothened trafficking and cell surface expression in Drosophila. PLoS Biol. 2012;10(1):e1001239.
Bijlsma MF, Spek CA, Zivkovic D, van de Water S, Rezaee F, Peppelenbosch MP. Repression of smoothened by patched-dependent (pro-)vitamin D3 secretion. PLoS Biol. 2006;4(8):e232.
Roberts B, Casillas C, Alfaro AC, Jagers C, Roelink H. Patched1 and Patched2 inhibit Smoothened non-cell autonomously. Elife. 2016;5:e17634.
Dwyer JR, Sever N, Carlson M, Nelson SF, Beachy PA, Parhami F. Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. J Biol Chem. 2007;282(12):8959–68.
Nachtergaele S, Mydock LK, Krishnan K, Rammohan J, Schlesinger PH, Covey DF, Rohatgi R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat Chem Biol. 2012;8(2):211–20.
Corcoran RB, Scott MP. Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc Natl Acad Sci U S A. 2006;103(22):8408–13.
Myers BR, Sever N, Chong YC, Kim J, Belani JD, Rychnovsky S, Bazan JF, Beachy PA. Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev Cell. 2013;26(4):346–57.
Nedelcu D, Liu J, Xu Y, Jao C, Salic A. Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nat Chem Biol. 2013;9(9):557–64.
Nachtergaele S, Whalen DM, Mydock LK, Zhao Z, Malinauskas T, Krishnan K, Ingham PW, Covey DF, Siebold C, Rohatgi R. Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. Elife. 2013;2:e01340.
Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci U S A. 2002;99(22):14071–6.
Sever N, Mann RK, Xu L, Snell WJ, Hernandez-Lara CI, Porter NA, Beachy PA. Endogenous B-ring oxysterols inhibit the Hedgehog component Smoothened in a manner distinct from cyclopamine or side-chain oxysterols. Proc Natl Acad Sci U S A. 2016;113(21). doi: 10.1073/pnas.1604984113.
Luchetti G, Sircar R, Kong JH, Nachtergaele S, Sagner A, Byrne EF, Covey DF, Siebold C, Rohatgi R. Cholesterol activates the G-protein coupled receptor Smoothened to promote morphogenetic signaling. Elife. 2016;5:e20304.
Byrne EF, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, Tully MD, Mydock-McGrane L, Covey DF, Rambo RP, et al. Structural basis of Smoothened regulation by its extracellular domains. Nature. 2016;535(7613):517–22.
Huang P, Nedelcu D, Watanabe M, Jao C, Kim Y, Liu J, Salic A. Cellular cholesterol directly activates Smoothened in hedgehog signaling. Cell. 2016;166(5):1176–87. e1114.
Xiao X, Tang JJ, Peng C, Wang Y, Fu L, Qiu ZP, Xiong Y, Yang LF, Cui HW, He XL, et al. Cholesterol modification of Smoothened is required for hedgehog signaling. Mol Cell. 2017;66(1):154–62. e110.
Cooper MK, Wassif CA, Krakowiak PA, Taipale J, Gong R, Kelley RI, Porter FD, Beachy PA. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. 2003;33(4):508–13.
Blassberg R, Macrae JI, Briscoe J, Jacob J. Reduced cholesterol levels impair Smoothened activation in Smith-Lemli-Opitz syndrome. Hum Mol Genet. 2016;25(4):693–705.
Xu L, Mirnics K, Bowman AB, Liu W, Da J, Porter NA, Korade Z. DHCEO accumulation is a critical mediator of pathophysiology in a Smith-Lemli-Opitz syndrome model. Neurobiol Dis. 2012;45(3):923–9.
Meljon A, Watson GL, Wang Y, Shackleton CH, Griffiths WJ. Analysis by liquid chromatography-mass spectrometry of sterols and oxysterols in brain of the newborn Dhcr7(Delta3-5/T93M) mouse: a model of Smith-Lemli-Opitz syndrome. Biochem Pharmacol. 2013;86(1):43–55.
Koide T, Hayata T, Cho KW. Negative regulation of Hedgehog signaling by the cholesterogenic enzyme 7-dehydrocholesterol reductase. Development. 2006;133(12):2395–405.
Lauth M, Rohnalter V, Bergstrom A, Kooshesh M, Svenningsson P, Toftgard R. Antipsychotic drugs regulate hedgehog signaling by modulation of 7-dehydrocholesterol reductase levels. Mol Pharmacol. 2010;78(3):486–96.
Bidet M, Joubert O, Lacombe B, Ciantar M, Nehme R, Mollat P, Bretillon L, Faure H, Bittman R, Ruat M, et al. The hedgehog receptor patched is involved in cholesterol transport. PLoS One. 2011;6(9):e23834.
Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280(5369):1603–07.
Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development. 1998;125(18):3553–62.
Incardona JP, Roelink H. The role of cholesterol in Shh signaling and teratogen-induced holoprosencephaly. Cell Mol Life Sci. 2000;57(12):1709–19.
Sugii S, Lin S, Ohgami N, Ohashi M, Chang CC, Chang TY. Roles of endogenously synthesized sterols in the endocytic pathway. J Biol Chem. 2006;281(32):23191–206.
Choudhury A, Sharma DK, Marks DL, Pagano RE. Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Mol Biol Cell. 2004;15(10):4500–11.
Chen H, Yang J, Low PS, Cheng JX. Cholesterol level regulates endosome motility via Rab proteins. Biophys J. 2008;94(4):1508–20.
Boehlke C, Bashkurov M, Buescher A, Krick T, John AK, Nitschke R, Walz G, Kuehn EW. Differential role of Rab proteins in ciliary trafficking: Rab23 regulates smoothened levels. J Cell Sci. 2010;123(Pt 9):1460–7.
Eggenschwiler JT, Espinoza E, Anderson KV. Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature. 2001;412(6843):194–8.
Evans TM, Ferguson C, Wainwright BJ, Parton RG, Wicking C. Rab23, a negative regulator of hedgehog signaling, localizes to the plasma membrane and the endocytic pathway. Traffic. 2003;4(12):869–84.
Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201–13.
Kanerva K, Uronen RL, Blom T, Li S, Bittman R, Lappalainen P, Peranen J, Raposo G, Ikonen E. LDL cholesterol recycles to the plasma membrane via a Rab8a-Myosin5b-actin-dependent membrane transport route. Dev Cell. 2013;27(3):249–62.
Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, Saier Jr MH. The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J Mol Microbiol Biotechnol. 1999;1(1):107–25.
Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of Smoothened. Nature. 2002;418(6900):892–7.
Johnson RL, Zhou L, Bailey EC. Distinct consequences of sterol sensor mutations in Drosophila and mouse patched homologs. Dev Biol. 2002;242(2):224–35.
Strutt H, Thomas C, Nakano Y, Stark D, Neave B, Taylor AM, Ingham PW. Mutations in the sterol-sensing domain of Patched suggest a role for vesicular trafficking in Smoothened regulation. Curr Biol. 2001;11(8):608–13.
Khaliullina H, Panakova D, Eugster C, Riedel F, Carvalho M, Eaton S. Patched regulates Smoothened trafficking using lipoprotein-derived lipids. Development. 2009;136(24):4111–21.
Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71(2):435–44.
Incardona JP, Gruenberg J, Roelink H. Sonic hedgehog induces the segregation of patched and smoothened in endosomes. Curr Biol. 2002;12(12):983–95.
Yavari A, Nagaraj R, Owusu-Ansah E, Folick A, Ngo K, Hillman T, Call G, Rohatgi R, Scott MP, Banerjee U. Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev Cell. 2010;19(1):54–65.
Jiang K, Liu Y, Fan J, Zhang J, Li XA, Evers BM, Zhu H, Jia J. PI(4)P promotes phosphorylation and conformational change of Smoothened through interaction with its C-terminal tail. PLoS Biol. 2016;14(2):e1002375.
Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, Jackson PK. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 2010;24(19):2180–93.
Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, Jackson PK. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell. 2013;152(1-2):210–23.
Patterson VL, Damrau C, Paudyal A, Reeve B, Grimes DT, Stewart ME, Williams DJ, Siggers P, Greenfield A, Murdoch JN. Mouse hitchhiker mutants have spina bifida, dorso-ventral patterning defects and polydactyly: identification of Tulp3 as a novel negative regulator of the Sonic hedgehog pathway. Hum Mol Genet. 2009;18(10):1719–39.
Norman RX, Ko HW, Huang V, Eun CM, Abler LL, Zhang Z, Sun X, Eggenschwiler JT. Tubby-like protein 3 (TULP3) regulates patterning in the mouse embryo through inhibition of Hedgehog signaling. Hum Mol Genet. 2009;18(10):1740–54.
Garcia-Gonzalo FR, Phua SC, Roberson EC, Garcia 3rd G, Abedin M, Schurmans S, Inoue T, Reiter JF. Phosphoinositides regulate ciliary protein trafficking to modulate hedgehog signaling. Dev Cell. 2015;34(4):400–9.
Chavez M, Ena S, Van Sande J, de Kerchove d’Exaerde A, Schurmans S, Schiffmann SN. Modulation of ciliary phosphoinositide content regulates trafficking and Sonic hedgehog signaling output. Dev Cell. 2015;34(3):338–50.
Jacob J, Briscoe J. Gli proteins and the control of spinal-cord patterning. EMBO Rep. 2003;4(8):761–5.
Matz-Soja M, Rennert C, Schonefeld K, Aleithe S, Boettger J, Schmidt-Heck W, Weiss TS, Hovhannisyan A, Zellmer S, Kloting N, et al. Hedgehog signaling is a potent regulator of liver lipid metabolism and reveals a GLI-code associated with steatosis. Elife. 2016;5:e13308.
Teperino R, Amann S, Bayer M, McGee SL, Loipetzberger A, Connor T, Jaeger C, Kammerer B, Winter L, Wiche G, et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell. 2012;151(2):414–26.
Hardie DG. AMPK--sensing energy while talking to other signaling pathways. Cell Metab. 2014;20(6):939–52.
Sim AT, Hardie DG. The low activity of acetyl-CoA carboxylase in basal and glucagon-stimulated hepatocytes is due to phosphorylation by the AMP-activated protein kinase and not cyclic AMP-dependent protein kinase. FEBS Lett. 1988;233(2):294–8.
Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13(4):376–88.
Carling D, Clarke PR, Zammit VA, Hardie DG. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur J Biochem. 1989;186(1-2):129–36.
Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25(9):545–55.
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26.
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–90.
Sharpe HJ, Wang W, Hannoush RN, de Sauvage FJ. Regulation of the oncoprotein Smoothened by small molecules. Nat Chem Biol. 2015;11(4):246–55.
Acknowledgements
The authors are grateful for helpful comments by the reviewer.
Author information
Authors and Affiliations
Contributions
JJ conceived the review and RB and JJ wrote the manuscript. Both authors read and approved the final version.
Corresponding author
Ethics declarations
Competing interests
JJ is funded by the National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) Oxford and by the Cancer Research UK (CRUK) Oxford Centre Development Fund. RB declares no competing interests. This work was supported by a grant from the Simons Foundation (202084).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Blassberg, R., Jacob, J. Lipid metabolism fattens up hedgehog signaling. BMC Biol 15, 95 (2017). https://doi.org/10.1186/s12915-017-0442-y
Published:
DOI: https://doi.org/10.1186/s12915-017-0442-y