
Abstract
Background
Rapid advancement of next generation sequencing technologies such as whole genome sequencing (WGS) has facilitated the search for genetic factors that influence disease risk in the field of human genetics. To identify rare variants associated with human diseases or traits, an efficient genome-wide binning approach is needed. In this study we developed a novel biological knowledge-based binning approach for rare-variant association analysis and then applied the approach to structural neuroimaging endophenotypes related to late-onset Alzheimer’s disease (LOAD).
Methods
For rare-variant analysis, we used the knowledge-driven binning approach implemented in Bin-KAT, an automated tool, that provides 1) binning/collapsing methods for multi-level variant aggregation with a flexible, biologically informed binning strategy and 2) an option of performing unified collapsing and statistical rare variant analyses in one tool. A total of 750 non-Hispanic Caucasian participants from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort who had both WGS data and magnetic resonance imaging (MRI) scans were used in this study. Mean bilateral cortical thickness of the entorhinal cortex extracted from MRI scans was used as an AD-related neuroimaging endophenotype. SKAT was used for a genome-wide gene- and region-based association analysis of rare variants (MAF (minor allele frequency) < 0.05) and potential confounding factors (age, gender, years of education, intracranial volume (ICV) and MRI field strength) for entorhinal cortex thickness were used as covariates. Significant associations were determined using FDR adjustment for multiple comparisons.
Results
Our knowledge-driven binning approach identified 16 functional exonic rare variants in FANCC significantly associated with entorhinal cortex thickness (FDR-corrected p-value < 0.05). In addition, the approach identified 7 evolutionary conserved regions, which were mapped to FAF1, RFX7, LYPLAL1 and GOLGA3, significantly associated with entorhinal cortex thickness (FDR-corrected p-value < 0.05). In further analysis, the functional exonic rare variants in FANCC were also significantly associated with hippocampal volume and cerebrospinal fluid (CSF) Aβ1–42 (p-value < 0.05).
Conclusions
Our novel binning approach identified rare variants in FANCC as well as 7 evolutionary conserved regions significantly associated with a LOAD-related neuroimaging endophenotype. FANCC (fanconi anemia complementation group C) has been shown to modulate TLR and p38 MAPK-dependent expression of IL-1β in macrophages. Our results warrant further investigation in a larger independent cohort and demonstrate that the biological knowledge-driven binning approach is a powerful strategy to identify rare variants associated with AD and other complex disease.
Similar content being viewed by others


Background
Rapid advances in next-generation sequencing technologies and bioinformatics tools over the past decade have made an important contribution to searching for disease susceptibility factors and understanding the impact of the genetic variation on human diseases [1, 2]. In particular, since the completion of the human genome project, whole genome sequencing (WGS) has been increasingly used as a tool to understand the complexity and diversity of genomes in disease by performing detailed evaluation of all genetic variation [3, 4].
Late-onset Alzheimer’s disease (LOAD) is the most prevalent form of age-related neurodegenerative disease and dementia [5]. Abnormal proteins forming histologically visible structures, amyloid plaques and neurofibrillary tangles, damage and destroy neurons and their connections [6]. With the increasing population of aging adults, it is predicted that the number of AD patients will triple in the United States by 2050 [7]. Models suggest that delaying the onset of AD by 5 years through early intervention could reduce the number of AD cases by nearly 50% [8, 9]. To develop effective therapeutic intervention to slow or prevent disease progression and to effectively target potential disease-modifying approaches, early biomarkers are needed to detect AD at pre-symptomatic stages with high accuracy and monitor the pathological progression. With an estimated heritability of about 80%, genetic factors play an important role in developing AD [10, 11]. Very recently, genetic association studies have used next-generation sequencing technologies to identify functional risk rare variants with moderate to large effects on LOAD risk within TREM2, ABCA7, UNC5C, AKAP9 and PLD3 genes [12,13,14].
For a rare-variant association analysis, gene- or region-based multiple-variant tests have been widely used due to improved power over single variant tests. There exist several different approaches in multiple-variant tests. Burden methods test the cumulative effect of variants within a knowledge-driven region such as genes and are easily applied to case–control studies as they assess the frequency of variant counts between these binary phenotypes. Burden tests, which collapse variants to a single genetic score, are powerful when the variants have the same effect direction with similar magnitudes [15]. When this assumption is violated, however, it can result in a significant loss of power. Variance component tests, such as sequence kernel association test (SKAT), were developed to overcome this limitation [16]. SKAT is a score-based variance component test that uses a multiple regression kernel-based approach to assess variant distribution and test for association. These are more powerful than Burden tests in the presence of opposite association directions or large numbers of non-causal variants [16].
A rare-variant study requires careful consideration, including choice of variant collapsing or binning approach for region-based association analysis. In this study, we propose a novel biological knowledge-driven binning approach (Bin-KAT) to identify trait- and disease-associated rare variants. Bin-KAT is a comprehensive, streamlined approach that unifies a genome-wide variant binning function in BioBin [17,18,19,20,21] and a dispersion-based association analysis tool such as SKAT [16, 22].
Methods
Study subjects and whole genome sequencing (WGS) analysis
This study utilized data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. The ADNI cohort consisted of cognitively normal older adults (CN), mild cognitive impairment (MCI) and early AD. We downloaded demographic information, raw MRI scan data, whole genome sequencing data and diagnostic information from the ADNI data repository (http://www.loni.usc.edu/ADNI/) [23]. All participants provided written informed consent and study protocols were approved by each participating sites’ Institutional Review Board. WGS was performed by Illumina on blood-derived genomic DNA samples obtained from 818 ADNI participants using paired-end 100-bp reads on the Illumina HiSeq2000 (www.illumina.com). As described previously in detail [24, 25], Broad GATK and BWA-mem were used to align raw sequence data to the reference human genome (human genome build 37) and call the variants.
Neuroimaging analysis
All available structural MRI scans at baseline acquired following the ADNI MRI protocol were downloaded from the ADNI data repository [26]. A widely employed automated MRI analysis technique, FreeSurfer (http://surfer.nmr.mgh.harvard.edu/), for automated segmentation and parcellation, was used to process MRI scans and extract mean volumes and cortical thicknesses (Euclidean distance between the grey/white boundary and the grey/cerebrospinal fluid boundary) for all target regions. In this analysis, we used the bilateral mean value of the entorhinal cortex thickness as an AD-related endophenotype as the entorhinal cortex is a region known to be affected early in AD.
Knowledge-driven binning approach
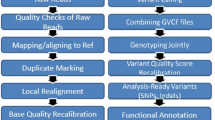
As a variant binning tool, BioBin aggregates variants into multiple user-selected features in a biologically informed manner using an internal biological data repository known as LOKI or the Library of Knowledge Integration. LOKI integrates multiple public databases including NCBI Entrez Gene, UCSC Genome Browser, Protein families (Pfam), Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, Genome Ontology (GO) and others, into one centralized data bank. Using these rich data sources, variants can be binned into various biological features such as genes, pathways, protein families, evolutionary conserved regions (ECRs), regulatory regions and others. The main utility of BioBin is a direct access to a comprehensive knowledge-guided binning approach for multiple biological features. Simultaneous to variant binning, a user can perform a phenotypic association analysis using selected burden tests (regression or the Wilcoxon rank sum) or dispersion tests (SKAT) directly within the framework of BioBin. Our knowledge-driven binning approach (Bin-KAT) was applied to determine the association of rare variants with LOAD-related neuroimaging endophenotype, entorhinal cortex thickness (Fig. 1), while adjusting for age, gender, years of education, intracranial volume (ICV) and MRI field strength. Functional exonic rare variants (minor allele frequency (MAF) < 0.05) extracted from the WGS data using ANNOVAR [27] were binned by five different biological features, genes, KEGG pathway, protein families, regulatory regions and ECRs (Fig. 1). A minimum bin size of 5 variants was used. Binned variants were weighted inversely proportional to their MAF using Madsen and Browning weighting [28].

Illustration of rare variant association analysis using Bin-KAT for neuroimaging genomics. First, rare variants were binned/collapsed based on biological knowledge, such as exon, gene, pathway, protein family, evolutionary conversed regions (ECR) or regulatory region, using BioBin. Then, statistical tests including a burden test and a dispersion test (SKAT), were incorporated into BioBin, called Bin-KAT [19]. Bin-KAT provides an option of performing unified rare variant association analysis methods in one tool to identify biologically-informed bins significantly associated with imaging endophenotypes of interest. VCF, variant call format
Results
Genome-wide gene-based association analysis of functional exonic rare variants with LOAD-related neuroimaging endophenotype
In order to remove spurious association in disease studies due to population stratification, a total of 750 non-Hispanic Caucasian ADNI participants who had both WGS data and MRI scans were used in this study [29]. The population demographics are shown in Table 1. From the WGS-identified variants, ANNOVAR identified 205,136 functional exonic variants. Among 205,136 variants, 188,508 rare variants (MAF < 0.05) were selected for the analysis. A genome-wide gene-based association analysis of rare variants with entorhinal cortex thickness using a burden-based approach did not identify any genes that exceeded a genome-wide significant threshold (FDR-corrected p-value < 0.05) (data not shown). However, a dispersion-based approach (SKAT) identified a gene, FANCC, which consisted of 16 functional exonic rare variants, achieved a genome-wide significant association with entorhinal cortex thickness (p-value < 2 x 10−6; FDR-corrected p-value < 0.05) (Fig. 2). To further investigate the effect of rare variants in FANCC on phenotypic variation, we re-ran SKAT for FANCC after removing one variant at a time and identified that rs1800361 out of 16 variants in FANCC had the strongest effect on entorhinal cortex thickness (Table 2). In addition, the functional exonic rare variants in FANCC were also associated with hippocampal volume and cerebrospinal fluid (CSF) Aβ1–42 (p-value < 0.05).

Manhattan plot of genome-wide gene-based rare variant association analysis for a LOAD-related neuroimaging endophenotype, entorhinal cortex thickness. –log10 p-value was plotted against the chromosomal location of each gene. FANCC exceeded the genome-wide significant threshold (FDR-corrected p-value = 0.05) (red line)
There were several genes marginally associated with entorhinal cortex thickness. Top 10 genes including FANCC were obtained based on SKAT p-values (Table 3). In particular, five genes (RFX7, SORCS2, FAF1, ABCA5 and NCF4) were marginally significant within FDR-corrected p-value < 0.1 (Table 3). To identify a functional relationship between top 5 genes, we performed the Integrated Multi-species Prediction (IMP) that combines biological evidence from multiple biological databases and provides a probability score that two genes are involved in a biological and functional relationship [30]. Figure 3 shows that FANCC, RFX7, FAF1 and ABCA5 are likely to be involved in the same biological process.

Functional networks based on top 5 genes associated with entorhinal cortex thickness. The Integrated Multi-species Prediction (IMP) performs a graphical search of a functional network to identify the genes most likely to participate in similar pathways as query genes including FANCC, RFX1, FAF1, ABCA5 and SORCS2. Nodes represent genes and edges represent the predicted probability that the connected genes are involved in the same biological process. Large nodes represent query genes and the color of the edge signifies the strength of the relationship confidence. Red edge represents higher confidence scores between nodes
Knowledge-based binning approach for an association analysis of rare variants
In addition to a gene rare variant analysis approach, our biological knowledge-based binning approach based on KEGG pathway, Pfam, ECRs and regulatory regions was performed. None of biologically-informed bins was significant when the burden-based approach was used (data not shown). However, the dispersion approach (SKAT) identified 7 evolutionary conserved regions, which were mapped to FAF1, RFX7, LYPLAL1 and GOLGA3, significantly associated with entorhinal cortex thickness (FDR-corrected p-value < 0.05) (Table 4).
Discussion
In this study we developed a novel knowledge-driven binning approach for rare-variant association analysis and then applied the approach to whole genome sequencing data to identify rare variants associated with a neuroimaging endophenotype related to LOAD. Our results showed that (1) the novel binning approach is useful to identify trait- and disease-associated rare variants; (2) a dispersion-based test (SKAT) outperforms a regression-based burden test [19]; and (3) quantitative traits (QT) as phenotypes substantially increase detection power for association analysis.
The biological knowledge-based binning approach identified rare variants in FANCC (Fanconi anemia complementation group C) as well as 7 evolutionary conserved regions significantly associated with a LOAD-related neuroimaging endophenotype, entorhinal cortex thickness. The entorhinal cortex (EC) is a region that is affected early in the progression of AD and one of the first sites of tau pathology, and the entorhinal cortex thickness was shown to predict cognitive decline in AD [31, 32].
Although the relationship between Fanconi anemia (FA) genes and AD has not been identified yet, there are some genetic modulators playing a role in FA and AD pathology. FA genes include several complementation groups [33, 34]. FA proteins form the complexes with each other against genotoxic stress for the survival of the hematopoietic and germ cells [33]. In addition to playing a role in the FA complex during homologous recombination repair, FANCC has the other crucial function in hematopoietic cells by protecting them from apoptosis [33, 35]. FANCC has been shown to modulate TLR and p38 MAPK-dependent expression of IL-1β in macrophages [36]. FANCC −/− mice produce 2.5 times more interleukin 1β (IL-1β) than wild type and in human CD14+ cells [37]. In addition to these roles of IL-1β and MAP kinases in the FA pathway, IL-1β and p38 MAPK and JNK were significantly related to Aβ-induced EC synaptic dysfunction by involving the receptor for advanced glycation end products (RAGE) signaling in microglia in AD mice model [38]. FANCC binds and regulates the phosphorylation of the Stathmin-1 (STMN1) that is crucial for the spindle organization during mitosis [39]. In addition, a microarray expression study showed that STMN1 is differentially expressed in AD and associated with calcium hemostasis in the human brain [40].
The evolutionary conserved regions (ECRs) we identified to be associated with entorhinal cortex thickness were also linked to the MAPK-p38 pathway [41, 42]. The ECRs are often required for basic cellular or metabolic function; finding ECRs is a useful method for identifying functional sequences in a genome. Several ECRs were identified to be associated with entorhinal cortex thickness including FAF1, which was found to activate the MAPK p38 signaling pathway [43]. FAF1 has also been found to be overexpressed in the frontal cortex of Parkinson’s disease (PD) as well as PD and AD patients [44]. GOLGA3 (golgin A3) has been found to have upregulated expression in AD possibly by promoting cell surface expression of the beta1-adrenergic receptor [45]. RFX7 plays an important role in the development of the neural tube during embryogenesis [46], and is highly expressed in various brain tissues [47]. Since the genes we mentioned above were related to the pathways common with AD pathology, these genes may be a potential target for future therapeutics to treat neurodegenerative disease and cognitive decline.
Conclusions
To conclude, our results warrant further investigation in a larger independent cohort and demonstrate that the knowledge-driven binning approach using Bin-KAT is a powerful strategy to identify rare variants associated with AD and other complex disease. Bin-KAT has previously shown to be successful in a multiple phenotype and multiple biological feature analysis [19]. This software package is open source and freely available from http://ritchielab.com/software/biobin-download.
References
Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46.
Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155(1):27–38.
Ng PC, Kirkness EF. Whole genome sequencing. Methods Mol Biol. 2010;628:215–26.
Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11(6):415–25.
Alzheimer’s A. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015;11(3):332–84.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39(3):409–21.
Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology. 2013;80(19):1778–83.
Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88(9):1337–42.
Wilson D, Peters R, Ritchie K, Ritchie CW. Latest advances on interventions that may prevent, delay or ameliorate dementia. Ther Adv Chronic Dis. 2011;2(3):161–73.
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–74.
Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(10):a006296.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–27.
Steinberg S, Stefansson H, Jonsson T, Johannsdottir H, Ingason A, Helgason H, Sulem P, Magnusson OT, Gudjonsson SA, Unnsteinsdottir U, et al. Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet. 2015;47(5):445–7.
Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, Harari O, Norton J, Budde J, Bertelsen S, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505(7484):550–4.
Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95(1):5–23.
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89(1):82–93.
Moore CB, Basile AO, Wallace JR, Frase AT, Ritchie MD. A biologically informed method for detecting rare variant associations. BioData Mining. 2016;9:27.
Kim D, Li R, Dudek SM, Wallace JR, Ritchie MD. Binning somatic mutations based on biological knowledge for predicting survival: an application in renal cell carcinoma. Pac Symp Biocomput. 2015:96–107.
Basile AO, Wallace JR, Peissig P, McCarty CA, Brilliant M, Ritchie MD. Knowledge driven binning and phewas analysis in Marshfield personalized medicine research project using biobin. Pac Symp Biocomput. 2016;21:249–60.
Moore CB, Wallace JR, Frase AT, Pendergrass SA, Ritchie MD. BioBin: a bioinformatics tool for automating the binning of rare variants using publicly available biological knowledge. BMC Med Genomics. 2013;6 Suppl 2:S6.
Moore CB, Wallace JR, Frase AT, Pendergrass SA, Ritchie MD. Using BioBin to explore rare variant population stratification. Pac Symp Biocomput. 2013:332–343.
Lee S, Wu MC, Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13(4):762–75.
Saykin AJ, Shen L, Foroud TM, Potkin SG, Swaminathan S, Kim S, Risacher SL, Nho K, Huentelman MJ, Craig DW, et al. Alzheimer’s disease neuroimaging initiative biomarkers as quantitative phenotypes: genetics core aims, progress, and plans. Alzheimers Dement. 2010;6(3):265–73.
Nho K, Horgusluoglu E, Kim S, Risacher SL, Kim D, Foroud T, Aisen PS, Petersen RC, Jack Jr CR, Shaw LM, et al. Integration of bioinformatics and imaging informatics for identifying rare PSEN1 variants in Alzheimer’s disease. BMC Med Genomics. 2016;9 Suppl 1:30.
Nho K, West JD, Li H, Henschel R, Bharthur A, Tavares MC, Saykin AJ. Comparison of multi-sample variant calling methods for whole genome sequencing. IEEE Int Conf Systems Biol. 2014;2014:59–62.
Jack Jr CR, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ JLW, Ward C, et al. The Alzheimer’s disease neuroimaging initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27(4):685–91.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5(2):e1000384.
Price AL, Zaitlen NA, Reich D, Patterson N. New approaches to population stratification in genome-wide association studies. Nat Rev Genet. 2010;11(7):459–63.
Wong AK, Park CY, Greene CS, Bongo LA, Guan Y, Troyanskaya OG. IMP: a multi-species functional genomics portal for integration, visualization and prediction of protein functions and networks. Nucleic Acids Res. 2012;40(Web Server issue):W484–490.
Velayudhan L, Proitsi P, Westman E, Muehlboeck JS, Mecocci P, Vellas B, Tsolaki M, Kloszewska I, Soininen H, Spenger C, et al. Entorhinal cortex thickness predicts cognitive decline in Alzheimer’s disease. J Alzheimers Dis. 2013;33(3):755–66.
Fu H, Hussaini SA, Wegmann S, Profaci C, Daniels JD, Herman M, Emrani S, Figueroa HY, Hyman BT, Davies P, et al. 3D visualization of the temporal and spatial spread of tau pathology reveals extensive sites of tau accumulation associated with neuronal loss and recognition memory deficit in aged tau transgenic mice. PLoS One. 2016;11(7):e0159463.
Bagby Jr GC. Genetic basis of fanconi anemia. Curr Opin Hematol. 2003;10(1):68–76.
Zhang X, Li J, Sejas DP, Rathbun KR, Bagby GC, Pang Q. The Fanconi anemia proteins functionally interact with the protein kinase regulated by RNA (PKR). J Biol Chem. 2004;279(42):43910–9.
Wang J, Otsuki T, Youssoufian H, Foe JL, Kim S, Devetten M, Yu J, Li Y, Dunn D, Liu JM. Overexpression of the fanconi anemia group C gene (FAC) protects hematopoietic progenitors from death induced by Fas-mediated apoptosis. Cancer Res. 1998;58(16):3538–41.
Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA. p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med. 2009;15(8):369–79.
Parajuli B, Sonobe Y, Horiuchi H, Takeuchi H, Mizuno T, Suzumura A. Oligomeric amyloid beta induces IL-1beta processing via production of ROS: implication in Alzheimer’s disease. Cell Death Dis. 2013;4:e975.
Origlia N, Bonadonna C, Rosellini A, Leznik E, Arancio O, Yan SS, Domenici L. Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J Neurosci. 2010;30(34):11414–25.
Magron A, Elowe S, Carreau M. The fanconi anemia C protein binds to and regulates stathmin-1 phosphorylation. PLoS One. 2015;10(10):e0140612.
Saetre P, Jazin E, Emilsson L. Age-related changes in gene expression are accelerated in Alzheimer’s disease. Synapse. 2011;65(9):971–4.
Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology. 2010;58(3):561–8.
Sun A, Liu M, Nguyen XV, Bing G. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp Neurol. 2003;183(2):394–405.
Juo P, Kuo CJ, Reynolds SE, Konz RF, Raingeaud J, Davis RJ, Biemann HP, Blenis J. Fas activation of the p38 mitogen-activated protein kinase signalling pathway requires ICE/CED-3 family proteases. Mol Cell Biol. 1997;17(1):24–35.
Betarbet R, Anderson LR, Gearing M, Hodges TR, Fritz JJ, Lah JJ, Levey AI. Fas-associated factor 1 and Parkinson’s disease. Neurobiol Dis. 2008;31(3):309–15.
Hicks SW, Horn TA, McCaffery JM, Zuckerman DM, Machamer CE. Golgin-160 promotes cell surface expression of the beta-1 adrenergic receptor. Traffic. 2006;7(12):1666–77.
Manojlovic Z, Earwood R, Kato A, Stefanovic B, Kato Y. RFX7 is required for the formation of cilia in the neural tube. Mech Dev. 2014;132:28–37.
Aftab S, Semenec L, Chu JS, Chen N. Identification and characterization of novel human tissue-specific RFX transcription factors. BMC Evol Biol. 2008;8:226.
Acknowledgments
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. Samples from the National Cell Repository for AD (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (AIG), were used in this study. Funding for the WGS was provided by the Alzheimer’s Association and the Brin Wojcicki Foundation.
Funding
Additional support for data analysis was provided by NLM R00 LM011384, NIA R01 AG19771, NIA P30 AG10133, NLM R01 LM011360, DOD W81XWH-14-2-0151, NCAA 14132004 and NCATS UL1 TR001108. This project was also funded, in part, under a grant with the Pennsylvania Department of Health (#SAP 4100070267). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. In addition, the publication charge for this article was funded by DK’s startup funding at Geisinger Health System.
Availability of data and materials
Demographic information, raw neuroimaging scan data, APOE and whole genome sequencing data, neuropsychological test scores and diagnostic information are available from the ADNI data repository (http://www.loni.usc.edu/ADNI/). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
Authors’ contributions
DK and KN designed and developed the research, and also performed the experiments. MDR, SL and AJS provided experienced guidance. AOB, LB, EH and SL collected medical literature and made up supporting materials to infer the results. DK, AOB and KN wrote the manuscript and all authors read the manuscript and approved it.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable
Ethics approval and consent to participate
Not applicable
About this supplement
This article has been published as part of BMC Medical Informatics and Decision Making Volume 17 Supplement 1, 2017: Selected articles from the 6th Translational Bioinformatics Conference (TBC 2016): medical informatics and decision making. The full contents of the supplement are available online at <https://bmcmedinformdecismak.biomedcentral.com/articles/supplements/volume-17-supplement-1>
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kim, D., Basile, A.O., Bang, L. et al. Knowledge-driven binning approach for rare variant association analysis: application to neuroimaging biomarkers in Alzheimer’s disease. BMC Med Inform Decis Mak 17 (Suppl 1), 61 (2017). https://doi.org/10.1186/s12911-017-0454-0
Published:
DOI: https://doi.org/10.1186/s12911-017-0454-0