
Abstract
Background
Sorafenib (Sor) is the only approved multikinase inhibitor indicated for the treatment of HCC. Previous studies have shown that amygdalin (Amy) possesses anticancer activities against several cancer cell lines; we suggested that these compounds might disrupt AMPK/mTOR and BCL-2. Therefore, the current study used integrated in vitro and in silico approaches to figure out Amy and Sor’s possible synergistic activity in targeting AMPK/mTOR and BCL-2 for anti-angiogenesis and apoptosis cell death in HepG2 cells.
Results
Notably, Amy demonstrated exceptional cytotoxic selectivity against HepG2 cells in comparison to normal WI-38 cells (IC50 = 5.21 mg/ml; 141.25 mg/ml), respectively. In contrast, WI-38 cells were far more sensitive to the toxicity of Sor. A substantial synergistic interaction between Amy and Sor was observed (CI50 = 0.56), which was connected to cell cycle arrest at the S and G2/M stages and increased apoptosis and potential necroptosis. Amy and Sor cotreatment resulted in the highest glutathione levels and induction of pro-autophagic genes AMPK, HGMB1, ATG5, Beclin 1, and LC3, suppressed the mTOR and BCL2 anti-apoptotic gene. Finally, the docking studies proposed that Amy binds to the active site of the AMPK enzyme, thus inhibiting its activity. This inhibition of AMPK ultimately leads to inhibition of mTOR and thus induces apoptosis in the HepG2 cells.
Conclusion
Although more in vivo research using animal models is needed to confirm the findings, our findings contribute to the evidence supporting Amy’s potential anticancer effectiveness as an alternative therapeutic option for HCC.
Similar content being viewed by others


Background
The most common malignant tumor, hepatocellular carcinoma (HCC), arises from well-differentiated hepatocytes [1]. It is the most common type of malignant tumor observed worldwide, and it usually results in death within 6–20 months [2, 3]. Biological occurrences connected to tumor development are among HCC’s distinguishing characteristics [4]. The distinguishing characteristics include, in brief, persistent proliferative signaling, evasion of growth suppressors, prevention of cell death, the establishment of replicative immortality, induction of angiogenesis, and activation of invasion and metastasis [5, 6]. The microenvironment of the cell and the interactions of many growth factors have a role in tumor development and survival [2, 7]. The most recent data on cancer stem cells (CSC) revealed, however, that liver cancer is linked to defective signaling pathways and its receptors, including transforming growth factor (TGF) [8], vascular endothelial growth factor, fibroblast growth factor, Wnt/mitogen-activated protein kinases, phosphoinositide 3-kinase, mechanistic target of rapamycin, and Hedgehog pathways [9].
Targeting malignant disorders by attacking protein kinases has become more popular recently [10]. A member of the family of PI3Ks/mTOR is a physiologically stable serine/threonine protein kinase [11]. It performs a variety of functions, including regulating metabolism, aging, and cell proliferation [12]. Solid tumors, including HCC, have been related to mTOR up-regulation, which is typically associated with a poor prognosis and fast recurrence [13, 14]. Furthermore, genomic investigations showed that the proteins PI3K and AKT, which are engaged in the mTOR pathway’s upstream signaling, are important actors in the unregulated network signaling pathways in HCC [15,16,17]. As a result, rapamycin/rapalogs and several mTOR second-generation blockers have been developed and are now being evaluated in clinical studies for the treatment of HCC [18, 19].
In this context, several members of our group have been trying to understand molecular pathogenesis, leading to improvements in the treatment of HCC. Radical liver removal, orthotropic liver transplantation, and locoregional therapy make up the main therapeutic modalities for HCC [20, 21]. However, systemic treatment, which includes molecular targeted therapy, nutraceuticals, and chemoprevention, is the only option for uncontrolled HCC. Sorafenib (Sor) can block receptor tyrosine kinases, which are involved in the development of new blood vessels, as well as cell proliferation as a result [22]. Sor has been shown in preclinical studies to block several receptors, including VEGFR13, PDGFR, cKIT, and FLT3 [23, 24]. However, the side effects of Sor treatment also include symptoms that are frequently brought on by drugs, such as hypertension, asthma, and esophageal irritation [25, 26].
Furthermore, bevacizumab and doxorubicin have not been coupled with any other standardized drugs [27]; only Sor has been done [28]. Sor combination therapy may therefore be more effective than using different chemotherapeutic drugs alone [29]. In that regard, several phytochemical components likewise share chemopreventive and angiopreventive qualities and ought to be tested in combination with chemotherapeutic substances to lessen drug-induced toxicity [30, 31].
Amygdalin (Amy) is a naturally occurring cyanogenic glycoside that may be found in a variety of fruits, such as the kernels of apricots, peaches, and bitter almonds [32]. Amy is not toxic by itself, but the β-glucosidase enzyme decomposes it into hydrocyanic acid (HCN), which stimulates the lysosome enzymes and raises the acidity of cancer cells, causing them to lyse [33]. β-glucosidase shows 1000–3000 times higher activity in tumor cells than in normal cells due to the presence of lactate generated during cancer cell anaerobic respiration [34]. HCN can also kill cancer cells by elevating the acidic content of the cell leading the lysosome to release its enzymatic content, causing the cells to lyse [35]. Moreover, detoxification of HCN to thiocyanate requires the mitochondrial enzyme rhodanese, which is more active in normal tissues but has lower activity in cancer cells. Thus, a combination of abundant cyanide liberating b-glucosidase activity together with a deficiency of the cyanide detoxifying rhodanese activity could provide a selective advantage for the killing of cancer cells by amygdalin without having plentiful harmful effects on normal cells [36]. Additionally, Amy prevents metastasis and inhibits mitochondrial cytochrome C oxidase [37]. It can also affect other signaling pathways [38]. Therefore, the unique combination of Sor and Amy-based anti-cancer activity may be a superior choice for HCC-targeted treatment [39].
Here, this study aimed to test the innovative activity of Amy and/or Sor against HCC using the HepG2 cell line. We suggested that these compounds might disrupt the prototypical survival pathway known as AMPK/mTOR and BCL-2, which is becoming more and more associated with the development of HCC carcinogenesis [38, 40]; additionally, we assessed the simulated binding process toward AMP-activated protein kinase (AMPK)/mTOR and B-cell lymphoma 2 (BCL-2) as the molecular target for inhibition using molecular docking, and we employed quantitative PCR to analyze the genes expression.
Methods
Chemicals and reagents
Amy was purchased from Sigma Aldrich, USA ( CAS-No. 29883-15-6), Sorafenib Tosylate 200 mg from Cipla, India, and dimethyl sulfoxide (DMSO) from Sigma–Aldrich ( CAS -No. 67-68-5), USA, 3-(4,5-dimethylthiazoil-2-yl)-2,5 diphenyltetrazolium bromide [MTT] from Sigma–Aldrich, USA (CAS-No. 298-93-1), Propidium iodide (CAS-No. 25535-16-4), Ethidium bromide (CAS-No. 1239-45-8), Acridine orange CAS-No. 65-61-2), and DAPI (CAS-No. 28718-90-3) were procured from Sigma Chemicals Co. (St. Louis, MO, USA). All other analytical grade chemicals used in the study were obtained from Biomed laboratories, Egypt. Stock solutions of Sor were freshly prepared in DMSO and Amy in DMEM media. Human hepatocellular carcinoma (HepG2) and normal lung WI-38 human cell lines were obtained from VACSERA, Dokki-Giza, Egypt.
Cell Culture and treatment
The HepG2 and normal lung WI-38 human cell lines were cultured in DMEM medium (Lonza, BioWhittaker®, USA ( CAS-No. 12–614) supplemented with 10% fetal bovine serum (FBS) (Sigma, USA) (CAS-No. 1943609-65-1), and 100 µg/ml penicillin, 100 µg/ml streptomycin (Lonza, BioWhittaker®, USA)( Cat. No. DE17-602E), at 37 °C in a humidified incubator under 5% CO2. Cells were subcultured when reaching 80%-90 confluency and were split in a 1:6 ratio before treatments that were routinely performed on 40–50% confluent cells.
Except for the morphology analysis that was assessed for 24 and 48 h, all treatments were performed for 48 h using the predetermined half IC50 dose (IC50/2) of the drugs, Amy was used at 2.6 mg/ml against HepG2 and 70.6 mg/ml against WI-38 cells. On the other hand, Sor 1.1 µM and 0.29 µM doses were used against HepG2 and WI-38 cells, respectively. Finally, for the combination treatments, 2.03629 mg/ml Amy and 0.40726 µM Sor were used. Cell culture experiments were performed in triplicates, and the results of three independent experiments were used for statistical analysis.
Cell viability assay
The effect of Amy & Sor on the viability of HepG2 and WI-38 cells was determined by the MTT colorimetric assay kit (Sigma-Aldrich, USA) (CAS-No. 298-93-1). Briefly, cells were seeded at 5 × 103 cells/well in 96-well plates at 37 °C and cultured overnight before treatment with varying concentrations of Amy (0.5–160 mg/ml) and Sor (0.125-8 µM) for 48 h. MTT working solution (100 µl) was added to each well and the plates were incubated in dark at 37 °C for 4 h. The medium was removed and the purple formazan crystals were dissolved by adding 100 µL/well of DMSO for 5 min. The optical density (OD) was measured at an absorbance value of 570 nm using a microplate reader (BIORAD PR4100, USA). Cell viability was calculated in treated cells compared to control untreated cells, which were considered 100% viability and were presented in graphs for the calculation of the 50% inhibitory concentration (IC50) [41].
Combination index (CI) assay
Using MTT assay data, the combination index (CI) was calculated by the Chou-Talalay method as described by Chou et al. using CompuSyn software (CompuSyn, Inc., Paramus, NJ, USA) [42]. The dose-effect curves for single and cotreatment were generated and the CI for every dose and the corresponding effect, fraction affected (Fa) were calculated. The resultant CI values reflect the potential interactions between two drugs. CI < 1 indicates synergism, CI = 1 indicates an additive effect and CI > 1 indicates antagonism. The Dose-reduction index (DRI) was calculated from the DRI equation and algorithm using CompuSyn software, (DRI50) values represent the magnitude of dose reduction obtained for the 50% growth inhibitory effect in Amy/Sor cotreatment compared to each drug alone that causes the same growth inhibition effect.
Evaluation of the antioxidant capacity for Amy and antioxidant markers
The 2, 2-diphenyl-1-picrylhydrazyl (DPPH) (CAS No. 1898-66-4) free radical scavenging method was used to measure the free radical scavenging abilities of Amy as described in a previously published study [43]. The DPPH scavenging activity was calculated using the formula:
The concentration of the sample leading to a 50% reduction of the initial DPPH concentration (EC50) was calculated for Amy and vitamin C from Sigma–Aldrich, USA (CAS No. 50-81-7) as a reference antioxidant.
Assessment of reduced glutathione (GSH) levels in both HepG2 & WI-38 cells lysate treated with half IC50 concentrations of Amy and/or Sor for 48 h was performed as described [44].
The concentration of GSH was calculated using a standard curve by the following equation:
GSH concentration (µmol/mg protein) = absorbance / (slope × protein concentration).
Similarly, the Malondialdehyde (MDA) level as a marker of lipid peroxidation was estimated in both HepG2 & WI-38 cell lysate as described in [45].
The MDA concentration was calculated as:
MDA (nM/ml) = (Absorbance/ molar absorptivity) X (1000/sample volume in µl.
Annexin-V assay
The cell death mechanism was evaluated by flow cytometry using the annexin-V & propidium iodide (PI) double staining apoptosis detection kit (Southern Biotech, Birmingham, AL, USA) following the manufacturer’s instructions. Cells were adjusted to 1 × 106 cells/ml and plated in 6-well plates and allowed to grow for 24 h. Cells were then treated with IC50/2 concentrations of Amy, Sor and in combo for 48 h before trypsinization, washing with PBS, and fixation with ethanol for 12 h. Cells were washed with ice-cold culture medium before staining with annexin V-FITC/PI solution and incubation on ice for 10 min. Finally, cells were analyzed using the Accuri C6 flow cytometer (Becton Dickinson, Sunnyvale, CA, USA) [46].
Cell cycle analysis
For the cell cycle phase distribution analysis assay, HepG2 and WI-38 cells were starved for 24 h before treatment with IC50/2 concentrations of Amy, Sor and their combination for 48 h. Treated cells were washed with PBS (Sigma - P2667) and fixed in the dark with 70% pre-cooled ethanol (Sigma –Aldrich, USA, CAS-No. 64-17-5) for 1 h before washing with PBS and subsequent treatment with RNase I at 37 °C for 30 min. Finally, cells were stained with propidium iodide (PI) (Sigma - P2667) at 4 °C for an additional 30 min and analyzed by an Accuri C6 flow cytometer [47].
RNA extraction and RT-PCR assessment
Total RNA was extracted from half of the IC50 concentration treated cells using the Thermo Fisher Scientific, Waltham, MA, USA, Gene JET RNA extraction kit (Cat#K0731), according to the manufacturer’s instructions. The purity and concentration of the extracted RNA were determined by Nanodrop Spectrophotometer (AnalytikJena Scandrop200, Germany) and cDNA was synthesized using SensiFAST™ cDNA Synthesis Kit, Thermo Co, BIO-6505, USA, according to the manufacturer’s protocol.
Quantitative RT-PCR was carried out utilizing specific primers for HMGB1, AMPK, mTOR, BCL2, ATG5, Beclin 1, and LC3 genes, primers sequences are listed in (Supplementary Table 1). The PCR reaction mixture consisted of 10µL SYBR green mix (SensiFAST SYBR No-ROX Mix), 2µL cDNA template, and 6.4 µL nuclease-free water. A rotor gene Q5plex detection system was used for amplification. The thermal cycling condition was as follows: an initial activation for 2 min at 95 °C, followed by 45 cycles of 95 °C for 5s and 62 °C for 10s followed by 72 °C for the 20s. The relative expression was calculated using the comparative 2−ΔΔCt method, GAPDH as an internal housekeeping gene [48].
In silico molecular docking analysis
Three-dimensional structure of human RAC(Rho family)-alpha serine/threonine-protein kinase (AKT1; (PDB ID: 6S9W), AMPK (PDB ID: 4CFF), DNA (cytosine-5)-methyltransferase 1 (DNMT1; (PDB ID: 4WXX), histone deacetylase 1 (HDAC1; (PDB ID: 4BKX), Jumonji domain containing 1 C (JMJD1C; (PDB ID: 2YPD), liver kinase B1 (LKB1; (PDB ID: 4ZDR), phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PK3CA; (PDB ID: 2RD0), and sirtuin 1 (SIRT1; (PDB ID: 5BTR) were retrieved from RCSB-PDB (https://www.rcsb.org/) database. All proteins were prepared for molecular docking by Molecular Operating Environment software (MOE, Chemical Computing Group). Amy and Sor’s three-dimensional structure were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/). Molecular docking between receptors and ligands has been performed using MOE software. The interactions between ligands and receptors were visualized using MOE software. Furthermore, ligand efficiency (kcal/mol), dissociation constant (pKd), and inhibition constant (pKi) were calculated by KDEEP (https://playmolecule.com/Kdeep/). Pharmacokinetics, including (ADME-Tox) absorption, distribution, metabolism, and excretion - toxicity of Amy and Sor were determined by ADME and Ames prediction that built in BIOVIA Discovery Studio 2016 (BIOVIA, Dassault Systèmes, France).
Statistical analysis
Data were analyzed using GraphPad Prism version 8.0 (GraphPad Software, Inc., La Jolla, CA, USA). Data were represented as mean ± SEM, and statistical comparisons between multiple groups were performed in oneway (ANOVA), followed by a Tukey’s post hoc test. For all tests, differences between means were determined by the least significance difference test with significance defined at P ≤ 0.05.
Results
Cytotoxicity, drug interaction, and selectivity of Amy and Sor
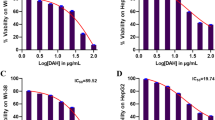
The cytotoxic potential of Amy and Sor was evaluated against HepG2 and WI-38 cells using an MTT test. 48 h were spent treating cells with Amy and Sor (range: Amy: 0.5–160 mg/ml; Sor: 0.125-8 µM). Both substances showed dose-dependent reductions in cell viability in both cell lines. Amy inhibited the growth of HepG2 (IC50 5.21 ± 0.09 mg/ml) and WI-38 (IC50 141.25 ± 0.23 mg/ml). Sor reduced the proliferation of WI-38 cells (IC50 0.59 ± 0.002 µM) and HepG2 cells (IC50 2.21 ± 0.06 µM) like that of HepG2 Fig. 1.

Effect of Sorafenib and Amygdalin on cell viability of HepG2 and WI-38 cells: Cells were treated with different concentrations of Sorafenib (0.125-8 µM) (A and C) and Amygdalin (0.5–160 mg/ml) (B and D) for 48 h and MTT assay was performed. Results were expressed as cell viability (% of control) and data are presented as the mean ± standard deviation, all experiments were repeated at least three times. (E-I) Graphic representations obtained from the CompuSyn Report (see supplementary materials 2) for Amy and Sor combinations indicating strong synergism between the two compounds when used in combo: (E) Dose-effect plot; (F) DRI plot; (G) Median-effect plot; (H) Isobolograms; and (I) Combination index blot
On HepG2 cells, the potential for pharmacological interactions (synergistic, additive, or antagonistic) between combinations of Amy and Sor were evaluated by MTT assay and analyzed by CompuSyn software (see supplementary materials 2 for complete report). The CompuSyn blots are illustrated in Fig. 1(E-I). Cells co-treated with different doses of Amy and Sor showed significantly decreased viability than the comparable single therapy. Additionally, the DRI50 for Amy and Sor were equivalent to 1.8 and 43.5, respectively (Table 1). When the two compounds were administered in combination at 2.036 mg/ml Amy and 0.40 µM Sor for 48 h, they resulted in a 0.5 fraction inhibition (Fa) and a strong synergistic interaction (CI50 = 0.56), the CI values ranged from 0.566 to 0.8 for the fractional inhibition of Fa = 0.50 ~ 0.97 (Table 2) (Supplementary File 1). The synergism was further substantiated by the isobolograms (Fig. 1H), and the Fa-DRI (Fig. 1F), and Fa-CI plots (Fig. 1I), which evaluated the possibility of dosage reduction and the impact of Amy and Sor’s co-treatment, respectively.
The Selectivity Index (SI) was calculated using the ratio of the IC50 values for Amy and Sor versus HepG2 and the normal WI-38 cells (Table 3). Amy was particularly toxic for HepG2 cells (SI = 27.1), but not toxic for WI-38 normal cells. However, both cell lines were severely damaged by Sor, especially the WI-38 cells (SI = 0.27).
Effect of Amy and Sor on the morphology of HepG2 and WI-38 cells
Under the inverted phase-contrast microscope, changes in HepG2 and WI-38 cell morphology were seen after being exposed to the IC50 concentrations of Amy or Sor for 24 and 48 h, respectively Fig. 2. Untreated control cells showed normal morphology, however, cells that had been exposed to Amy and/or Sor showed abnormal morphology and characteristic signs of cell death, such as fragmented nuclei, rounded membrane deformations, decreased cell density, and the emergence of clusters of floating dead cells.

Representative photographs showing morphological changes in HepG2 and WI-38 cells exposed to IC50/2 concentration of Amygdalin or Sorafenib and combination treatment for 24 and 48 h. Images were taken using an inverted phase contrast microscope at ×100. Scale bar 50 μm
Amy induces apoptosis and ameliorates the Sor-induced necrosis in HepG2 cells
The loss in cell viability could be brought about by an apoptotic response to Amy and Sor, as suggested by the MTT assay and microscopic inspection. To further establish the cell death process (apoptosis vs. necrosis), both HepG2 and WI-38 cells were treated with half IC50 doses of each chemical for 48 h. Figure 3.

Representative dot plots of cell apoptosis: (A) HepG2 and (C) WI-38 cells treated with IC50/2 concentration of amygdalin or sorafenib alone and in combination for 48 h before double staining with annexin V-FITC/PI and analysis by flowcytometry. The percentage of live cells (AnnexinV-FITC− /PI −), early apoptotic (AnnexinV-FITC+ /PI −), late apoptotic (AnnexinV-FITC+ /PI +), and necrotic cells (AnnexinV-FITC− /PI +) were measured, represented graphically in (B) and (D). Data are represented as mean ± SEM (n = 3; *significantly different as compared to the untreated control group; *** (P ≤ 0.001), ** (P ≤ 0.01),* (P ≤ 0.005)
In untreated control, HepG2 cells, the percentages of viable, early, late, and necrotic cells were 99.8%, 0.2%, 0.0%, and 0.0%, respectively. The proportion of late apoptotic cells, however, significantly increased following 48 h of treatment with Amy, Sor and in combination, reaching 40.1%, 6.5%, and 47.4%, respectively. Similar to this, the proportion of necrotic cells was 43.0% in Amy-treated cells, 71.8% in Sor-treated cells, and 45.2% in Amy/Sor-cotreated cells. According to the cytotoxic synergistic impact, only 5.4% of viable cells were found in the Amy/Sor combination therapy, compared to 15.7% in the Amy and 20.3% in the Sor single treatment .
Compared to HepG2 cells, WI-38 normal cells responded to treatment somewhat differently. In general, WI-38 cells revealed more live and early apoptotic cells than HepG2 cells did when given the same therapy. When compared to the control (99.3%), a single treatment with Sor generated the lowest percentage of viable cells (50.2%) and the highest late apoptotic (9.0%) and necrotic cells (40.2%). Our findings show that Amy and Sor work together to kill cancer cells by triggering late apoptosis in HepG2 cells (Supplementary Fig. 1).
Effect of Amy and Sor on the cell cycle distribution
Using flow cytometry, the distribution of the cell cycle phase was examined in Fig. 4. After treatment with Amy (11.1 ± 0.11%) and combined Amy/Sor (11.2% ±0.11), HepG2 cells showed a considerably high accumulation of cells, indicating a cell cycle arrest at the S phase after treatment with Amy (11.1 ± 0.11%) and combined Amy/Sor (11.2%±0.11) as compared to control (4.8%±0.11). Amy’s treatment of HepG2 cells resulted in a notable five-fold rise in the population of G2/M cells (15.3% ±0.05) as compared to the control (3% ±0.3). After combining Amy/Sor and Sor’s single treatments, the normal WI-38 cell line showed the greatest levels of cell cycle arrest in the G2/M and S phases (Supplementary Fig. 2).

Effect of amygdalin, sorafenib, and their combination on cell cycle. Flow cytometry analysis of (A) HepG2; (C) WI-38 cells after treatment with IC50/2 concentration for each drug for 48 h. (B, D) Quantitative data analysis for the cell population (% of total) in Sub-G1 G0/G1, S, and G2/M phases. Results are presented as mean ± SEM (n = 3; *significantly different as compared to the untreated control group; *** (P ≤ 0.001), ** (P ≤ 0.01), and * (P ≤ 0.05)
Effect of Amy and Sor on signaling pathways and apoptosis-autophagy-related marker genes
RT-PCR was used to analyze the impact of Amy and Sor on the expression of the genes for AMPK and mTOR as well as many apoptosis-autophagy-related indicators, including HMGB1, BCL2, LC3, Beclin 1, and ATG5. In HepG2 cells, as depicted in Fig. 5A, combined Amy/Sor treatment caused the most prominent upregulation of AMPK, HMGB1, Beclin 1, and ATG5 expression, whereas mTOR and BCL2 expressions were most strongly suppressed when compared to control and single Amy or Sor treated HepG2 cells. As opposed to control, single Amy, or combination Amy/Sor therapy, Sor single treatment substantially (p < 0.05) caused the highest expression of AMPK, LC3, Beclin 1, and ATG5, along with the highest suppression for mTOR and BCL2 genes in WI-38 cells Fig. 5B.

Effect of Amygdalin or Sorafenib single and combined treatment on expression of apoptosis-autophagy related genes: (A) HepG2; (B) WI-38 cells. Results are presented as mean ± SEM (n = 3). * (P ≤ 0.05), ** (P ≤ 0.01) *** (P ≤ 0.001) compared to control
Free radical scavenging capacity and oxidative stress markers
The reaction of the dry DPPH dissolved in methanol to various doses of vitamin C and Amy (5–75 mg/ml and 2.5–25 µg/ml, respectively) is depicted in Fig. 6A; the amount of DPPH that Amy was able to scavenge increased dose-dependently. Amy and vitamin C each had an EC50 of 15.64 mg/ml and 178.5 g/ml, respectively. As such, treated cells had much higher GSH levels than the untreated control cells. HepG2 and WI-38 cells with Amy/Sor combination therapy had the greatest GSH levels, as demonstrated in Fig. 6B. However, in HepG2 and WI-38 cells, the combination of Amy and Sor therapy resulted in the most significant reduction in MDA levels when compared to the control. In contrast, HepG2 (1.89 nM/mg ± 0.05) and WI-38 (3.12 nM/mg ± 0.1) cells treated with Sor alone had significantly higher MDA levels than control cells Fig. 6C.

Scavenging activity of AMG and effect of AMG and SOR single and combined treatments on oxidative stress markers: (A) DPPH scavenging activity of AMG versus the standard vitamin C; (B) GSH and (C) MDA concentration in control and treated HepG2 and WI-38 cells. * (P ≤ 0.05), ** (P ≤ 0.01) and *** (P ≤ 0.001) means significantly different compared to control. All analyses were performed in triplicate and data are expressed as mean ± SD.
Molecular docking analyses predict the cellular target proteins of Amy and Sor
Amy and Sor were screened for probable targets using the Swiss target prediction algorithm. These proteins have been identified as having a high binding probability, and virtual screening was used to further narrow them down to those with the highest affinity and most promise for modulating the processes associated with cancer progression. The results of the screening suggest that these eight proteins [AKT1, AMPK1, DMNT1, HDAC1, JMJDIC, LKB1, PK3CA, and SIRT1] may have the potential to effectively target cancer progression. The measurement of a compound’s free binding energy (measured in Kcal/mol) allows molecular docking to forecast the compounds that have the highest chance of forming a strong bond with a protein. Each receptor was allowed to dock with several ligand poses, which were then analyzed based on the binding energy of each docking position. Amy has been molecularly docked to PI3K and mTOR proteins to examine its potential impact as an AMPK/PI3K/mTOR inhibitor. Our findings in this investigation suggested that Amy/Sor had greater binding energies for AKT1 (-7.12 kcal/mol and − 7.05 kcal/mol, respectively), which made them good candidates for the antagonistic treatment of angiogenesis and cell proliferation. Interestingly, the interaction between Amy and the ATP binding site AKT resembles the Sor ligand-protein complex in certain ways Fig. 7A. Furthermore, Amy also showed binding energies with a variety of receptors, including HDAC1 (-5.63 kcal/mol), JMJD1C (-6.8 kcal/mol), and LKB1 (-5.79 kcal/mol). However, Amy and Sor have demonstrated strong binding energy with AMPK (-5.75 and − 6.43 kcal/mol), DNMT1 (-5.89 and − 6.50 kcal/mol), PK3CA (-6.33 and − 7.03 kcal/mol), and SIRT1 (-6.66 and − 6.35 kcal/mol), respectively (Fig. 7B-H). Furthermore, the dissociation constant (pKd) and inhibition constant (pKi) of Amy and Sor toward the target proteins are represented in (Table 4). Amy and Sor were determined by ADME and Ames prediction and the results were tabulated in (Table 5).

Molecular docking analysis for the visualization of binding poses of amygdalin and sorafenib in the binding sites of (A) AKT1 (PDB ID: 6S9W); (B) HDAC1 (PDB ID: 4BKX); (C) JMJD1C (PDB ID: 2YPD); (D) LKB1 (PDB ID: 4ZDR); (E) AMPK (PDB ID: 4CFF); (F) DNMT1 (PDB ID: 4WXX); (G) PK3CA (PDB ID: 2RD0) and (H) SIRT1 (PDB ID: 5BTR). Molecular docking and interactions between ligands and receptors have been performed using InstaDock and BIOVIA discovery studio visualizer software, respectively. Amygdalin and sorafenib three-dimensional structure were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/)
Discussion
The development of novel chemotherapeutic medicines is crucial in the fight against liver cancer [49], which is regarded as the third greatest cause of mortality worldwide [50]. For patients with advanced-stage HCC, Sor has been the most important molecular targeted medicine [51], but it hasn’t responded to either curative interventional or chemotherapies [52], can’t extend overall survival past three months, and over 50% of patients experience severe clinical side effects [53]. Additionally, Sor medication resistance significantly diminishes the treatment’s efficacy in patients [54]; Sor dose attenuation is required due to all of these considerations [55]. As a result, phytoconstituents that exhibit antioxidant, anti-cancerous, and hepatoprotective properties are of significant interest to oncologists [56]. The ADMET results of the current study also, stated the toxicity of Sor despite its anticancer potentials. Therefore, the idea behind this combination is to reduce the toxic effects of Sor by introducing a natural product such as Amy that may be better tolerated by the body. This could potentially increase the effectiveness of the combination while reducing the potential side effects. Several in vitro and in vivo studies on Amy have been conducted to gain insight into its biological function and potential therapeutic targets for HCC [57, 58]. In this work, Amy and Sor’s antiproliferative activities against HepG2 cancer cells and the non-cancerous WI-38 cell line were assessed both singly and in combination to determine whether Amy is potentially cancer-selective and whether it is safe for normal cells; a review of the tested medications’ ability to cause autophagy-induced programmed cell death, which has not previously been properly evaluated.
The MTT assay is a useful test that accurately determines the number of viable cells and may be used to assess the cytotoxicity of substances used in the treatment of cancer [59]. The MTT results show that Amy decreases the HepG2 cell line’s viability in a dose-dependent manner. Similar to our study, other researchers have also documented the cytotoxicity effect of Amy in a variety of cancer cells, including oral cancer cell line [60], breast cancer [61], and human cervical cancer [62]. This effect could be explained by the morphological changes in HepG2 cells that lose their epithelial shape, which makes them appear smaller, spherical, with a disfiguration in the cell membrane, and detached from the surface when compared to untreated ones, which is a sign of cell death as described in the literature.
Our result revealed that Sor showed a low selectivity index (SI) value (0.27) versus the 100 folds higher SI value observed for Amy (27.1). As previously clarified in many published literature the higher the (SI) value, the greater drug selectivity as it reflects a higher drug normal IC50 relative to cancer; where (SI) value less than 2; suggested general drug toxicity [63,64,65]; such results confirmed Sor toxic effect; representing Amy as a good choice therapy with high selectivity for HCC accomplished by no toxic side effects.
Our data showed that our co-treatment had a strong synergistic effect, as indicated by the CI analysis, which had a value equal to (0.65), indicating strong synergism. This synergistic effect was beneficial for reducing the doses of both Sor and Amy, as demonstrated by the DRI50 value, which calculates how many folds the dose of each drug in a synergistic combination may be reduced, with DRI50 1 being more favorable. In addition, a significant dosage decrease was noted in Sor, where the DRI50 value was (43.5 with a lowered dose from 17.75 µM to 0.74 µM), and the DRI50 value for Amy was (1.8 with a reduced dose from 3.74 mg/L to 2.04 mg/L). However, this potency reduction could be the result of Amy’s naturally multi-targeting bioactive characteristics [66]. In this study, the neutralizing effect of Amy/Sor was seen in both MDA and GSH levels, with considerable augmentation of GSH in cells treated with Amy/Sor cotreatment (6.94 & 7.64), followed by single Amy (4.19 & 6.85), and accomplished with Sor (2.28 & 2.03 in HepG2 and WI-38 cells, respectively. However, MDA significantly decreased in cells treated with Amy (0.04 & 0.26), while it increased in cells treated with Sor (1.89 & 3.12) in HepG2 and WI-38 cells, respectively. This indicates that Amy has a better effect on Sor’s toxic effects when combined, which is a promising attribute in overcoming Sor’s toxic effects. These results were consistent with several previous investigations [67, 68].
Cell cycle analysis is a crucial test that demonstrates the proportion of cells that accumulate in each phase during cell proliferation after exposure to any harmful substance [69]. To ascertain if Amy/Sor affects cell cycle progression and/or the activation of apoptosis, Annexin V-FITC/PI labelling was carried out on HepG2 cells treated with Amy/Sor. Our data showed that all groups slowed the progression of the HepG2 cell cycle in the S and G2/M phases. However, single Amy and/or Sor is the most stressful effect among the groups, inducing the death of HepG2 cells with a drastic increase in the population of Sub-G cells. Besides, the greatest incidence of S and G2/M arrest was seen by Amy or Amy/Sor with the least stressful effect on normal cells. However, various research indicates that Amy and Sor alone were able to halt the cell cycle at various phases after treating distinct cell types [70, 71].
According to our findings, Amy and/or Sor caused HepG2 cells to undergo both necrosis and apoptosis. Cotreatment markedly increased apoptosis, followed by Amy and Sor. Additionally, Sor had the highest rate of necrotic cell death, whereas Amy had the lowest rate. This finding might be explained by MTT and CI data, which show that the combination of Amy and/or Sor played crucial roles in the reduction of HepG2 cell proliferation via both apoptosis and necrosis pathways with the least toxic impact in comparison to a single Sor treatment. The antiproliferative and docking mechanism of Amy further supports these findings.
The ability of cancer cells to avoid apoptosis, or programmed cell death, is one of their well-known traits [72]. Additionally, in cancer cells, autophagy suppresses tumorigenesis by inhibiting cancer-cell survival and inducing cell death. Consequently, further gene analysis for intrinsic and extrinsic genes that regulate apoptosis and autophagy was carried out in our study. The results showed that the Amy/Sor co-therapy led to the upregulation of AMPK, HMGB1, Beclin 1, and ATG5 expression while simultaneously significantly suppressing the expression of mTOR and BCL2. These results may be supported by the fact that mTOR/BCL-2, which is classified as an anti-apoptotic protein because of its function in the production of cell death, controls the apoptotic pathway. Here, we show that Amy causes the HepG2 cell line to go into apoptosis by down regulating BCL-2. Other investigations in numerous cancerous cells have demonstrated that Amy can cause apoptosis via lowering mTOR/BCl-2 [61, 73]. However, sustaining apoptosis depends on the equilibrium between cell division and death [74].
New targeted therapeutics that can either induce death or make cancer cells more susceptible to known cytotoxic drugs have been developed as a result of our growing understanding of the processes of intrinsic and extrinsic apoptotic signaling [75]. Amy causes apoptosis, demonstrating that it blocks the AMPK/mTOR pathway’s activation. The results of this study revealed that the activation of the AMPK signaling pathway was meritoriously inhibited in HepG2 cells and that the translocation of mTOR and BCL2 was significantly suppressed in Amy/Sor-treated cells, suggesting that the AMPK inhibition mechanism may be involved in apoptosis. This behaviour may be explained by the critical function of AMPK as a metabolic sensor and regulation of cell growth [76]; besides, AMPK negatively regulates the mTOR signal pathway, resulting in the inhibition of cancer proliferation and growth [77]; by incorporating signals from the PI3K/Akt pathway [11], it controls cell survival, proliferation, and angiogenesis [78]; additionally, HCC typically has elevated mTOR, which is linked to an early recurrence and a worse prognosis [79]; treatment for HCC has been proposed as the inactivation of mTOR to limit cancer cell growth [80].
Consequently, these results are consistent with our theory that Amy induces autophagy through AMPK/mTOR/BCL2 signaling and that autophagy is a critical factor in Amy-mediated cell death [81]. Figure 8 recapitulates the proposed antiproliferative and apoptotic action of Amy on HepG2 cells by up-regulation expression of AMPK, HMGB1, Beclin 1, LC3, and ATG5, and down-regulation expression of mTOR/BCL2 genes that were induced by Amy relative to control and simulate the induction of intrinsic and extrinsic apoptosis pathway in the treated liver cancer cell line.

A schematic diagram shows the mode of action of Amy on the HepG2 cell line. Treatment of the HepG2 cell line with Amy resulted in potential cytotoxicity, and significant down-regulation for the AMPK/mTOR/BCL-2 pathway, with induction of apoptosis and autophagy by extrinsic and intrinsic pathways
Interestingly, the proposed antiproliferative and apoptotic action of Amy on HepG2 has been confirmed by molecular docking of anticipated cellular proteins like AKT1, AMPK, DNMT1, HDAC1, JMJD1C, LKB1, PK3CA, and SIRT1 as possible targets of both Amy and Sor. These target proteins play crucial roles in the junction of apoptotic and autophagy crosstalk. Most of the selected proteins in the in-silico study could be inhibited by Amy leading to downregulation of AMPK and subsequently downregulated mTOR. By inhibiting PI3K/AKT, Amy disrupts the pathway that would normally activate mTOR, meaning that mTOR is unable to activate the downstream processes that would lead to cancer progression. In addition, by downregulating AMPK, Amy reduces the amount of AMP in the cell, which in turn reduces the amount of mTOR available to activate the downstream pathways. However, molecular docking proposed the inhibitory action of Amy as follows; the AMPK gene was upregulated as a result of Amy’s inhibition of (a) AKT activity (a repressor for AMPK) by docking the AKT protein and indirectly by docking the PI3K (an enhancer for AKT); (B) Methylation of the AMPK and LKB1 (activator for c) promoter area by docking the DNMT1 enzyme, resulting in direct and indirect increased expression of AMPK. (C) Histone demethylase JMJDIC (the negative regulator for AMPK) by direct docking into the active site. JMJDIC suppresses the CAMKK2 gene, which ordinarily results in the overexpression of AMPK; (D) Binding of DBC1 inhibitor to SIRT1 (AMPK activator), causing sequential activation of SIRT1and LKB1 which in turn causes AMPK activation.
Conclusion
In this study, we showed that Amy and/or Sor have an anti-proliferative impact and apoptotic action on hepatocellular carcinoma HepG2 cells. Together, the AMPK/mTOR inhibitory signalling pathway contributes significantly to HCC. This study reveals the potential apoptotic anti-HCC properties of Amy and/or Sor via inhibition of AMPK/mTOR. Cytotoxic activity of Amy and/or Sor exhibited that Amy inhibited the growth of HepG2 (IC50: 5.21–0.09 mg/ml). Furthermore, a strong synergistic interaction between Amy and Sor (CI50 = 0.56) was detected; additionally, the DRI50 for Amy and Sor were equal to 1.8 and 43.5, respectively. For investigation of the apoptotic activity, Amy significantly stimulated apoptotic HCC cell death; after 48 h of treatment with Amy and/or Sor, the percentage of late apoptotic cells increased remarkably to 44.9%, 34.8%, and 59%, respectively. However, the greatest levels of mTOR and BCL2 suppression are concurrent with the elevation of AMPK, HMGB1, Beclin 1, LC3, and ATG5 expression in Amy and/or Sor-treated HepG2 cells. In this work, the AMPK/mTOR signalling pathway of Amy was examined using integrated techniques in vitro and in silico. Our findings taken together provide more evidence for Amy’s potential anticancer efficacy as a different therapy option for HCC, although additional in vivo studies in animal models are required to corroborate the results.
Data Availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
Loesch R et al. Deleting the β-catenin degradation domain in mouse hepatocytes drives hepatocellular carcinoma or hepatoblastoma-like tumor growth. J Hepatol (2022).
Chen Q, et al. Molecular imaging of tumor microenvironment to assess the effects of locoregional treatment for hepatocellular carcinoma. Hepatol Commun. 2022;6:652–64.
Reinders MTM et al. Safety and efficacy of holmium-166 radioembolization in hepatocellular carcinoma–the HEPAR primary study. J Nucl Med (2022).
Zhang Y, et al. A systems biology-based investigation into the therapeutic effects of Gansui Banxia Tang on reversing the imbalanced network of hepatocellular carcinoma. Sci Rep. 2014;4:1–10.
Ross SA, Davis CD. MicroRNA, nutrition, and cancer prevention. Adv Nutr. 2011;2:472–85.
Sasahira T, Kirita T. Hallmarks of cancer-related newly prognostic factors of oral squamous cell carcinoma. Int J Mol Sci. 2018;19:2413.
Wang J, Li D, Cang H, Guo B. Crosstalk between cancer and immune cells: role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019;8:4709–21.
Mishra L, et al. Liver stem cells and hepatocellular carcinoma. Hepatology. 2009;49:318–29.
Yang L, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5:8.
Roskoski R Jr. Properties of FDA-approved small molecule protein kinase inhibitors: a 2020 update. Pharmacol Res. 2020;152:104609.
Owusu-Brackett N, Shariati M, Meric-Bernstam F. Role of pi3k/akt/mtor in cancer signaling. in Predictive Biomarkers in Oncology 263–270 (Springer, 2019).
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44.
Bhat M, Sonenberg N, Gores GJ. The mTOR pathway in hepatic malignancies. Hepatology. 2013;58:810–8.
Chen R, et al. The role of methyltransferase NSD2 as a potential oncogene in human solid tumors. Onco Targets Ther. 2020;13:6837.
Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314–41.
Yu L, Wei J, Liu P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Seminars in Cancer Biology (Elsevier, 2021).
Ferrín G, Guerrero M, Amado V, Rodríguez-Perálvarez M. De la Mata, M. activation of mTOR signaling pathway in hepatocellular carcinoma. Int J Mol Sci. 2020;21:1266.
McCubrey JA, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012;3:1068.
Kattan SW, et al. Molecular docking, anti-proliferative activity and induction of apoptosis in human liver cancer cells treated with androstane derivatives: implication of PI3K/AKT/mTOR pathway. J Steroid Biochem Mol Biol. 2020;198:105604.
Zimmerman MA, et al. Recurrence of hepatocellular carcinoma following liver transplantation: a review of preoperative and postoperative prognostic indicators. Arch Surg. 2008;143:182–8.
Landman MP, Feurer ID, Pinson CW, Moore DE. Which is more cost-effective under the MELD system: primary liver transplantation, or salvage transplantation after hepatic resection or after loco‐regional therapy for hepatocellular carcinoma within Milan criteria? HPB. 2011;13:783–91.
Pauty J, et al. A vascular endothelial growth factor-dependent sprouting angiogenesis assay based on an in vitro human blood vessel model for the study of anti-angiogenic drugs. EBioMedicine. 2018;27:225–36.
Huynh H. Molecularly targeted therapy in hepatocellular carcinoma. Biochem Pharmacol. 2010;80:550–60.
Huynh H, et al. Targeting receptor tyrosine kinase pathways in hepatocellular carcinoma. Anti-Cancer Agents Med Chem (Formerly Curr Med Chem Agents). 2011;11:560–75.
Cho Y-K, Park D-H, Jeon I-C. Medication trends for age-related macular degeneration. Int J Mol Sci. 2021;22:11837.
Randrup Hansen C, Grimm D, Bauer J, Wehland M, Magnusson NE. Effects and side effects of using sorafenib and sunitinib in the treatment of metastatic renal cell carcinoma. Int J Mol Sci. 2017;18:461.
Shih T, Lindley C. Bevacizumab: an angiogenesis inhibitor for the treatment of solid malignancies. Clin Ther. 2006;28:1779–802.
Marsh AM, Lo L, Cohen RA, Feusner JH. Sorafenib and bevacizumab for recurrent metastatic hepatoblastoma: stable radiographic disease with decreased AFP. Pediatr Blood Cancer. 2012;59:939–40.
Ibrahim N, Yu Y, Walsh WR, Yang J-L. Molecular targeted therapies for cancer: sorafenib monotherapy and its combination with other therapies. Oncol Rep. 2012;27:1303–11.
Ali R, et al. Is amygdalin outcomes weighing detriments of Sorafenib Treatment in female mice with kidney Injury Induced by Ehrlich Ascites Carcinoma Model? Preliminary study. Biochem Lett. 2021;17:0.
Nasser HM, El-Naggar SA, El-Sayed Rizk ME-SR, Elmetwalli A, Salama AF. Effect of Sorafenib on Liver Biochemistry prior to vitamin B17 Coadministration in Ehrlich Ascites Carcinoma mice Model: preliminary phase study. Biochem Lett. 2021;17:40–9.
Dowling II, Eliche CE, B., Bagasra O. Effects of Amygdalin on prostate cancer. Herbal Medicines 591–609 (Elsevier, 2022).
Kolesar E, et al. Assessment of rabbit spermatozoa characteristics after amygdalin and apricot seeds exposure in vivo. Toxicol Rep. 2018;5:679–86.
Zielińska A, Płonka-Czerw J, Nowak A, Kuśmierz D. Effect of amygdalin on MCF-7, MDA-MB-231 and T-47D breast cancer cells in the in vitro study. Postepy Hig Med Dosw. 2022;76:132–42.
Blaheta RA, Nelson K, Haferkamp A, Juengel E. Amygdalin, quackery or cure? Phytomedicine. 2016;23:367–76.
Abboud MM, Awaida A, Alkhateeb W, H. H., Abu-Ayyad AN. Antitumor action of amygdalin on human breast cancer cells by selective sensitization to oxidative stress. Nutr Cancer. 2019;71:483–90.
Shi J, et al. Recent updates and future perspectives about amygdalin as a potential anticancer agent: a review. Cancer Med. 2019;8:3004–11.
Figurová D, et al. Inflammation, it’s regulation and antiphlogistic effect of the cyanogenic glycoside amygdalin. Molecules. 2021;26:5972.
Attia AA, et al. Amygdalin potentiates the anti-cancer effect of Sorafenib on Ehrlich ascites carcinoma and ameliorates the associated liver damage. Sci Rep. 2022;12:1–9.
Liu W, Jin W, Zhu S, Chen Y, Liu B. Targeting regulated cell death (RCD) with small-molecule compounds in cancer therapy: a revisited review of apoptosis, autophagy-dependent cell death and necroptosis. Drug Discov Today (2021).
Chung DM, Kim JH, Kim JK. Evaluation of MTT and Trypan Blue assays for radiation-induced cell viability test in HepG2 cells. Int J Radiat Res. 2015;13:331.
Chou T-C. Drug combination studies and their synergy quantification using the Chou-Talalay MethodSynergy quantification method. Cancer Res. 2010;70:440–6.
Patel Rajesh M, Patel Natvar J. In vitro antioxidant activity of coumarin compounds by DPPH, Super oxide and nitric oxide free radical scavenging methods. J Adv Pharm Educ Res. 2011;1:52–68.
Giustarini D, et al. Assessment of glutathione/glutathione disulphide ratio and S-glutathionylated proteins in human blood, solid tissues, and cultured cells. Free Radic Biol Med. 2017;112:360–75.
Tsikas D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal Biochem. 2017;524:13–30.
Pietkiewicz S, Schmidt JH, Lavrik IN. Quantification of apoptosis and necroptosis at the single cell level by a combination of Imaging Flow Cytometry with classical annexin V/propidium iodide staining. J Immunol Methods. 2015;423:99–103.
Nunez R. DNA measurement and cell cycle analysis by flow cytometry. Curr Issues Mol Biol. 2001;3:67–70.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2 – ∆∆CT method. Methods. 2001;25:402–8.
Cragg GM, Pezzuto JM. Natural products as a vital source for the discovery of cancer chemotherapeutic and chemopreventive agents. Med Princ Pract. 2016;25:41–59.
Shedid M, Abdelmonem M, Boraik A, Elmetwalli A, Esmael A. Role of plasma miRNA-501 expression Profile as a marker of Hepatocellular Carcinoma (HCC) Progression with Hepatitis C virus infection. Am J Clin Pathol. 2019;152:134.
Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15:599–616.
Kudo M. Treatment of advanced hepatocellular carcinoma with emphasis on hepatic arterial infusion chemotherapy and molecular targeted therapy. Liver cancer. 2012;1:62–70.
Colagrande S, Regini F, Taliani GG, Nardi C, Inghilesi AL. Advanced hepatocellular carcinoma and sorafenib: diagnosis, indications, clinical and radiological follow-up. World J Hepatol. 2015;7:1041.
Cheng Z, Wei-Qi J, Jin D. New insights on sorafenib resistance in liver cancer with correlation of individualized therapy. Biochim Biophys Acta (BBA)-Reviews Cancer. 2020;1874:188382.
Wang S, et al. Inhibition of EZH2 attenuates sorafenib resistance by targeting NOTCH1 activation-dependent liver cancer stem cells via NOTCH1-related MicroRNAs in hepatocellular carcinoma. Transl Oncol. 2020;13:100741.
Rizeq B, et al. The power of phytochemicals combination in cancer chemoprevention. J Cancer. 2020;11:4521.
Al-Khafaji K, Tok TT. Molecular dynamics simulation, free energy landscape and binding free energy computations in exploration the anti-invasive activity of amygdalin against metastasis. Comput Methods Programs Biomed. 2020;195:105660.
Elmalla A, Elmetwalli A, Rizk ME-S, Salama AF. The effect of vitamin B17 on Cardiomyopathy against Ehrlich Tumor Development in Female mice. Biochem Lett. 2021;17:69–76.
Meerloo J, Van, Kaspers GJL, Cloos J. Cell sensitivity assays: the MTT assay. in Cancer cell culture 237–245 (Springer, 2011).
Sireesha D, Reddy BS, Reginald BA, Samatha M, Kamal F. Effect of amygdalin on oral cancer cell line: an in vitro study. J Oral Maxillofac Pathol JOMFP. 2019;23:104.
Moradipoodeh B, Jamalan M, Zeinali M, Fereidoonnezhad M, Mohammadzadeh G. In vitro and in silico anticancer activity of amygdalin on the SK-BR-3 human breast cancer cell line. Mol Biol Rep. 2019;46:6361–70.
Choi SP, Song YK, Kim KJ, Lim HH. Amygdalin extract from armeniacae semen induces apoptosis through bax-dependent caspase-3 activation in human cervical cancer cell line ME-180. J Korean Med. 2005;26:130–42.
Nabende PN, Karanja SM, Mwatha JK, Wachira SW. Anti-proliferative activity of Prunus africana, Warburgia stuhlmannii and Maytenus senegalensis extracts in breast and colon cancer cell lines. Eur J Med Plants. 2015;5:366.
Murati T, et al. Comparative assessment of Prunus spinosa L. flower extract in non-neoplastic hepatocytes and hepatoblastoma cells. South Afr J Bot. 2019;123:36–42.
Tietze LF, Feuerstein T. Enzyme and proton-activated prodrugs for a selective cancer therapy. Curr Pharm Des. 2003;9:2155–75.
Roy A, Datta S, Bhatia KS, Jha P, Prasad R. Role of plant derived bioactive compounds against cancer. South Afr J Bot (2021).
Mamdouh AM, Khodeer DM, Tantawy MA, Moustafa YM. In-vitro and in-vivo investigation of amygdalin, metformin, and combination of both against doxorubicin on hepatocellular carcinoma. Life Sci. 2021;285:119961.
Mosayyebi B, Mohammadi L, Kalantary-Charvadeh A, Rahmati M. Amygdalin decreases Adhesion and Migration of MDA-MB-231 and MCF-7 breast Cancer cell lines. Curr Mol Pharmacol. 2021;14:667–75.
Kim JA, Åberg C, Salvati A, Dawson KA. Role of cell cycle on the cellular uptake and dilution of nanoparticles in a cell population. Nat Nanotechnol. 2012;7:62–8.
Markowitsch SD, et al. Artesunate inhibits growth of sunitinib-resistant renal cell carcinoma cells through cell cycle arrest and induction of ferroptosis. Cancers (Basel). 2020;12:3150.
Cook A, Siddiqi SA, Phytochemicals. Current Understandings of the Modern Therapeutic Approaches for Hepatocellular Carcinoma. in Phytochemicals Targeting Tumor Microenvironment in Gastrointestinal Cancers 303–329 (Springer, 2020).
Wang Y, Kanneganti T-D. From pyroptosis, apoptosis and necroptosis to PANoptosis: a mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J. 2021;19:4641–57.
Aamazadeh F, Ostadrahimi A, Rahbar Saadat Y, Barar J. Bitter apricot ethanolic extract induces apoptosis through increasing expression of Bax/Bcl-2 ratio and caspase-3 in PANC-1 pancreatic cancer cells. Mol Biol Rep. 2020;47:1895–904.
Noguchi M, et al. Autophagy as a modulator of cell death machinery. Cell Death Dis. 2020;11:1–12.
Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17:395–417.
Juszczak F, Caron N, Mathew AV, Declèves A-E. Critical role for AMPK in metabolic disease-induced chronic kidney disease. Int J Mol Sci. 2020;21:7994.
Jia J, et al. Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy. 2019;15:169–71.
Simioni C, Martelli AM, Zauli G, Melloni E, Neri LM. Targeting mTOR in acute lymphoblastic leukemia. Cells. 2019;8:190.
Dong P, et al. Dampened VEPH1 activates mTORC1 signaling by weakening the TSC1/TSC2 association in hepatocellular carcinoma. J Hepatol. 2020;73:1446–59.
Luo Y-D, et al. p53 haploinsufficiency and increased mTOR signalling define a subset of aggressive hepatocellular carcinoma. J Hepatol. 2021;74:96–108.
Sebastian A, Sebastian SLA, W., Björn S. Role of AMPK-mTOR-Ulk1/2 in the regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol Cell Biol. 2012;32:2–11.
Acknowledgements
Not applicable.
Funding
Not applicable.
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
Author contributions: Conceptualization, T.E.S., A.E., and A.F.S.; Formal analysis, T.E.S., N.M.E, A.H.E.F; Investigation, A.F.S., A.E.S.M., T.E.S.; Project administration, A.F.S., A.N.A.B., A.E.S.M., T.E.S.; Software, A.H.E.F; Validation N.M.E., A.E., A.N.A.B., A.F.S; Visualization, N.M.E., A.E., A.F.S; Writing—original draft, T.E.S., A.F.S., N.M.E.; Writing—review and editing, A.E., A.F.S., T.E.S.; All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethical approval and Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
El-Sewedy, T., Salama, A.F., Mohamed, A.E. et al. Hepatocellular Carcinoma cells: activity of Amygdalin and Sorafenib in Targeting AMPK /mTOR and BCL-2 for anti-angiogenesis and apoptosis cell death. BMC Complement Med Ther 23, 329 (2023). https://doi.org/10.1186/s12906-023-04142-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12906-023-04142-1