
Abstract
Background
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder clinically characterized by loss of memory and cognition. Cholinergic deficit and oxidative stress have been implicated in the pathogenesis of AD. Therefore, inhibition of acetylcholinesterase and oxidation are the two promising strategies in the development of drug for AD. Phyllanthus acidus, belonging to the family Euphorbiaceae, is a tree and has been used in traditional medicine to treat several pain, inflammatory and oxidative stress related disorders such as rheumatism, bronchitis, asthma, respiratory disorder, also important to promote intellect and enhance memory, thus supporting its possible anti-Alzheimer’s properties. In this study, P. acidus was evaluated for its cholinesterase inhibitory and antioxidant activities.
Methods
In this study, we evaluated the antioxidant potential and neuroprotective activity of P. acidus by assessing total phenol content (FCR assay), total flavonoid content, total antioxidant capacity, Fe3+ reducing power capacity, DPPH (2, 2-diphenyl-1-picrylhydrazyl) and hydroxyl radical scavenging capacity, lipid peroxidation inhibition activity & metal chelating activity. In addition acetylcholinestrase (AChE) and butyrylcholinestrase (BChE) inhibitory activities were performed using Ellman’s method.
Results
Total phenolic content and total flavonoid content of the extract were 116.98 mg of gallic acid equivalent and 168.24 mg of quercetin equivalent per gm of dried extract. The methanolic extract of P. acidus (MEPA) showed considerable total antioxidant activity and reducing capacity. In DPPH scavenging assay and hydroxyl radical scavenging assay, the MEPA showed 84.33 % and 77.21 % scavenging having IC50 of 15.62 and 59.74 μg/ml respectively. In lipid peroxidation inhibition activity MEPA showed moderate inhibition of peroxidation at all concentrations with IC50 value of 471.63 μg/ml and exhibited metal chelating activity with IC50 value 308.67 μg/ml. The MEPA exhibited inhibition of rat brain acetylcholinesterase and human blood butyrylcholinesterase in a dose dependent manner and the IC50 value was found to be 1009.87 μg/ml and 449.51 μg/ml respectively.
Conclusion
These results of the present study reveal that MEPA has considerable amount of antioxidant activity as well as anti-acetylcholinesterase and anti-butyrylcholinesterase activity which suggest its effectiveness against Alzheimer's disease and other neurodegenerative disorders.
Similar content being viewed by others

Background
It is highly impossible to consider a biological life without oxygen and this valuable oxygen is metabolized and produce free radicals (FR) in human body by oxidative process having an extensive effects on human health [1, 2]. FR and its by-product reactive oxygen species (ROS) are continuously produced in human body [3, 4]. In some cases (like alcohol, exposure to chemicals, stress, tobacco and UV exposure) there is an excess production of ROS. In this condition, bodily produced antioxidants are insufficient and produce imbalance called oxidative stress [5]. Super oxide anion (O −2 ⋅), hydroxyl radical (⋅OH) and hydrogen peroxide (H2O2) are the primary production of ROS is regulated by an enzymatic antioxidant system and the intake of vitamins related to the daily diet [6]. Over production of ROS is responsible for oxidative damage of macromolecules such as DNA, proteins, lipids, carbohydrates etc. and this damage to DNA also may produce cancer [7, 8]. As a result our body cannot protect from ROS by the primary defense system and ROS continuously oxidize the cells producing secondary ROS that leads to the oxidative chain reactions and resulting cells destruction called oxidative damage [4]. This oxidative damage can causes various acute and chronic neurodegenerative diseases related to aging such as Alzheimer, Parkinson, osteoarthritis, atherosclerosis, myocardial infarction and age related muscular degeneration [9, 10]. This oxidative damage by FR and ROS is blocked by the Antioxidants [11]. Antioxidants are the substances which neutralize body produced FR by donating one of their own electron and preventing cellular and membrane damage. Antioxidants act by several ways such as by preventing the propagation of oxidative chain reactions, by scavenging free radicals, by regulating gene expression, by being part of the redox reaction and by preventing FR formations [12–15]. To reduce the effects of oxidation both synthetic and natural antioxidants are used [2]. BHT and BHA are highly effective synthetic antioxidants but have toxic and side effects on human [16, 17]. Nitric oxide (.NO), superoxide anion (O2−), hydrogen peroxide (H2O2) and hypochloride ion (OCl−) are the natural antioxidants produced by phagocytes as protective agents against cell infection in immune responses [18]. Carotenoids polyphenols, bioflavonoids, vitamin C (ascorbic acid), vitamin E are the major natural antioxidants reported [19].
AD is a neurological disorder associated with memory loss, cognitive dysfunction, behavioral turbulence and abnormalities in activities of daily life [20–22]. AD is frequently founded in elderly people and characterized by malfunctioning of different biochemical pathways [23]. AD has been associated with a significant decrease in the amount of acetylcholine (ACh) by breaking down of ACh [24–26]. ACh is a neurotransmitter that transmits signal in the synapse, after delivering signal ACh is hydrolyzed and given choline and acetyl group in a reaction catalyzed by the enzyme AChE and its pharmacological action is done primarily by acetylcholinesterase (AChE) and secondary by butyrylcholinesterase (BChE) [27]. Over activity of AChE and BChE enzymes are responsible for the development of different neurological disorder like AD, Parkinson’s disease etc. [24, 28]. The most successful way to get rid of this problem is “cholinergic hypothesis” and the approving drugs are working to increase the ACh level in the brain [23] that will improve cognitive function [29]. AChE inhibitors tacrine, donepezile, rivastigmine, and galanthamine are only the approved drugs for the treatment of AD although having numerous side effects [30]. The mechanism based inhibitors due to its role in the hydrolysis of the neurotransmitter Ach is an attractive target for the rational drug design and for the discovery of new drugs for AD [31].
Medicinal plants have been used from ancient to the present time for the remedy of disease of human being. Galanthamine is an anticholinesterase alkaloid isolated from snowdrop approved for the treatment of AD [32]. P. acidus (from the family Euphorbiaceae) plant is also one of the important plants having various medicinal properties such as antioxidants and anti-inflammatory effects. Many crude plants found having antioxidant properties and among the compounds phenolic and flavonoid attracted as significant choice for being used as antioxidants [2]. Traditionally, P. acidus is used in the treatment of fever, respiratory disorders, diabetes, bronchitis, inflammation, several pains etc. and also helpful to cure cough, psoriasis, sudorific, to improve eyesight and memory [33]. Methanolic extract of fruits and leaves was reported to show antimicrobial effect [34]. Petroleum ether extract of fruits was reported to show cytotoxic, antibacterial and antioxidant activities [35]. The fruits and leaves of the plant yielded promising hepatoprotective activity [36]. The methanolic fruit extract of the plant reported to show antibacterial, cytotoxic and antioxidant properties [37]. But no AD related activity of methanolic fruit extract of this plant has been done yet. Thus, our main objective of the present study was to evaluate the antioxidant and neuroprotective potential of P. acidus to treat the AD and other neurodegenerative diseases.
Methods
List of chemicals
Folin–Ciocalteu reagent, Methanol, Gallic acid, Ascorbic acid, DPPH, 2-deoxy-D-ribose, Thiobarbituric acid (TBA), (+)-Catechin, 5,5´-dithio-bis-(2-nitro) benzoic acid (DTNB), Acetylthiocholine iodide, S-Butyrylthiocholine iodide, Donepezil, Ferrozine monosodium,Trichloro acetic acid (TCA) and Triton X-100 were purchased from Sigma chemical company, USA. Butylated hydroxyl toluene (BHT) and Tris–HCl buffer were purchased from Merck, Germany.
Collection of plant
The fruits of P.acidus were collected from Kapasia, in the district Gazipur of Bangladesh in August 2014, and identified by an expert taxonomist from the Bangladesh national herbarium. A voucher specimen (DACB, ACCESSION NUMBER:40181) was preserved in the national herbarium, Dhaka, Bangladesh for future reference.
Preparation of extract
The collected fresh fruit of P. acidus weighing 5 kg was then washed properly to remove dirty materials and shade dried for several days with occasional sun drying. These were then dried in an oven for 24 h at considerably low temperature for better grinding. The grounded powder (500 g) was macerated with methanol (2.5 L) and extracted by cold extraction process. Finally 15 g methanol extract was obtained after evaporating the filtrate.
Determination of phytoconstituents
Determination of total phenolics
Total phenolic content of P. acidus was determined according to the method of Singleton V. L. et al., [38] with minor modifications using Folin-Ciocalteu reagent. Each test tube contained 0.5 ml of plant extract or standard solution at different concentrations, 2.5 ml of Folin–Ciocalteu reagent solution (10 times diluted with water) and 2.5 ml of Sodium carbonate (7.5 %) solution. After adding all of the reagents the test tubes were incubated for 25 min at 25 °C to complete the reaction and the absorbance of the solution was measured at 760 nm. A standard curve was prepared using gallic acid as standard (Y = 0.0151x + 0.059, R2 = 0.9913) for expressing the total content of phenolic compounds in plant extract and shown as mg of gallic acid equivalent (GAE)/gm of dried extractives.
Determination of total flavonoids
Total flavonoid content was determined by the aluminum chloride colorimetric method described by Barrera et al., [35], and quercetin was used as standard. Briefly, 1.0 ml of plant extract or standard of different concentration were added to 3 ml of methanol, 0.2 ml of 10 % AlCl3, 0.2 ml of 1 M potassium acetate and 5.6 ml of distilled water. After incubation for 25 min the absorbance was taken at 420 nm. A quercetin standard curve was prepared (Y = 0.009x + 0.036, R2 = 0.972) to express the result as mg of quercetin equivalent(QE)/g of dried extractives.
Antioxidant ability assay
Determination of total antioxidant capacity
Total antioxidant capacity was determined according to the method as described by Prieto P. et al., [39] with some modifications. In this experiment, 0.5 ml of MEPA or standard (ascorbic acid) of different concentration (100 – 600 μg/ml) was added to 3 ml of reaction mixture (containing 0.6 M sulphuric acid, 28 mM sodium phosphate and 1 % ammonium molybdate) into the test tube. After incubating for15 min at 90°C for completing the reaction followed by cooling at room temperature the absorbance was measured at 695 nm. Ascorbic acid (AA) was used as standard in this study.
Reducing power capacity assessment
The reducing power was evaluated by the method of Oyaizu [37]. In this method, various concentrations of MEPA or standard solutions (1.0 ml) were mixed with 2.5 ml of potassium buffer (0.2 M, pH 6.6) and 2.5 ml of Potassium ferricyanide [K3Fe (CN) 6] (1 %) solution. After 30 min incubation at 500 C, 2.5 ml of trichloro acetic acid (10 %) solution was added into the test tube. The total mixture was centrifuged at 3000 g for 10 min. Then 2.5 ml supernatant solution was withdrawn from the mixture and mixed with 2.5 ml of distilled water and 0.5 ml of FeCl3 (0.1 %) solution. Then the absorbance of the solution was measured at 700 nm and AA was used as standard.
Determination of DPPH radical scavenging activity
DPPH radical scavenging activity was determined according to the method as described by Choi et al., [40]. 2 ml of methanolic solution of plant extract or standard (BHT) at different concentration was mixed with 3 ml (0.02 %) of methanol solution of DPPH. After incubation for 30 min at dark place the absorbance was taken at 517 nm against methanol as blank.
Determination of hydroxyl radical scavenging assay
Hydroxyl radical scavenging activity of different concentrations of MEPA was determined by the method of Elizabeth et al., [41]. Hydroxyl radical was generated by the Fe3+-ascorbate-EDTA-H2O2 system (the Fenton reaction). 1 ml of reaction mixture was made by adding 2-deoxy-D-ribose (2.8 mM), KH2PO4-KOH buffer (20 mM, pH 7.4), FeCl3 (100 μM), EDTA (100 μM), H2O2 (1.0 mM), AA (100 μM) and various concentrations of the test sample or reference compound [(+)- catechin)]. After incubation for 1 h at 37 °C, 0.5 ml of the reaction mixture was mixed with 1 ml of 2.8 % TCA and 1 ml of 1 % aqueous TBA then the mixture incubated at 90 °C for 15 min to develop the color. After cooling, the mixture’s absorbance was measured at 532 nm against an appropriate blank solution.
Determination of metal chelating activity assay
The chelating activity of MEPA for ferrous ion (Fe2+) was measured according to the method of J. Sabate [42], using ferrozine (substrate) and ferrous chloride (FeCl2). In this method, 0.5 ml of extract or standard was added to 1.6 ml of FeCl2(2mM). After incubation for 30 s, 0.1 ml ferrozine (5 mM) was added and kept 10 min at room temperature then the absorbance of the Fe2+ –Ferrozine complex was measured at 562 nm. A typical blank solution contained all reagents except plant extract or standard (BHT) solution.
Determination of lipid peroxidation inhibition activity
The inhibition of lipid peroxidation activity was evaluated according to the method as described by Liu et al., [43], with a slight modification. The adult long Evan rats weighing 150 g were anesthetized with sodium phenobarbitone. The brain of rats were dissected and homogenized with a homogenizer in ice-cold Phosphate buffer (50 mM, pH 7.4) to produce a 1/10 homogenate. The homogenate was centrifuged at 10,000 g for 20 min at 4 °C. The supernatant was used as liposome for in vitro lipid peroxidation assay. The ability of MEPA to inhibit lipid peroxidation was studied by incubating rat brain homogenates treated with hydrogen peroxide (10 μM) and different concentrations of extract or standard solution. Hydrogen peroxide induced lipid peroxidation in rat brain homogenates. 1 ml of 0.15 M KCl and 0.5 ml of liposome containing brain homogenate were added with different concentrations of plant extract or standard solution. The reaction was started by adding 100 μl of 0.2 mM ferric chloride with the above mentioned mixture then incubated at 37 ° C for 30 min. The reaction was stopped by adding 2 ml of 0.25 N HCl, 15 % TCA, 0.5 % BHT and 0.38 % TBA solution. Lipid peroxides reacted with TBA to form a pink product, thiobarbituric acid reacting substances (TBARS), measurable colorimetrically at 532 nm. The difference between the control and the test sample is the measurement of decrease in TBARS formation, reflecting reduced hydroxyl radical induced lipid peroxidation. (+)-Catechin was used as standard for comparison.
Determination of AChE inhibitory activity
The AChE inhibitory activity was performed according to the colorimetric method of Ellman’s et al., [40, 44] using acetylthiocholine iodide as a substrate. For the enzyme source, the rat brains were homogenized in a homogenizer with 5 volumes of a homogenization buffer [10 mMTris-HCl (pH 7.2), which contained 1 M NaCl, 50 mM MgCl2 and 1 % Triton X-100], and centrifuged at 10,000 g for 30 min. The resulting supernatant was used as an enzyme source. All of the extraction steps were carried out at 4 °C. Protein concentration was determined using the BCA kit (bicinchoninic acid; Sigma Co., USA) with bovine serum albumin (BSA) as a protein standard. The rates of hydrolysis by AChE were monitored spectrophotometrically. Each MEPA or standard solution (500 μl) was mixed with an enzyme solution (500 μl). After incubation at 37 °C for 15 min the absorbance was measured at 405 nm immediately after adding an Ellman’s reaction mixture (3.5 ml; 0.5 mM acetylthiocholine iodide, 1 mM DTNB) in a 50 mM sodium phosphate buffer (pH 8.0) to the above reaction mixture. Reading was repeated for 10 min at 2 min intervals to verify that the reaction occurred linearly. The blank reaction was measured by substituting saline for the enzyme. Donepezil was used as standard.
Determination of BChE inhibitory activity
The BChE assay was performed according to the colorimetric method of Ellman’s et al., [40, 44], with some modifications using s-butyrylthiocholine iodide as a substrate. For the enzyme source, the human blood was homogenized in a homogenizer with 5 volumes of a homogenization buffer [10 mM Tris–HCl (pH 7.2), which contained 1 M NaCl, 50 mM MgCl2 and 1 % Triton X-100], and centrifuged at 10,000 g for 30 min. The resulting supernatant was used as an enzyme source. All of the extraction steps were carried out at 4 °C. The rates of hydrolysis by BChE were monitored spectrophotometrically. Each MEPA or standard solution (500 μl) was mixed with an enzyme solution (50 μl) and incubated at 37 °C for 15 min. Absorbance at 405 nm was read immediately after adding an Ellman’s reaction mixture (3.5 ml; 0.5 mM S-butyrylthiocholine iodide, 1 mM DTNB) in a 50 mM sodium phosphate buffer (pH 8.0) to the above reaction mixture. Reading was repeated for 10 min at 2 min intervals to verify that the reaction occurred linearly. The blank reaction was measured by substituting saline for the enzyme. Donepezil was used as standard.
Calculations and statistical analysis
The percentage inhibitions or scavenging of DPPH radicals, hydroxyl radicals, metal chelating, lipid peroxidation, AChE and BChE inhibitory activity of the MEPA were calculated by using the formula:
Where,
A0 is the absorbance of the control, and
A1 is the absorbance of the extract/standard.
The IC50value (the concentration of the extract required to scavenge 50 % of radicals or to inhibit 50 % of enzyme activity) was calculated for the standard and MEPA. The IC50 values of different studies shown in Table 1.
Statistical analyses were carried out in triplicate. All results are expressed as mean ± standard deviation (SD) values average from 3 independent experiments. Free R-software version 2.15.1 (https://cran.r-project.org/bin/windows/base/old/2.15.1/) and Microsoft Excel 2007 (Roselle, IL, USA) were used for the statistical and graphical evaluations.
Results and discussions
Determination of phytoconstituents
Total phenolics
Due to presence of hydroxyl group phenolic compounds possess primary antioxidant property with free radical neutralizing activity [45]. Total phenolic content of MEPA was determined using Folin-Ciocalteu reagent. Total phenolic content of the sample was calculated on the basis of the standard curve for gallic acid and the results were expressed as mg of gallic acid equivalent (GAE)/gm of dried extractives. The phenolic content of MEPA was 58.12 mg of GAE / gm of dried extract. In a previous study the phenolic content of methanolic fruit extract of Phyllanthus emblica L. (Family- Euphorbiaceae) was found to be 120.9 mg of GAE / gm of dried extract which is higher in amount than MEPA [46]. According to epidemiological studies prolonged use of polyphenol enriched diet can provide protection from many chronic diseases including AD [47].
Total flavonoids
Flavonoids are known as reputed antioxidants due to their radical scavenging, metal ion chelating and lipid peroxidation inhibiting activities [48, 49]. Flavonoids may prevent or slow the progression of AD by interfering with the generation and polymerization of amyloid-β peptides into neurotoxic oligomeric aggregates and also by reducing aggregation of tau proteins [50]. Total flavonoids content of MEPA was determined using much known aluminum chloride colorimetric method. Flavonoid content of the samples was calculated on the basis of the standard curve for quercetin and the results were expressed as mg of quercetin equivalent (QE)/g of dried extractives. The flavonoid content of MEPA was 168.24 mg of QE/g of dried extract which is very high in comparison with the flavonoid content of methanolic fruit extract of Phyllanthus emblica L. reported earlier [46].
Antioxidant activity
Total antioxidant capacity
Total antioxidant capacity can be useful to determine nutritional interventions with antioxidant enriched foods on risk and prevention of AD [51]. Total antioxidant capacity was assessed by phosphor-molybdenum method. The phosphor-molybdenum method was based on the reduction of Mo (V1) to Mo (v) by the antioxidant compound and the formation of green phosphate/ Mo (v) complex with a maximal absorption at 695 nm. The MEPA showed considerable antioxidant activity compared to ascorbic acid (AA). At the concentration 100 μg/ml, the absorbance of MEPA and AA were 0.283 and 1.535 respectively. The extract was found to increase the total antioxidant activity with the increasing concentration of the extract. At 600 μg/ml concentration, the absorbance of MEPA and AA were 1.986 and 3.930 respectively depicted in Fig. 1. Total antioxidant capacity of methanol and water extracts of Phyllanthus niruri and Phyllanthus urinaria leaves were performed in a previous study showing that MEPA has less activity than two extracts of these plants [52].

Total antioxidant activity of the MEPA and ascorbic acid at different concentrations
Reducing power capacity
The reducing capacity of a compound is a significant indicator of its potential antioxidant activity because of its ability to break the free radical chain through donation of a hydrogen atom. The Fe3+ reducing power of MEPA was determined by the method of Oyaizu [37]. The MEPA showed reducing activity less than that of AA which is a reference antioxidant. The extract showed increased reducing activity with the increasing concentration of the extract. At concentration 100–600 μg/ml the absorbance of MEPA and AA were 0.045 - 0.872 and 0.984 - 3.950 respectively which shown in Fig. 2. In an earlier study the reducing power capacity of methanolic fruit extract of Phyllanthus emblica L. was performed and the result is quite similar like MEPA which demonstrate that the MEPA has considerable iron reducing capacity [46].

Reducing power capacity of the MEPA and ascorbic acid at different concentrations
DPPH radical scavenging activity
Radical scavenging activities are very important to prevent the deleterious role of free radicals in AD. The model of scavenging the stable DPPH radical is a widely used method to evaluate the free radical scavenging ability of various samples including plant extracts [53]. DPPH antioxidant assay is based on the ability of DPPH, a stable free radical, to decolorize in the presence of antioxidants. The DPPH radical contains an odd electron, which is responsible for the absorbance at 517 nm and also for a visible deep purple color. When DPPH accepts an electron donated by an antioxidant compound, the DPPH is decolorized, which can be quantitatively measured from the change in absorbance and % of scavenging activity is calculated. The activity was increased by increasing the concentration of the sample extract. The antioxidant activity of the extractive of P. acidus was evaluated by DPPH radical scavenging activity. The results of DPPH radical scavenging activity of MEPA and butylated-hydroxyl-toluene (BHT) (standard) are shown in Fig. 3, and the scavenging activity of the MEPA was less than that of BHT. The IC50 of BHT and MEPA were 3.48 μg/ml and 15.62 μg/ml respectively shown in Fig. 4. In comparison with a former published work [46], the IC50 value of MEPA is about equal in value to the earlier report and it is very close to that of standard BHT. These results suggest that MEPA has high radical scavenging activity.

DPPH radical scavenging activity of MEPA and BHT at different concentrations

IC50 (μg/ml) values of BHT and MEPA for DPPH radical scavenging activity
Hydroxyl radical scavenging activity
Hydroxyl radicals are the major reactive oxygen species causing lipid oxidation and enormous biological damage [43]. By incubating Fe3+-EDTA with H2O2 and ascorbic acid at pH 7.4, hydroxyl radicals can be generated in free solution and detected by their ability to degrade 2-deoxy-2-ribose into fragments that on heating with TBA at low pH forms a pink chromogen [43, 54]. In the hydroxyl radical scavenging activity the ability of P. acidus to remove the formed hydroxyl radical in solution was evaluated quantitatively by colorimetric method as included in materials and methods. When plant extractive and the reference compound, (+)-catechin, added to the reaction mixture they removed hydroxyl radicals from the sugar and prevented degradation. Removal of hydroxyl radicals from the reaction mixture decolorized the pink chromogen which can be quantitatively measured from the change in absorbance at 532 nm. The results demonstrate that the MEPA possesses hydroxyl radical scavenging activity in comparison with the standard (+)-catechin and the activity increased by increasing the concentration of the plant extractive which shown in Fig. 5. IC50 value of MEPA and (+)-catechin were 59.74 μg/ml and 15.20 μg/ml respectively shown in Fig. 6. The hydroxyl radical scavenging activity of methanolic fruit extract of Phyllanthus emblica L. was performed in a previous study and the result demonstrated that MEPA has lower hydroxyl radical scavenging activity than Phyllanthus emblica L [46].

Hydroxyl radical scavenging activity of MEPA and (+)-catechin at different concentrations

IC50 (μg/ml) values of (+)-catechin and MEPA for Hydroxyl radical scavenging activity
Metal chelating activity
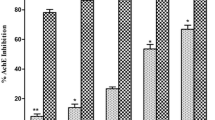
Free radical reactions can be catalyzed by Fe2+ which may lead to oxidative damage of brain found in AD patients. Therefore, substances having metal chelating activity may be used to delay the rate of progression and onset of AD [55]. The chelating activity of MEPA for ferrous ions was measured according to the method as described by J. Sabate [42]. In this method free iron binds in the blood stream and enhancing metal elimination in the urine which reduces the damage done to various organs and tissues, such as the liver and nerves and estimates chelating using Ferrozine (substrate) and ferrous chloride (FeCl2). The inhibitory activity of MEPA increased with increasing concentration and the highest activity (53.03 %) was obtained at 600 μg/ml concentration but at the same concentration BHT (Standard) gave 98.84 % inhibitory activity that shown in Fig. 7. The IC50 value of BHT and MEPA were 16.21 μg/ml and 308.67 μg/ml respectively shown in Fig. 8. In comparison with a previous report on metal chelating activity of methanolic fruit extract of Phyllanthus emblica L. and with standard BHT, MEPA demonstrated lower ferrous ion chelating activity [46].

Metal chelating activity of MEPA and BHT at different concentrations

IC50 (μg/ml) values of BHT and MEPA for Metal chelating activity
Lipid peroxidation inhibition activity
Reactive oxygen species produced by ultraviolet light, ionizing radiation, chemical reactions, and metabolic processes have numerous pathological effects, such as causing lipid peroxidation, protein peroxidation, DNA damage, and cellular degeneration related to a variety of diseases including Alzheimer’s disease [54, 56]. Lipid peroxidation has been reported to be elevated in the brain of AD. During lipid peroxidation, low molecular weight end products, generally malonaldehyde, are formed by oxidation of poly-unsaturated fatty acids that may react with two molecules of thiobarbituric acid to give a pinkish red chromogen [57]. In the lipid peroxidation inhibition activity, the activity of MEPA against non-enzymatic lipid peroxidation in rat brain homogenate was evaluated. Addition of Fe2+- ascorbate to the brain homogenate caused an increase in lipid peroxidation which can be quantitatively measured from the change in absorbance at 532 nm and % of inhibition activity of different concentrations were shown in Fig. 9. The IC50 value of (+)-catechin and MEPA were 58.20 μg/ml and 471.63 μg/ml respectively shown in Fig. 10. The lipid peroxidation inhibition activity of methanolic fruit extract of Phyllanthus emblica L. was performed in an earlier study and the results show that MEPA has considerable inhibitory activity against lipid peroxidation [46].

Lipid peroxidation inhibition activity of MEPA and (+)-catechin at different concentrations

IC50 (μg/ml) values of (+)-catechin and MEPA for Lipid peroxidation inhibition activity
AChE inhibitory activity
Preventing breaking down of ACh is responsible for the elevation of ACh level in the synaptic cleft by inhibition of AChE is the most significant changes observed in AD [42]. The inhibitory activity of MEPA against rat brain AChE was determined by Ellman’s method [44]. This method estimates the level of AChE using acetylthiocholine iodide (substrate) and DTNB. The enzymatic activity was measured by the yellow color compound produced by thiocholine when it reacts with dithiobisnitro benzoate ion. The inhibitory activity of MEPA increased with increasing concentration and the highest activity was obtained at 1000 μg/ml which is 49.52 % but, at the same conditions donepezil gave 97.51 % inhibitory activity shown in Fig. 11. The IC50 value of donepezil and MEPA were 31.83 ± 0.49 μg/ml and 1009.87 ± 19.27 μg/ml respectively shown in Fig. 12. The AChE inhibitory activity of aqueous extract of Emblica officinalis (Family: Euphorbiaceae) was reported in an earlier study [58]. The comparison between IC50 values of aqueous extract of Emblica officinalis and MEPA demonstrates that it has lower acetylcholinesterase inhibitory activity than Emblica officinalis [58].

AChE inhibitory activity of MEPA and Donepezil at different concentrations

IC50 (μg/ml) values of Donepezil and MEPA for AChE inhibitory activity
BChE inhibitory activity
The inhibitory activity of MEPA against human blood BChE was determined by Ellman’s method [44] with some modifications. This method estimates BChE using s-butyrylythiocholine iodide (substrate) and DTNB. The enzymatic activity was measured by the yellow color compound produced by thiocholine when it reacts with dithiobisnitro benzoate ion. The inhibitory activity of MEPA increased with increasing concentration and the highest activity was obtained at 1000 μg/ml which is 54.78 % but, at the same conditions donepezil gave 97.92 % inhibitory activity that shown in Fig. 13. The IC50 value of donepezil (Standard) and MEPA were 16.72 ± 0.21 μg/ml and 499.51 ± 7.42 μg/ml respectively shown in Fig. 14. The comparison between IC50 values of aqueous extract of Emblica officinalis and MEPA demonstrates that it has lower BChE inhibitory activity than Emblica officinalis [58].

BChE inhibitory activity of MEPA and Donepezil at different concentrations

IC50 (μg/ml) values of Donepezil and MEPA for BChE inhibitory activity
Conclusion
This present study gathered experimental evidence that the methanol extract obtained from fruits of P. acidus presented substantial amount of polyphenols and flavonoids and exhibited potential antioxidants and radical scavenging activities which are comparable with the standard (references) drugs by scavenging various free radicals as well as effectively inhibits AChE and BChE activity. Therefore, the plant has promising compounds to be tested as potential drugs for the treatment of diseases resulting from oxidative stress like AD. Due to the presence of significant antioxidant activity of this plant, further studies are underway for isolation and identification of lead compound(s) to prevent the AD and other neurodegenerative disorders.
References
Roy P, Amdekar A, Kumar A, Singh V. Preliminary study of the antioxidant properties of flowers and roots of Pyrostegiavenusta (Ker Gawl) Miers. BMC Complement Altern Med. 2011;11:69.
Ayaz M, Junaid M, Ahmed M, Ullah F, Sadiq A, Ahmad S, Imran M. Phenolic contents, antioxidant and anticholinesterase potentials of crude extract, subsequent fractions and crude saponins from Polygonum hydropiper L. BMC Complement Altern Med. 2014;14:145.
Jeong JM, Kang SK, Lee IH, Lee JY, Jung H, Choi CH. Antioxidant and Chemosensitizing Effects of Flavonoids with Hydroxy and or Methoxy Groups and Structure-Activity Relationship. J Pharm Pharmaceut Sci. 2007;10:537–46.
Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907.
Royer M, Stevanovic T. Study of Coryluscornuta Twig Extracts. Antioxidant, Radical Scavenging, Anti-Enzymatic Activities and Cytotoxicity. Intl J Biot Well Ind. 2012;1:67–84.
Aiyegoro OA, Okoh AI. Preliminary phytochemical screening and In vitro antioxidant activities of the aqueous extract of Helichrysum longifolium DC. BMC Complement Altern Med. 2010;10:21–8.
Gul MZ, Ahmad F, Kondapi AK, Qureshi IA, Ghazi IA. Antioxidant and antiproliferative activities of Abrus precatorius leaf extracts - an in vitro study. BMC Complement Altern Med. 2013;13:53.
Lavrik IN, Eils R, Fricker N, Pforr C, Krammer PH. Understanding apoptosis by systems biology approaches. Mol Biosyst. 2009;5:1105–11.
Rahman I, Biswas SK, Kirkham PA. Regulation of inflammation and redox signaling by dietary polyphenols. Biochem Pharmacol. 2006;72:1439–52.
Zheng W, Wang SY. Antioxidant activity and phenolic compounds in selected herbs. Agric Food Chem. 2001;49:5165–70.
Braca A, Tommasi ND, Bari LD, Pizza C, Politi M, Morelli I. Antioxidant principles from Bauhinia terapotensis. J Natu Prod. 2001;64:892–5.
Sies H. Oxidative stress: Introductory remarks. (Sies H, Ed.). London: Academic; 1985. p. 1–8.
Toborek M, Wasik T, Drozdz M, Klin M, Magner-Wrobel K, Kopieczna-Gzrebieniak E. Effect of hemodialysis on lipid peroxidation and antioxidant system in patients with chronic renal failure. Metabolism. 1992;41:1229–32.
Klahr S. Oxygen radicals and renal diseases. Miner Electrolyte Metab. 1997;23:140–3.
Gulcin I, Bursa E, Şehitoglu MH, Bilsel M, Goren AC. Polyphenol contents and antioxidant activity of lyophilized aqueous extract of propolis from Erzurum, Turkey. Food Chem Tech. 2010;48:2227–38.
Gulcin I, Topal F, Cakmakcı R, Bilsel M, Goren AC, Erdogan U. Pomological Features, Nutritional Quality, Polyphenol Content Analysis, and Antioxidant Properties of Domesticated and 3 Wild Ecotype Forms of Raspberries (Rubusidaeus L.). J Food Sci. 2011;76:585–93.
Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J Physiol Pharmacol. 2003;54:469–87.
Ahmad M, Saeed F, Mehjabeen M, Jahan N. Evaluation of Insecticidal and Anti-oxidant activity of Selected Medicinal Plants. J Pharm Phyt. 2013;2:153–8.
Atta-ur R, Wahab A, Nawas S, Choudhary M. New cholinesterase inhibiting bisbenzylisoquinoline alkaloids from Cocculus pendulus. Chem Pharm Bull. 2004;52:802–6.
Ahmad W, Ahmad B, Ahmad M, Iqbal Z, Nisar M, Ahmad M. In-vitro inhibition of acetylcholinesterase, buty-rylcholinesterase and lipoxygenase by crude extract of Myricaria elegans Royle. J Biol Sci. 2003;11:1046–9.
Jann MW. Preclinical pharmacology of metriphonate. Pharmacotherapy. 1998;18:55–67.
Aisen PS, Davis KL. The search for disease-modifying treatment for Alzheimer’s Disease. Neurology. 1997;48:35–41.
Sung SY, Kang SY, Lee KY, Park MJ, Kim JH, Park JH, Kim YC, Kim J, Kim YC. (+)-α-Viniferin, a stilbenetrimer from Caranga chamlague inhibits acetylcholinesterase. Biol Pharm Bull. 2002;25:125–7.
Bachman DL, Wolf PA, Linn RT. Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham study. Neurology. 1992;42:115–9.
Terry RD. Senile dementia of Alzheimer’s type. Ann Neurol. 1983;14:497–506.
Kalauni SK, Choudhary MI, Khalid A, Manandhar MD, Shaheen F, Atta-ur R, Gewali MB. New cholinesterase inhibiting steroidal alkaloids from the leaves of Sarcococca coriacea of Nepalese origin. ChemPharm Bull. 2002;50:1423–6.
Heinrich M, Teoh HL. Galanthamine from snowdrop-the development of a modern drug against Alzheimer’s disease from local Caucasian knowledge. J Ethnopharmacol. 2004;92:147–62.
Voet D, Voet JG, John W, Jhon S. Serine proteases. In Biochemistry, 2nd edition, USA 1995, 390.
Silman I, Sussman JL. Acetylcholinesterase “Classical” and “non-classical” functions and pharmacology. Curr Opin Pharmacol. 2005;5:293–302.
Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66:137–47.
Chakraborty R, De B, Devanna N, Sen S. Antiinflammatory, antinociceptive and antioxidant activities of Phyllanthus acidus L. extracts. A P J Trop Biomed. 2012;2:S953-S961.
Jagessar RC, Marsa A, Gomesb G. Selective Antimicrobial properties of Phyllanthus acidus leaf extract against Candida albicans, Escherichia coli and Staphylococcus aureus using Stokes Disc diffusion, Well diffusion, Streak plate and a dilution method. Nat Sci. 2008;6:24–38.
Habib MR, Sayeed MA, Rahman MM, Hasan MR, Saha A. In vitro evaluation of cytotoxic, antibacterial, antioxidant and phytochemical screening of petroleum ether extract of Phyllanthus acidus. Int J Pharm Sci Res. 2011;2:875–81.
Lee CY, Peng WH, Cheng HY, Lai MT, Chiu TH. Hepatoprotective effect of Phyllanthus in Taiwan on acute liver damage induced by carbon tetrachloride. Am J Chin Med. 2006;34:471–82.
Rahman MM, Habib MR, Hasan SMR, Sayeed MA, Rana MS. Antibacterial, cytotoxic and antioxidant potential of methanolic extract of Phyllanthus acidus L. Int J Drug Dev Res. 2011;3:154–61.
Singleton VL, Rossi JA. Colorimetry of total phenolics with phosphomolybdic phosphotungstic acid reagents. Am J Enol Vitic. 1965;16:144–58.
Zhishen J, Mengcheng T, Jianming W. The determination of flavonoid contents in mulberry and their scavengingeffects on superoxide radicals. Food Chem. 1999;64:555–9.
Prieto P, Pineda M, Aguilar M. Spectrophotometric quantitation of antioxidant capacity through the formation of a phosphomolybdenum complex, specific application to the determination of vitamin E. Anal Biochem. 1999;269:337–41.
Oyaizu M. Studies on products of browning reactions, antioxidant activities of products of browning reaction prepared from glucose amine. Jap J Nutr. 1986;44:307–15.
Choi HY, Jhun EJ, Lim BO. Application of flow injection-chemilumineacence to the study of radical scavenging activity in plants. Phytother Res. 2000;14:250–3.
Elizabeth K, Rao MNA. Oxygen radical scavenging activity of curcumin. Int J Pharmaceut. 1990;58:237–40.
LIU F. Antioxidative and free radical scavenging activities of selected medicinal herbs. Life Sci. 2000;66:725–35.
Ellman GL, Courtney KD, Andres V, Feather-stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95.
Doctor BP, Toker L, Roth E, Silman I. Microtiter assay for acetylcholinesterase. Anal Biochem. 1987;166:399–403.
Sabate J. The contribution of vegetarian diets to healt and disease: A paradiagram Shift. Am J Clin Nutr. 2003;78:502–7.
Aurand LW, Boone NH, Gidding GG. Superoxide and singlet oxygen in milk lipid peroxidation. J Dairy Sci. 1977;60:63–369.
Halliwell B. Free radicals, antioxidants, and human disease: curiosity, cause, or consequence. Lancet. 1994;344:721–4.
Kumarasamy Y, Byres M, Cox PJ, Sarkar SD. Screening seeds of some Scottish plants for free radical scavenging activity. Phytothe Res. 2007;21:615–21.
Asaduzzaman M, Uddin MJ, Kader MA, Alam AHMK, Rahman AA, Rashid M, Kato K, Tanaka T, Takeda M, Sadik G. In vitro acetylcholinesterase inhibitory activity and the antioxidant properties of Aegle marmelos leaf extract, implications for the treatment of Alzheimer’s disease. Psychogeriatrics. 2014;14:1–10.
Dhalwal K, Shinde VM, Namdeo AG. Antioxidant profile and HPTLC densitometric analysis of umbelliferone and psoralen in Aegle marmelos. Pharma Biol. 2008;46:266–72.
Liua X, Zhaoa M, Wanga J, Yangb B, Jiangb Y. Antioxidant activity of methanolic extract of emblica fruit (Phyllanthus emblica L.) from six regions in China. J F Comp Anal. 2008;21:219–28.
Arts ICW, Hollman PCH. Polyphenols and disease risk in epidemiologic studies. Am J Clin Nutr. 2005;81:317S–25S.
Oboh G, Puntel RL, Rocha JBT. Hot pepper (Capsicum annuum, Tepin and Capsicum chinese, Habanero) prevents Fe2+-induced lipid peroxidation in brain in vitro. Food Chem. 2007;102:178–85.
Baptista FI, Henriques AG, Silva AMS, Wiltfang J, da Cruz e Silva OA. Flavonoids as Therapeutic Compounds Targeting Key Proteins Involved in Alzheimer’s Disease. ACS Chem Neurosci. 2014;5:83–92.
Kusano C, Ferrari B. Total Antioxidant Capacity: a biomarker in biomedical and nutritional studies. J Cell Mol Biol. 2008;7:1–15.
Nimmi I, Jahan IA, Hossain MH, Uddin MB, Rana MS, Haq MM. A Comparative Study on Antioxidant Properties of Two Phyllanthus Species Growing in Bangladesh. Dhaka Univ J Pharm Sci. 2012;11:191–7.
Bartzokis G, Sultzer D, Cummings J, Holt LE, Hance DB, Henderson VW, Mintz J. In Vivo Evaluation of Brain Iron in Alzheimer Disease Using Magnetic Resonance Imaging. Arch Gen Psychiatry. 2000;57:47–53.
Shekhar C, Kumar S. Dual-cholinesterase inhibition by an aqueous extract of Emblica officinalis fruit and its mode of inhibition by kinetic study. Adv Biomed Pharma. 2015;2:7–12.
Acknowledgement
The authors acknowledge the Department of Pharmacy, Southeast University, Dhaka, Bangladesh for financial support. We also thank to Dr. S.M. Abdur Rahman, Professor, Faculty of Pharmacy, University of Dhaka to provide laboratory facilities and National Herbarium, Dhaka, Bangladesh for the identification of the plant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MM and MA designed the study and carried out the tests under the supervision of MMR. MSH carried out the lipid peroxidation inhibition assay. JS carried out acetylcholinesterase and butyrylcholinesterase inhibitory activities. SMAR helped to coordinate the biological assay and draft the manuscript. MR checked the grammatical errors and corrected the final manuscript. All authors read and approved the final manuscript.
Md. Moniruzzaman and Md. Asaduzzaman contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Moniruzzaman, M., Asaduzzaman, M., Hossain, M.S. et al. In vitro antioxidant and cholinesterase inhibitory activities of methanolic fruit extract of Phyllanthus acidus. BMC Complement Altern Med 15, 403 (2015). https://doi.org/10.1186/s12906-015-0930-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12906-015-0930-y